

Abstract
Microbial sulfate reduction is an important metabolic activity in petroleum hydrocarbon (PHC)-contaminated aquifers. We quantified carbon source-enhanced microbial SO42− reduction in a PHC-contaminated aquifer by using single-well push-pull tests and related the consumption of sulfate and added carbon sources to the presence of certain genera of sulfate-reducing bacteria (SRB). We also used molecular methods to assess suspended SRB diversity. In four consecutive tests, we injected anoxic test solutions (1,000 liters) containing bromide as a conservative tracer, sulfate, and either propionate, butyrate, lactate, or acetate as reactants into an existing monitoring well. After an initial incubation period, 1,000 liters of test solution-groundwater mixture was extracted from the same well. Average total test duration was 71 h. We measured concentrations of bromide, sulfate, and carbon sources in native groundwater as well as in injection and extraction phase samples and characterized the SRB population by using fluorescence in situ hybridization (FISH) and denaturing gradient gel electrophoresis (DGGE). Enhanced sulfate reduction concomitant with carbon source degradation was observed in all tests. Computed first-order rate coefficients ranged from 0.19 to 0.32 day−1 for sulfate reduction and from 0.13 to 0.60 day−1 for carbon source degradation. Sulfur isotope fractionation in unconsumed sulfate indicated that sulfate reduction was microbially mediated. Enhancement of sulfate reduction due to carbon source additions in all tests and variability of rate coefficients suggested the presence of specific SRB genera and a high diversity of SRB. We confirmed this by using FISH and DGGE. A large fraction of suspended bacteria hybridized with SRB-targeting probes SRB385 plus SRB385-Db (11 to 24% of total cells). FISH results showed that the activity of these bacteria was enhanced by addition of sulfate and carbon sources during push-pull tests. However, DGGE profiles indicated that the bacterial community structure of the dominant species did not change during the tests. Thus, the combination of push-pull tests with molecular methods provided valuable insights into microbial processes, activities, and diversity in the sulfate-reducing zone of a PHC-contaminated aquifer.
Dissimilatory microbial SO42− reduction is an important metabolic activity in many reduced environments, such as marine sediments (22), anaerobic sludge (28), and contaminated aquifers (25). This process is mediated by a metabolically diverse group of microorganisms: the sulfate-reducing bacteria (SRB) (31, 35, 51). SRB were found to grow on environmental contaminants such as petroleum hydrocarbon (PHC) constituents (e.g., benzene, toluene, ethylbenzene, xylenes, naphthalene, phenanthrene, and alkanes) and halogenated compounds (15, 55). A survey of 38 PHC-contaminated aquifers revealed that on average, SO42− reduction was responsible for 70% of PHC attenuation (53).
During the degradation of PHC, low-molecular-weight organic acids such as acetate, propionate, and butyrate are intermediates (11), which in turn may serve as carbon sources for SRB. Unfortunately, little is known about the role that such low-molecular-weight organic acids may play in the community pathway of PHC degradation in contaminated aquifers. In marine sediments, organic acids derived primarily from fermentation serve as the SRB's main carbon source (33, 45). Numerous studies have shown that all SRB genera preferentially degrade certain organic acids and cannot degrade others (19, 24, 50-52). Only a few SRB genera are known to readily degrade a wide range of organic acids; e.g., Desulforhabdus amnigenus is able to consume lactate, acetate, butyrate, and propionate (14). In general, lactate seems to be the most generic carbon source for SRB.
In the laboratory, microbial SO42− reduction was investigated by using batch and column studies (13, 20, 37, 48). However, over the last decade it has become increasingly apparent that an accurate assessment of SO42− reduction in aquifers requires appropriate in situ test methods (17, 26, 27). Recently, single-well “push-pull” tests (PPTs) have been used for the in situ quantification of microbial activities in PHC-contaminated aquifers (21, 36, 41). In a PPT, a test solution that contains a nonreactive, conservative tracer and one or more reactive solutes (reactants) is injected (pushed) into the aquifer through an existing well. During an initial incubation period (i.e., a rest phase without pumping), indigenous microorganisms ideally consume reactants and generate metabolic products. Thereafter, the test solution-groundwater mixture is extracted (pulled) from the same location, and the concentrations of tracer, reactants, and products are analyzed. Rates of microbial activities are then determined by comparing the breakthrough curves of tracer and reactants (18, 44).
Recently, we successfully employed PPTs to quantify microbial SO42− reduction concomitant with PHC degradation in a contaminated aquifer (42). Stable sulfur isotope fractionation in extracted, unconsumed SO42− during those tests provided strong evidence that SO42− reduction was microbially mediated.
To our knowledge, no study has been published specifically addressing the diversity of SRB in PHC-contaminated aquifers. Nevertheless, this information is essential for our understanding of the biogeochemical processes that are intimately linked to bacterial diversity. Direct information on SRB communities may be obtained by using laboratory molecular methods such as fluorescence in situ hybridization (FISH) (4) and PCR with subsequent denaturing gradient gel electrophoresis (DGGE) (30). These methods have been used previously to investigate SRB communities in marine sediments (12), seawater (47), and anaerobic bioreactors (49).
Unfortunately, we do not know whether the introduction of reactants during PPTs changes the microbial community in the subsurface. If it does, rate coefficients determined by using this method may not reflect the activities of the native community. Analysis of the suspended population during a PPT may provide some information on this issue, even though suspended and attached populations may be dissimilar (1).
The purpose of this research was to assess SRB diversity in a PHC-contaminated aquifer by using macroscopic measurements of activities as well as molecular analyses. We quantified carbon source (acetate, propionate, butyrate, and lactate) -enhanced microbial SO42− reduction by using PPTs and related SO42− and carbon source consumption to the presence of certain SRB genera. These findings were compared with results from molecular analyses (FISH and PCR-DGGE), which we used to assess suspended SRB diversity and to monitor population changes of suspended bacteria during PPTs. Stable sulfur isotope analyses of extracted, unconsumed SO42− were used to determine isotope enrichment factors, which served as indicators of microbial SO42− reduction.
MATERIALS AND METHODS
Field site.
