

Abstract
The brown-rot basidiomycete Gloeophyllum trabeum uses a quinone redox cycle to generate extracellular Fenton reagent, a key component of the biodegradative system expressed by this highly destructive wood decay fungus. The hitherto uncharacterized quinone reductase that drives this cycle is a potential target for inhibitors of wood decay. We have identified the major quinone reductase expressed by G. trabeum under conditions that elicit high levels of quinone redox cycling. The enzyme comprises two identical 22-kDa subunits, each with one molecule of flavin mononucleotide. It is specific for NADH as the reductant and uses the quinones produced by G. trabeum (2,5-dimethoxy-1,4-benzoquinone and 4,5-dimethoxy-1,2-benzoquinone) as electron acceptors. The affinity of the reductase for these quinones is so high that precise kinetic parameters were not obtainable, but it is clear that kcat/Km for the quinones is greater than 108 M−1 s−1. The reductase is encoded by a gene with substantial similarity to NAD(P)H:quinone reductase genes from other fungi. The G. trabeum quinone reductase may function in quinone detoxification, a role often proposed for these enzymes, but we hypothesize that the fungus has recruited it to drive extracellular oxyradical production.
The basidiomycetes that cause brown rot of wood are major recyclers of lignocellulose in terrestrial ecosystems and also cause the most destructive type of decay found in wooden structures (7, 17, 22). Although much remains unknown about their biodegradative mechanisms, there is growing evidence (2, 8, 11, 13, 21) that these fungi produce extracellular hydroxyl radicals (•OH), which attack wood polymers rapidly (6). In biological systems, •OH is generally produced by the Fenton reaction (i.e., when Fe2+ reduces H2O2). Therefore, to attack wood via Fenton chemistry, brown-rot fungi need mechanisms to reduce the sources of oxygen and iron which they generally encounter, O2 and Fe3+.
Recent work has shown that the brown-rot basidiomycete Gloeophyllum trabeum has such a mechanism (12, 14, 18). This fungus secretes 2,5-dimethoxyhydroquinone and 4,5-dimethoxycatechol, both of which reduce extracellular Fe3+. These one-electron oxidoreductions generate semiquinone radicals, which reduce either Fe3+ or O2 in a second one-electron step that produces 2,5-dimethoxy-1,4-benzoquinone (2,5-DMBQ) and 4,5-dimethoxy-1,2-benzoquinone (4,5-DMBQ). G. trabeum then reduces these quinones to regenerate the hydroquinone and catechol that are needed to produce additional Fenton reagent. A similar redox cycle may contribute to biodegradation by some white-rot fungi (9).
It is unclear how G. trabeum reduces the quinones that it produces; indeed, almost nothing is known about biodegradative enzymes in this highly destructive fungus. In this study we characterized the major quinone reductase, as well as the gene and mRNA that encode this enzyme, from G. trabeum cultures that express high levels of hydroquinone biosynthesis and extracellular Fenton chemistry.
MATERIALS AND METHODS
Organism.
Static cultures of G. trabeum ATCC 11539 were grown at 31°C in the medium previously described (12). For experiments on quinone production and reduction by the intact fungus, we used mycelial mats from 5-ml cultures that were grown in 125-ml Erlenmeyer flasks for 1 to 10 days. For experiments on quinone reductase fractionation and purification, we used 50-ml cultures, each containing a 7-day mycelium with a dry weight of 107 ± 9 mg, that were grown in 2.8-liter Fernbach flasks.
Reagents.
4,5-DMBQ was prepared by oxidizing and methylating phenol as described previously (12, 19). 2,5-DMBQ was purchased from TCI America (Portland, Oreg.). Other chemicals, all reagent grade, were obtained from Sigma/Aldrich or Pharmacia.
Production of extracellular quinones by G. trabeum.
Each assay was done in triplicate as follows. The extracellular medium from three cultures of the same age was pooled, oxidized by adding FeCl3 to a final concentration of 0.1 mM, and at filtered. A portion of the sample was then assayed for 2,5-DMBQ and 4,5-DMBQ by high-performance liquid chromatography (HPLC) as described previously (12).
Reduction of quinones by G. trabeum.
Each assay was done in triplicate as follows. Three G. trabeum mycelial mats of the same age were placed in a glass vial that contained 2,6-dimethoxy-p-benzoquinone (2,6-DMBQ) or 2,5-DMBQ (250 μM) in 20 ml of 25 mM sodium oxalate (pH 4.0). The solution was recirculated through a 1-cm-path-length quartz flow cell at a rate of 6 ml/min and at the ambient temperature. The decrease in absorbance of the solution was monitored at 394 nm for 2,6-DMBQ (ɛ = 0.64 mM−1 cm−1) and at 380 nm for 2,5-DMBQ (ɛ = 0.31 mM−1 cm−1). Previous work has shown that very little of the quinone is taken up by the mycelium in such experiments and therefore that the decrease in visible absorbance serves as a reliable measure of quinone reduction (12, 14).
Subcellular fractionation of quinone reductase activity.
The mycelial mats from 12 Fernbach cultures were harvested on day 7, collected by filtration through miracloth, and rinsed with ice-cold Milli-Q water. All subsequent steps were done at 0 to 4°C. The mycelium was combined with 150 ml of homogenization buffer, which consisted of 50 mM sodium citrate (pH 6.0), 300 mM sucrose, 5 mM EDTA, 5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and 1 mM benzamidine. The mixture was disrupted three times for 1 min with a 10-min cooling period between bursts by using an ice-jacketed glass bead homogenizer (Bead-Beater; Biospec Products, Bartlesville, Okla.). The homogenate was centrifuged for 20 min at 10,000 × g to remove mitochondria and cell wall material, and the supernatant fraction was retained. A measured portion of the supernatant fraction was ultracentrifuged for 60 min at 100,000 × g to separate the soluble fraction from the crude microsomal fraction, and the microsomal pellet was resuspended in 50 mM sodium citrate (pH 6.0) that contained 300 mM sucrose and 5 mM KCl. The three fractions were assayed for quinone reductase activity as described below.
Enzyme purification.
All enzyme purification steps were done at 0 to 5°C unless stated otherwise. The mycelial mats from 25 Fernbach cultures were harvested on day 7 as described above and frozen for 1 h at −75°C. The frozen mycelium was suspended in 150 ml of buffer that contained 100 mM sodium citrate (pH 6.0), 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 1 mM benzamidine. The mixture was homogenized as described above, and the glass beads were rinsed with additional buffer to give about 200 ml of crude extract.
The extract was centrifuged at 14,000 × g for 20 min, and the supernatant fraction was loaded on a column of Phenyl Sepharose 6 (fast flow grade; 18.5 by 2.5 cm; Sigma) that had been equilibrated beforehand with 