The study was conducted in a heating oil-contaminated aquifer in Studen, Switzerland, which is undergoing remediation by monitored natural attenuation and was characterized in detail by Bolliger et al. (7). The PPTs described in this paper were conducted in monitoring well PS3, which is located within the contaminant source zone (free-phase PHC present). Well PS3 is constructed of polyvinyl chloride casing (11.5-cm inner diameter) and partially penetrates the aquifer to a depth of ≈1 m below the groundwater table. Groundwater in PS3 exhibited reduced conditions and contained up to 1 mg of dissolved PHC liter−1 (7). Previous studies have shown that PS3 is located within a transition zone where both SO42−-reducing and methanogenic conditions are found (7, 8).
PPTs and sample collection procedures.
To quantify rates of microbial SO42− reduction concomitant with the degradation of either propionate, butyrate, lactate, or acetate, we performed four PPTs (PPTpr, PPTbu, PPTla, and PPTac, respectively) over a 6-month period from May to October 2000 in a fashion similar to that described by Schroth et al. (42). Test solutions were prepared by collecting groundwater in 500-liter plastic carboys and adding Br− (as KBr) as a nonreactive, conservative tracer and SO42− (as K2SO4) as a reactant to achieve final concentrations of ≈0.5 mM Br− and ≈1.0 mM SO42− (Table 1). As carbon sources, we added either propionate, butyrate, acetate (prepared from their respective sodium salts), or lactate (prepared from a 50% dl-sodium lactate solution) to achieve final concentrations of ≈2.0 mM. In all PPTs, the carboys were continuously sparged with nitrogen gas to minimize O2 dissolution from air into test solutions during preparation and subsequent injection.
TABLE 1.
Summary of experimental conditions during four PPTs performed to evaluate microbial SO42− reduction concomitant with carbon source degradation in a PHC-contaminated aquifer
| Test | Carbon source injected | Carbon source concn (mM) | SO42− injection concn (mM) | Br− injection concn (mM) | Injection vol (liters) | Injection duration tinj (h) | Initial incubation period (h) | Total extracted vol (liters) | Total test duration (h) | Groundwater temp (°C) | SO42− background concn (mM) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PPTpr | Propionate | 1.89 | 0.92 | 0.47 | 1,000 | 0.72 | 21.9 | 1,000 | 70.4 | 12.5 | 0.05 |
| PPTbu | Butyrate | 2.19 | 1.02 | 0.51 | 1,000 | 1.67 | 24.6 | 1,000 | 72.9 | 12.7 | 0.03 |
| PPTla | Lactate | 2.05 | 1.04 | 0.53 | 1,000 | 0.95 | 21.0 | 1,000 | 70.0 | 13.1 | 0.04 |
| PPTac | Acetate | 2.36 | 1.15 | 0.58 | 1,000 | 0.93 | 21.9 | 1,000 | 71.3 | 14.5 | 0.02 |
For each PPT, injection of 1,000 liters of test solution into PS3 began at time t = 0 h and was completed within 0.72 to 1.67 h by using gravity drainage. After an average initial incubation period of 22 h, we extracted a total of 1,000 liters of test solution-groundwater mixture (batches of 300 to 400 liters each per day) during further incubation for 2 additional days. The average total test duration was 71 h.
Water samples for chemical and biological analyses were obtained during the collection of groundwater in carboys (background concentrations), injection of test solutions (injection concentrations), and at regular intervals during the extraction phases of the PPTs. Specifically, samples for the analysis of Br−, SO42−, and organic acids were filtered in the field by using 0.45-μm polyvinylidene fluoride filters (Millipore, Bedford, Mass.) and stored in 12-ml plastic vials. Samples for the determination of CH4 concentrations were collected without headspace in 117-ml serum bottles by using butyl rubber stoppers. Samples for sulfur isotope measurements in unconsumed SO42− were collected in 1-liter glass bottles acidified with 2 ml of 32% HCl. All samples were stored at 4°C prior to analysis.
For total cell counts and FISH, 50 ml of unfiltered water was collected in sterile Falcon tubes. During PPTac, unfiltered water samples (250 ml each) were collected in sterilized glass bottles for use in subsequent PCR-DGGE analysis. All samples for biological analyses were immediately placed on ice until further processing in the laboratory.
Analytical methods.
Bromide, SO42−, and organic acid concentrations were determined by using a DX-320 ion chromatograph system equipped with an electrical conductivity detector and an EG40 eluent gradient generator (Dionex, Sunnyvale, Calif.). The following KOH eluent gradient was used: 0 to 7 min, 1 mM KOH; 7 to 25 min, 1 mM to 25 mM KOH; 25 to 28 min, 25 mM to 60 mM KOH; 28 to 28.1 min, 60 mM to 1 mM KOH; and 28.1 to 32 min, 1 mM KOH. Methane was quantified by using the headspace method described by Bolliger et al. (7).
Stable sulfur isotope measurements were conducted as described previously (42). Data are reported in the conventional δ notation relative to the Vienna-Canyon Diabolo Troilite (V-CDT) standard by using:
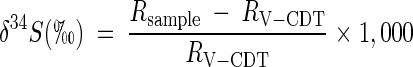 |
(1) |
where Rsample and RV-CDT are the 34S/32S sulfur isotope ratios in the sample and V-CDT standard, respectively.
Determination of isotope enrichment factors.
Sulfur isotope fractionation was quantified by computing isotope enrichment factors ɛ (in per mille). In a closed system, enrichment factors can be determined by fitting Rayleigh distillation equations to experimental data (29). Specifically, enrichment factors of extracted, unconsumed SO42− may be determined from measured δ34S values by using (10):
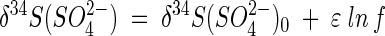 |
(2) |
where f is the fraction of extracted, unconsumed SO42− and δ34S(SO42−)0 is the initial isotope composition of SO42− in the injected test solution. We corrected values of δ34S(SO42−) to account for the isotope composition of background SO42− (by using Br− breakthrough data as a measure of dilution between test solutions and native groundwater).
Determination of first-order rate coefficients.
First-order rate coefficients for SO42− reduction and carbon source degradation were determined from SO42− and carbon source consumption by the method of Haggerty et al. (18). This method is based on an analysis of tracer and reactant transport in the diverging-converging radial flow field surrounding a monitoring well during a PPT. Assuming a first-order type reaction d Cr /d t = −k Cr, where Cr is the reactant concentration and t is the time, the rate coefficient k can be determined from (18):
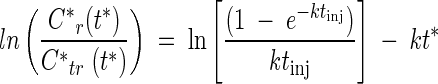 |
(3) |
where C* is relative concentration (i.e., measured concentration divided by the concentration in the injected test solution), subscripts r and tr denote reactant and tracer, respectively, t* is time elapsed since the end of the test solution injection, and tinj is the duration of the test solution injection. A nonlinear least-squares routine was used to fit equation 3 (both slope and intercept) to experimental breakthrough data to obtain estimates of first-order rate coefficients. The standard deviation of k was computed from the variance of the estimated k as described by Schroth et al. (41).
Calculation of stoichiometric ratios.
Theoretical stoichiometric ratios (moles of carbon source per mole of SO42−) for incomplete carbon source degradation (to acetate) may be obtained from the following reactions:
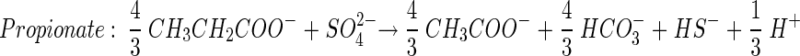 |
(4) |
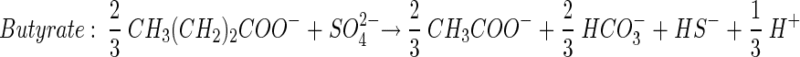 |
(5) |
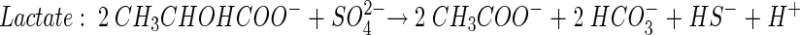 |
(6) |
The theoretical stoichiometric ratio for acetate degradation was obtained from:
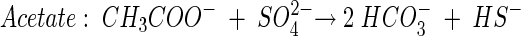 |
(7) |
Furthermore, we obtained theoretical stoichiometric ratios for complete carbon source degradation of propionate, butyrate and lactate by combining equations 4 to 6 individually with equation 7.
Actual stoichiometric ratios were calculated individually for each water sample by using measured concentrations of reactants and tracer. Alternatively, we calculated stoichiometric ratios from total masses recovered during the PPTs. In these calculations we assumed that added SO42− was used exclusively for the degradation of added carbon sources during the PPTs.
Cell counts and in situ hybridization.
Cell counts and FISH were performed on samples collected during PPTla and PPTac. Total cell counts were conducted by using 4′,6′-diamidino-2-phenylindole (DAPI) staining (54). For in situ hybridization, we used the indocarbocyanine (Cy3)-labeled 16S rRNA oligonucleotide probes (all purchased from MWG Biotech, Ebersberg, Germany) EUB338 to target Bacteria (3), Arch915 (46) for Archaea, SRB385 (2) plus SRB385-Db (35) for SRB, DSV698 plus DSV1292 for Desulfovibrio, DSB985 for Desulfobacter, and probe 660 for Desulfobulbus (28). Water samples for DAPI and FISH counts were processed within a few hours after sampling by centrifugation at 2,500 × g for 5 min and resuspension of the debris or cell pellet in 1 ml of 4% paraformaldehyde in phosphate-buffered saline (130 mM NaCl, 7 mM Na2HPO4, 3 mM NaH2PO4).
Samples were further processed as described before (54); 20 μl from each fixed and dispersed sample was spotted onto ethanol-washed slides. Drying, hybridizations with oligonucleotide probes, DAPI staining and washing were performed under standard conditions (54). Formamide concentrations in the hybridization mix were 30% for probe EUB338, 20% for Arch915, SRB385, SRB385-Db, and DSB985, 35% for DSV698 and DSV1292, and 60% for probe 660. Sodium chloride concentrations in the wash buffer were 112 mM for probe EUB338, 250 mM for Arch915, SRB385, SRB385-Db, and DSB985, 88 mM for DSV698 and DSV1292, and 15.6 mM for probe 660. The slides were mounted, and visually detectable cells were counted (54). Counting results were corrected by subtracting autofluorescent cells.
DNA extraction and PCR-DGGE.
To concentrate suspended bacterial cells, groundwater (250 ml) was filtered through 0.22-μm polyvinylidene fluoride filters (Millipore, Bedford, Mass.), followed by storage of the filters in 1.5 ml of lysis buffer (50 mM Tris [pH 8], 50 mM EDTA, 50 mM NaCl) at −20°C. Following the addition of ≈0.7 g of glass beads (0.10 to 0.11 mm in diameter) to the lysis buffer or filters, DNA was extracted by bead beating in a FastPrep 120 (Savant Instruments, Inc., Holbrook, N.Y.) for 15 s at 4.5 m s−1. After brief centrifugation to settle the filter pieces, the buffer-DNA supernatant was transferred into a new tube, and the extraction of the filters was repeated with 0.5 ml of lysis buffer. Approximately 10 mg of lysozyme ml−1 was added to the combined buffer-DNA solutions, and the samples were incubated at room temperature for 10 min. Then 100 μl of sodium dodecyl sulfate (SDS) (20%) and 20 μl of proteinase K (100 μg ml−1) were added to each sample, followed by incubation for 30 min at 37°C and 10 min at 55°C. DNA was further purified by chloroform-phenol extraction and isopropanol precipitation (38), and resuspended in 100 μl of water. Further cleanup of the DNA was performed by using the QIAquick gel extraction kit (Qiagen AG, Basel, Switzerland). DNA was quantified by measuring absorbance at 260 nm and stored at −20°C.
PCR of partial 16S rRNA genes was performed by using two sets of primers. Primers UNIV 907 r-gc (5′-GC clamp-CCG TCA ATT CCT TTR AGT TT-3′) and SRB 385-f (5′CCT GAC GCA GCG ACG CCG-3′) (5) were used to amplify approximately 523 bp of the 16S rRNA gene from SRB as well as some gram-positive bacteria and other δ-proteobacteria (39). A second set of primers, BAC 968 f-gc (5′-GC clamp-AAC GCG AAG AAC CTT AC-3′) and BAC 1401 r (5′-CGG TGT GTA CAA GAC CC-3′), were used to amplify approximately 434 bp of the 16S rRNA gene sequence from most Bacteria (16). DGGE of PCR products was performed in a denaturing gradient of 30 to 55% at 200 V for 3 h as described previously (43). DNA banding patterns were digitized and photographed by using the GelDoc 2000 system and QuantityOne software (Bio-Rad Laboratories, Hercules, Calif.).
RESULTS
PPTs.
Breakthrough curves for Br−, SO42−, and carbon sources showed a gradual decline in C* during PPT extraction phases as extracted test solution was increasingly diluted with native groundwater (Fig. 1). Relative SO42− and carbon source concentrations were smaller than relative Br− concentrations during all PPT extraction phases. This difference is significant, since the error of the IC measurements is smaller than 5%.
FIG. 1.

Extraction phase breakthrough curves for Br−, SO42−, propionate, butyrate, lactate, and acetate during (a) PPTpr, (b) PPTbu, (c) PPTla, and (d) PPTac.
To account for SO42− contained in native groundwater (on average 0.035 mM), the relative SO42− concentrations shown were corrected by using Br− breakthrough curves as a measure of the dilution of test solution with native groundwater, assuming constant SO42− background concentrations for each test (41). Acetate, propionate, butyrate, and lactate concentrations were below the detection limit (≈5 μM) in native groundwater from well PS3. During the extraction phases of all PPTs, we recovered 43% ± 7% (average ± standard deviation) of the injected Br− mass and 33% ± 5% of the injected SO42− mass (computed by integrating solute breakthrough curves shown in Fig. 1). Furthermore, 43% of propionate, 35% of butyrate, 39% of lactate, and 21% of acetate were recovered. Acetate was also detected during the extraction phase of PPTpr (Fig. 1a). Concentrations of acetate decreased linearly from 40 μM in the beginning to 10 μM at the end of PPTpr. In all other PPTs, no intermediate organic acids were detected. Methane concentrations were 0.48 ± 0.61 mM in native groundwater of well PS3 and 0.27 ± 0.19 mM in the injection solutions and remained essentially constant during all PPT extraction phases, with concentrations at 0.45 ± 0.26 mM (not shown).
Rate coefficients and stoichiometric ratios.
Computed values of k for SO42− reduction (ksulfate) were lowest for PPTbu and highest for PPTla and PPTpr (Table 2). Standard deviations ranged between 4.2 and 7.8% of the value of ksulfate. For carbon source degradation, k values were highest for PPTac and lowest for PPTla with intermediate values for PPTpr and PPTbu and standard deviations between 3.6 and 10.0% of the respective first-order rate coefficient.
TABLE 2.
First-order rate coefficients k, stoichiometric ratios for SO42− reduction concomitant with the degradation of carbon sources, and isotope enrichment factors during microbial SO42− reduction in four PPTs
| Test | First-order rate coefficient k ± SD (day−1)
|
Stoichiometric ratiosa
|
Isotope enrichment factor ɛ ± SD (‰) | |||
|---|---|---|---|---|---|---|
| SO42− | Carbon source | Actual ± SD | Theoretical
|
|||
| Completeb | Incompletec | |||||
| PPTpr | 0.29 ± 0.02 | 0.19 ± 0.02 | 1.51 ± 0.25 | 0.57 | 1.33 | 20.2 ± 1.2 |
| PPTbu | 0.19 ± 0.01 | 0.14 ± 0.01 | 1.63 ± 0.25 | 0.40 | 0.66 | 25.7 ± 1.8 |
| PPTla | 0.32 ± 0.03 | 0.13 ± 0.01 | 0.97 ± 0.15 | 0.66 | 2.00 | 16.1 ± 0.8 |
| PPTac | 0.24 ± 0.01 | 0.60 ± 0.06 | 3.49 ± 0.51 | 1.00 | — | 20.8 ± 1.2 |
Expressed as moles of carbon source utilized per mole of SO42−.
Complete, complete oxidation to CO2.
Incomplete, incomplete oxidation to acetate.
Actual stoichiometric ratios changed little during the PPTs, with only a small increase, and therefore only average values are displayed (Table 2). Per 1 mol of SO42−, 1.51 ± 0.25 mol of propionate, 1.63 ± 0.25 mol of butyrate, 0.97 ± 0.15 mol of lactate, and 3.49 ± 0.51 mol of acetate were consumed. Similarly, when stoichiometric ratios were calculated from total masses recovered during the PPTs (see previous section), 1.49 mol of propionate, 1.66 mol of butyrate, 0.98 mol of lactate, and 3.44 mol of acetate were consumed per 1 mol of SO42−. Actual stoichiometric ratios were higher than theoretical stoichiometric ratios, assuming both complete and incomplete oxidation for all carbon sources, except for lactate, for which the actual value was between the two theoretical values.
If we assume complete oxidation of the carbon sources by SRB, the stoichiometry of the reactions would indicate that SO42− reduction accounted for 41% ± 12% of degraded propionate, 25% ± 4% of the butyrate, 69% ± 11% of the lactate, and 29% ± 5% of the acetate. These numbers represent the minimum amount of carbon sources that were consumed by SRB when assuming that injected SO42− was used exclusively for degradation of these carbon sources. If we assume incomplete oxidation of the carbon sources by SRB, the stoichiometry of the reactions would indicate that SO42− reduction accounted for 96% ± 27% of the degraded propionate, 41% ± 7% of the butyrate, and 210% ± 33% of the lactate. Assimilation of carbon and sulfur by SRB was not taken into account in these calculations, since it was assumed to be low (51).
Isotope enrichment factors.
Sulfur isotope fractionation in unconsumed SO42− was observed during extraction phases of all PPTs. Values of δ34S(SO42−) were 10.7‰ ± 0.2‰ in test solutions [δ34S(SO42−)0] and increased up to 20.4‰ ± 5.0‰ on average during PPT extraction phases. Computed isotope enrichment factors (equation 2) ranged from 16.1‰ ± 0.8‰ to 25.7‰ ± 1.8‰ (Table 2).
Cell counts and in situ hybridization.
Total cell numbers in PPTla and PPTac (DAPI) ranged from 6.6 × 104 to 1.51 × 105 cells ml−1. Percentages of cells hybridizing with probe EUB338 ranged from 16 to 33% and with probe Arch915 from 27 to 44% of total (DAPI-stained) cells during PPTla (Fig. 2a) and PPTac (Fig. 2b). Probes SRB385 and SRB385-Db together detected between 11 and 24% of total cells (Fig. 2). In both PPTs, lower percentages of Bacteria and SRB were determined in the background than during extraction phases.
FIG. 2.

DAPI fractions of cells in (a) PPTla and (b) PPTac hybridizing with fluorescent probes. BG, background. Error bars indicate 1 standard deviation.
Hybridizations with the genus-specific SRB probes showed that all of the targeted genera were present. Percentages of Desulfobulbus spp. ranged from 2.6 to 8.0% of total cells, Desulfovibrio spp. from 2.6 to 7.6%, and Desulfobacter spp. from 0.0 to 6.0%. Proportions of SRB genera changed little during both PPTs. However, Desulfovibrio spp. and Desulfobulbus spp. signals increased during PPTla, and those of all three genera increased during PPTac compared to the background. The three genera investigated make up 30 to 59% of all SRB (as determined by probes SRB385 plus SRB385-Db) in PPTac and 56 to 104% of all SRB in PPTla.
PCR-DGGE.
DGGE of PCR products generated from both primer sets resulted in distinct profiles, which exhibited approximately eight dominant bands each and an underlying smear of DNA that represented nondominant phylotypes (Fig. 3). Apart from slight band intensity differences, little change could be detected between the DGGE profiles of DNA extracted from native groundwater from well PS3 (background) and samples taken during PPTac, regardless of the primer set used (as an example, profiles from the 1.0 extracted volume/injected volume sample are shown in Fig. 3).
FIG. 3.

DGGE profiles of DNA extracted from groundwater during PPTac, generated with (a) SRB and (b) EUB primer sets. BG, background; 1.0 = 1.0 extracted volume/injected volume sample.
DISCUSSION
PPTs.
Lower relative SO42− and carbon source concentrations compared to relative Br− concentrations throughout all PPTs (Fig. 1) indicated that SO42− and carbon sources were consumed during those tests, presumably due to microbial activity. Differences between recovered relative Br− and SO42− masses (10.4% ± 2.3% on average) and differences between relative Br− and carbon source masses (propionate 9%, butyrate and lactate 6%, and acetate 15%) illustrate that total test durations (Table 1) were sufficiently long to allow detectable SO42− and carbon source consumption during the tests. Conversely, mass recovery of injected Br− tracer in the tests (36 to 52%) was poor. This was due to a fairly high average pore water velocity (≈0.4 m day−1) at the site (42). Thus, during the PPTs, a significant portion of test solution migrated beyond the radius of influence of PS3. However, it is important to note that complete tracer mass recovery is not required during PPTs for an accurate quantification of rate coefficients (18). Sulfide and Fe(II) concentrations were routinely measured in all PPTs according to Schroth et al. (42). However, precipitation of these ions as FeS or FeCO3 obscures true sulfide and Fe(II) concentrations, rendering these data useless for quantification purposes.
Rate coefficients and stoichiometric ratios.
We determined values of ksulfate that were up to seven times higher than ksulfate values obtained from PPTs in the same location in which only SO42− was added (42). Therefore, addition of carbon sources in the current PPTs substantially enhanced microbial SO42− reduction in the vicinity of well PS3. Hence, the rate coefficients we measured do not represent indigenous conditions, even though they are within the range of values reported elsewhere in the literature (for a discussion, see Schroth et al. [42]). A variety of environmental factors may evoke variations of ksulfate between tests, e.g., groundwater temperature. However, in this case groundwater temperature variations cannot explain differences in ksulfate because the temperature varied only by ±1.3°C between tests (Table 1). Therefore, differences between ksulfate in the PPTs seemed to be due largely to distinct carbon source degradability. Hence, ksulfate values must be discussed in combination with rate coefficients for carbon sources (Fig. 4) and stoichiometries (Table 2).
FIG. 4.

First-order rate coefficients for SO42− reduction and carbon source degradation during PPTpr, PPTbu, PPTla, and PPTac. Error bars indicate 1 standard deviation.
Lactate enhanced SO42− reduction the most, and hence, SRB preferred lactate over the other added carbon sources. This finding agrees with the results of other researchers, who found that most SRB are able to grow on lactate (19, 24, 50-52). Conversely, the acetate degradation rate coefficient was by far the highest for all the carbon sources, but SO42− reduction accounted for only ≈30% of total acetate degradation in PPTac. Therefore, most of the acetate was probably consumed by Archaea that were an important part of the suspended population (Fig. 2). Their activity was indicated by the presence of CH4.
If we assume complete degradation of the carbon sources by SRB, the observed SO42− consumption would only account for a part of the total carbon source consumption during the PPTs. Therefore, incomplete degradation of propionate, butyrate, and lactate by SRB may have been a dominant degradation pathway (equations 4 to 6). Fermentation processes (acetogenesis) may also have been important for propionate, butyrate, and lactate degradation. For example, even when we assume incomplete degradation of butyrate by SRB, this only accounts for 41% of total butyrate degraded during PPTbu. Fermentation may well have been responsible for the missing 59% (23).
Isotope enrichment factors.
Enrichment factors computed for our tests (Table 2) were in the same range as those determined previously in the same aquifer during PPTs without carbon source addition (22.8‰ ± 1.7‰ and 20.2‰ ± 2.8‰) (42). However, in this study, variations in ɛ between tests were higher. Interestingly, our data also agreed with enrichment factors determined for a mixed, toluene-degrading, SO42−-reducing batch culture for which the inoculum was collected from the same aquifer (19.8 to 28.2‰) (9). Furthermore, enrichment factors agreed well with ɛ values obtained by others for microbial SO42− reduction in different environments, and they were within a range that is indicative of microbial SO42− reduction (for a discussion, see Schroth et al. [42]). Thus, these data clearly suggest that observed SO42− consumption during the PPTs was microbially mediated.
In situ hybridization.
Numbers of suspended cells associated with the domain Bacteria (EUB338, 16 to 33%) during PPTla and PPTac were similar to Bacteria numbers determined in a previous study for the same aquifer (13 to 32%) (8). Archaea numbers, however, were higher (Arch915, 27 to 44% in our samples compared to 9 to 31% previously [8]). Counts of SRB (SRB385 plus SRB385-Db) detected in this study (11 to 24%) were higher than those detected by other authors using the same method in different environments, e.g., in activated sludge, anaerobic biofilms, and bulk soil (1 to 12% of total bacteria) (5, 28, 54). The reasons for this difference may include the environment that we investigated and our choice of probes (SRB385 and SRB385-Db in combination), which may likely have resulted in higher detection rates. Moreover, we are aware that both probes also detect a range of other anaerobic bacteria (28). Thus, the numbers reported here likely overestimate true SRB numbers.
For both Bacteria and SRB and during both tests, differences between numbers of cells in extraction phase samples compared to background samples were in the same range. Therefore, the increase in the Bacteria numbers may have been due to an increase in SRB numbers. This agrees with an increase in counts with the genus-specific SRB probes during the tests. Interestingly, counts of Desulfovibrio (2.6 to 7.6%) and Desulfobacter (0 to 6%) in this PHC-contaminated environment were in the same range as was found in activated sludge, another freshwater environment (2.8 to 5.2% and 1.8%, respectively [28]). However, the same authors detected Desulfobulbus numbers below the detection limit (<0.1%), compared to 2.6 to 8% in our samples, which may be due to the different environments examined. Differences in cell counts between the tests, especially with regard to Desulfobacter spp., are difficult to interpret and may be caused by naturally occurring fluctuations of the populations due to changes of environmental conditions.
PCR-DGGE.
The presence of several bands in DGGE profiles indicated a diverse bacterial population in groundwater near well PS3. Although we cannot statistically test the significance of this observation, our results suggest that the suspended bacterial community of the dominant species remained the same during PPTac (Fig. 3). At first, this finding does not seem to agree with the FISH results, as FISH indicated higher SRB and Bacteria activity during extraction phases compared to background samples. However, FISH detects the active portion of the microbial population, which may change during a PPT (increase in RNA content), while DGGE profiles reveal the population patterns, which appeared to remain unaltered.
Comparison of chemical data with molecular analyses.
Although the carbon sources that we added in the current PPTs may not be important SRB substrates in situ in this aquifer, their consumption in the tests may be related to the presence of certain SRB genera and hence provide information on the bacterial community. Since most SRB are unable to readily degrade all of the added carbon sources (19, 24, 50-52), substantial enhancement of SO42− reduction in all of our tests compared to tests without carbon source addition (42) suggests that a diverse SRB population is present. This agrees with our results from FISH and DGGE. More specifically, consumption of acetate, propionate, and lactate coupled to SO42− reduction suggests the presence of Desulfobacter, Desulfobulbus, and Desulfovibrio, respectively, because these genera were commonly found to be associated with the degradation of the respective carbon sources in many different environments (32, 34, 40) and also degrade them in pure culture (51). Indeed, by using FISH we demonstrated the presence of these three genera.
Interestingly, acetate as a bacterial metabolite was detected during PPTpr, suggesting incomplete degradation of propionate. This agrees with the presence of Desulfobulbus, an incomplete propionate oxidizer (51). However, fermentation of propionate to acetate may also have occurred. Butyrate, on the other hand, is degraded by none of the three genera mentioned, suggesting the presence of additional SRB.
Nevertheless, the comparison of results from molecular analyses with measurements of macroscopic activities is complicated by the fact that with the former we targeted only the suspended bacteria population, while the latter reflects both attached and suspended populations. Although we are aware that suspended microbial communities may not accurately reflect the overall microbial population (1), others have indicated that, in contaminated aquifers, the difference between structures of suspended and attached microbial populations may not be significant (6). This will remain an issue of further study.
Conclusions.
In this study, we presented a novel combination of single-well PPTs with molecular microbiological methods. Molecular and chemical data complemented each other and provided valuable insights into microbial processes and activities in the SO42−-reducing zone of a PHC-contaminated aquifer. SRB from this freshwater environment were able to use a variety of organic carbon sources, which is indicative of a diverse SRB population, as many SRB are specialized for only a few carbon sources. Molecular data confirmed substantial diversity of suspended SRB. Activities of SRB were considerably enhanced by addition of organic carbon sources, which was corroborated by higher FISH detection rates during PPTs compared to native groundwater. Results from DGGE indicated that within the time frame of our experiments (4 days), the introduction of reactants during PPTs did not change the suspended microbial community of the dominant species. In future studies we will focus on community members responsible for PHC degradation and characterization of populations that are attached to the aquifer solid matrix.
Acknowledgments
We thank J. P. Clément (Amt für Gewässerschutz und Abfallwirtschaft, Kanton Bern, Switzerland) for cooperation at the field site. Helpful comments by two anonymous reviewers were greatly appreciated.
This study was funded by the Swiss National Science Foundation, Priority Program Environment, and by the Swiss Agency for the Environment, Forests and Landscape (BUWAL).
REFERENCES
- 1.Alfreider, A., M. Krössbacher, and R. Psenner. 1997. Groundwater samples do not reflect bacterial densities and activity in subsurface systems. Water Res. 31:832-840. [Google Scholar]
- 2.Amann, R. I., B. J. Binder, R. J. Olson, S. W. Chisholm, R. Devereux, and D. A. Stahl. 1990. Combination of 16S ribosomal-RNA-targeted oligonucleotide probes with flow-cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56:1919-1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amann, R. I., L. Krumholz, and D. A. Stahl. 1990. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J. Bacteriol. 172:762-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amann, R. I., W. Ludwig, and K.-H. Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amann, R. I., J. Stromley, R. Devereux, R. Key, and D. A. Stahl. 1992. Molecular and microscopic identification of sulfate-reducing bacteria in multispecies biofilms. Appl. Environ. Microbiol. 58:614-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bekins, B. A., E. M. Godsy, and E. Warren. 1999. Distribution of microbial physiologic types in an aquifer contaminated by crude oil. Microb. Ecol. 37:263-275. [DOI] [PubMed] [Google Scholar]
- 7.Bolliger, C., P. Höhener, D. Hunkeler, K. Häberli, and J. Zeyer. 1999. Intrinsic bioremediation of a petroleum hydrocarbon-contaminated aquifer and assessment of mineralization based on stable carbon isotopes. Biodegradation 10:201-217. [DOI] [PubMed] [Google Scholar]
- 8.Bolliger, C., F. Schönholzer, M. H. Schroth, D. Hahn, S. Bernasconi, and J. Zeyer. 2000. Characterizing intrinsic bioremediation in a petroleum hydrocarbon-contaminated aquifer by combined chemical, isotopic and biological analyses. Bioremediation J. 4:359-371. [Google Scholar]
- 9.Bolliger, C., M. H. Schroth, S. M. Bernasconi, J. Kleikemper, and J. Zeyer. 2001. Sulfur isotope fractionation during microbial sulfate reduction by toluene degrading bacteria. Geochim. Cosmochim. Acta 65:3289-3298. [Google Scholar]
- 10.Böttcher, M. E., S. M. Sievert, and J. Kuever. 1999. Fractionation of sulfur isotopes during dissimilatory reduction of sulfate by a thermophilic gram-negative bacterium at 60°C. Arch. Microbiol. 172:125-128. [DOI] [PubMed] [Google Scholar]
- 11.Cozzarelli, I. M., M. J. Baedecker, R. P. Eganhouse, and D. F. Goerlitz. 1994. The geochemical evolution of low-molecular-weight organic acids derived from the degradation of petroleum contaminants in groundwater. Geochim. Cosmochim. Acta 58:863-877. [Google Scholar]
- 12.Devereux, R., M. D. Kane, J. Winfrey, and D. A. Stahl. 1992. Genus- and group-specific hybridization probes for determinative and environmental studies of sulfate-reducing bacteria. Syst. Appl. Microbiol. 15:601-609. [Google Scholar]
- 13.Edwards, E. A., L. E. Wills, M. Reinhard, and D. Grbic-Galic. 1992. Anaerobic degradation of toluene and xylene by aquifer microorganisms under sulfate-reducing conditions. Appl. Environ. Microbiol. 58:794-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elferink, S. J. W. H. O., R. N. Maas, H. J. M. Harmsen, and A. J. M. Stams. 1995. Desulforhabdus amnigenus gen-nov sp-nov, a sulfate reducer isolated from anaerobic granular sludge. Arch. Microbiol. 164:119-124. [PubMed] [Google Scholar]
- 15.Ensley, B. D., and J. M. Suflita. 1995. Metabolism of environmental contaminants by mixed and pure cultures of sulfate-reducing bacteria, p. 336. In L. L. Barton (ed.), Sulfate-reducing bacteria. Plenum Press, New York, N.Y.
- 16.Felske, A., B. Engelen, U. Nübel, and H. Backhaus. 1996. Direct ribosome isolation from soil to extract bacterial rRNA for community analysis. Appl. Environ. Microbiol. 62:4162-4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillham, R. W., R. C. Starr, and D. J. Miller. 1990. A device for in situ determination of geochemical transport parameters. 2. Biochemical reactions. Ground Water 28:858-862. [Google Scholar]
- 18.Haggerty, R., M. H. Schroth, and J. D. Istok. 1998. Simplified method of “push-pull” test data analysis for determining in situ reaction rate coefficients. Ground Water 36:314-324. [Google Scholar]
- 19.Hanselmann, K. W., J. P. Kaiser, M. Wenk, R. Schon, and R. Bachofen. 1995. Growth on methanol and conversion of methoxylated aromatic substrates by Desulfotomaculum orientis in the presence and absence of sulfate. Microbiol. Res. 150:387-401. [Google Scholar]
- 20.Hunkeler, D., D. Jörger, K. Häberli, P. Höhener, and J. Zeyer. 1998. Petroleum hydrocarbon mineralization in anaerobic laboratory aquifer columns. J. Contaminant Hydrol. 32:41-61. [Google Scholar]
- 21.Istok, J. D., M. D. Humphrey, M. H. Schroth, M. R. Hyman, and K. T. O'Reilly. 1997. Single-well, “push-pull” test for in situ determination of microbial activities. Ground Water 35:619-631. [Google Scholar]
- 22.Jørgensen, B. B. 1977. The sulfur cycle of a coastal marine sediment (Limfjorden, Denmark). Limnol. Oceanogr. 22:814-832. [Google Scholar]
- 23.Kleerebezem, R., and A. J. M. Stams. 2000. Kinetics of syntrophic cultures: A theoretical treatise on butyrate fermentation. Biotechnol. Bioeng. 67:529-543. [DOI] [PubMed] [Google Scholar]
- 24.Kuever, J., M. Konneke, A. Galushko, and O. Drzyzga. 2001. Reclassification of Desulfobacterium phenolicum as Desulfobacula phenolica comb. nov and description of strain Sax(T) as Desulfotignum balticum gen. nov., sp nov. International J. Syst. Evol. Microbiol. 51:171-177. [DOI] [PubMed] [Google Scholar]
- 25.Lovley, D. R. 1997. Potential for anaerobic bioremediation of BTEX in petroleum-contaminated aquifers. J. Ind. Microbiol. Biotechnol. 18:75-81. [Google Scholar]
- 26.Madsen, E. L. 1991. Determining in situ biodegradation - facts and challenges. Environ. Sci. Technol. 25:1663-16673. [Google Scholar]
- 27.Madsen, E. L. 1998. Epistemology of environmental microbiology. Environ. Sci. Technol. 32:429-439. [Google Scholar]
- 28.Manz, W., M. Eisenbrecher, T. R. Neu, and U. Szewzyk. 1998. Abundance and spatial organization of Gram-negative sulfate-reducing bacteria in activated sludge investigated by in situ probing with specific 16S rRNA targeted oligonucleotides. FEMS Microbiol. Ecol. 25:43-61. [Google Scholar]
- 29.Mariotti, A., J. C. Germon, P. Hubert, P. Kaiser, R. Letolle, A. Tardieux, and P. Tardieux. 1981. Experimental determination of nitrogen kinetic isotope fractionation: some principles; illustration for the denitrification and nitrification processes. Plant Soil 62:413-430. [Google Scholar]
- 30.Muyzer, G., and K. Smalla. 1998. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek 73:127-141. [DOI] [PubMed] [Google Scholar]
- 31.Odom, J. M., and R. Singleton, Jr. 1993. The sulfate-reducing bacteria: contemporary perspectives. Springer-Verlag New York Inc., New York, N.Y.
- 32.Parkes, R. J., N. J. E. Dowling, D. C. White, R. A. Herbert, and G. R. Gibson. 1993. Characterization of sulphate-reducing bacterial populations within marine and estuarine sediments with different rates of sulphate reduction. FEMS Microbiol. Ecol. 102:235-250. [Google Scholar]
- 33.Parkes, R. J., G. R. Gibson, I. Mueller-Harvey, W. J. Buckingham, and R. A. Herbert. 1989. Determination of the substrates for sulphate-reducing bacteria within marine and estuarine sediments with different rates of sulphate reduction. J. Gen. Microbiol. 135:175-187. [Google Scholar]
- 34.Purdy, K. J., D. B. Nedwell, T. M. Embley, and S. Takii. 1997. Use of 16S rRNA-targeted oligonucleotide probes to investigate the occurrence and selection of sulfate-reducing bacteria in response to nutrient addition to sediment slurry microcosms from a Japanese estuary. FEMS Microbiol. Ecol. 24:221-234. [Google Scholar]
- 35.Rabus, R., M. Fukui, H. Wilkes, and F. Widdel. 1996. Degradative capacities and 16S rRNA-targeted whole-cell hybridization of sulfate-reducing bacteria in an anaerobic enrichment culture utilizing alkylbenzenes from crude oil. Appl. Environ. Microbiol. 62:3605-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reinhard, M., S. Shang, P. K. Kitanidis, W. Orwin, G. D. Hopkins, and C. A. Lebron. 1997. In situ BTEX biotransformation under enhanced nitrate- and sulfate-reducing conditions. Environ. Sci. Technol. 31:28-36. [Google Scholar]
- 37.Rueter, P., R. Rabus, H. Wilkes, F. Aeckersberg, F. A. Rainey, H. W. Jannasch, and F. Widdel. 1994. Anaerobic oxidation of hydrocarbons in crude oil by new types of sulphate-reducing bacteria. Nature 372:455-458. [DOI] [PubMed] [Google Scholar]
- 38.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 39.Santegoeds, C. M., T. G. Ferdelman, G. Muyzer, and D. de Beer. 1998. Structural and functional dynamics of sulfate reducing populations in bacterial biofilms. Appl. Environ. Microbiol. 64:3731-3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sass, H., H. Cypionka, and H. D. Babenzien. 1997. Vertical distribution of sulfate-reducing bacteria at the oxic-anoxic interface in sediments of the oligotrophic Lake Stechlin. FEMS Microbiol. Ecol. 22:245-255. [Google Scholar]
- 41.Schroth, M. H., J. D. Istok, G. T. Conner, M. R. Hyman, R. Haggerty, and K. T. O'Reilly. 1998. Spatial variability in in situ aerobic respiration and denitrification rates in a petroleum-contaminated aquifer. Ground Water 36:924-937. [Google Scholar]
- 42.Schroth, M. H., J. Kleikemper, C. Bolliger, S. M. Bernasconi, and J. Zeyer. 2001. In-situ assessment of microbial sulfate reduction in a petroleum-contaminated aquifer by using push-pull tests and stable sulfur isotope analyses. J. Contaminant Hydrol. 51:179-195. [DOI] [PubMed] [Google Scholar]
- 43.Sigler, W. V., C. H. Nakatsu, Z. J. Reicher, and R. F. Turco. 2001. Fate of the biological control agent Pseudomonas aureofaciens TX-1 after application to turfgrass. Appl. Environ. Microbiol. 67:3542-3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snodgrass, M. F., and P. K. Kitanidis. 1998. A method to infer in situ reaction rates from push-pull experiments. Ground Water 36:645-650. [Google Scholar]
- 45.Sørensen, J., D. Christensen, and B. B. Jørgensen. 1981. Volatile fatty acids and hydrogen as substrates for sulfate-reducing bacteria in anaerobic marine sediment. Appl. Environ. Microbiol. 42:5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stahl, D. A., and R. I. Amann. 1991. Development and application of nucleic acid probes, p. 205-248. In E. Stackebrandt and M. Goodfellow (ed.), Nucleic acid techniques in bacterial systematics. Wiley, New York, N.Y.
- 47.Teske, A., C. Wawer, G. Muyzer, and N. B. Ramsing. 1996. Distribution of sulfate-reducing bacteria in a stratified fjord (Mariager fjord, Denmark) as evaluated by most-probable-number counts and denaturing gradient gel electrophoresis of PCR-amplified ribosomal DNA fragments. Appl. Environ. Microbiol. 62:1405-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thierrin, J., G. B. Davis, C. Barber, B. M. Patterson, F. Pribac, T. R. Power, and M. Lambert. 1993. Natural degradation rates of BTEX compounds and naphthalene in a sulphate reducing groundwater environment. Hydrol. Sci. 38:309-322. [Google Scholar]
- 49.Wawer, C., and G. Muyzer. 1995. Genetic diversity of Desulfovibrio spp in environmental-samples analyzed by denaturing gradient gel-electrophoresis of [NiFe] hydrogenase gene fragments. Appl. Environ. Microbiol. 61:2203-2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Widdel, F. 1992. The genus Desulfotomaculum, p. 1792-1799. In A. Balows (ed.), The prokaryotes: a handbook on the biology of bacteria: ecophysiology, isolation, identification, applications, vol. 4. Springer, New York, N.Y. [Google Scholar]
- 51.Widdel, F. 1988. Microbiol. and ecology of sulfate- and sulfur-reducing bacteria, p. 469-585. In A. J. B. Zehnder (ed.), Biology of anaerobic microorganisms. John Wiley & Sons, Inc., New York, N.Y.
- 52.Widdel, F., and F. Bak. 1992. Gram-negative mesophilic sulfate-reducing bacteria, p. 3352-3378. In A. Balows (ed.), The prokaryotes: a handbook on the biology of bacteria: ecophysiology, isolation, identification, applications, vol. 4. Springer, New York, N.Y. [Google Scholar]
- 53.Wiedemeier, T. H., H. S. Rifai, C. J. Newell, and J. T. Wilson. 1999. Natural attenuation of fuels and chlorinated solvents in the subsurface. John Wiley & Sons, Inc., New York, N.Y.
- 54.Zarda, B., D. Hahn, A. Chatzinotas, W. Schönhuber, A. Neef, R. I. Amann, and J. Zeyer. 1997. Analysis of bacterial community structure in bulk soil by in situ hybridization. Arch. Microbiol. 168:185-192. [Google Scholar]
- 55.Zhang, X., and L. Y. Young. 1997. Carboxylation as an initial reaction in the anaerobic metabolism of naphthalene and phenanthrene by sulfidogenic consortia. Appl. Environ. Microbiol. 63:4759-4764. [DOI] [PMC free article] [PubMed] [Google Scholar]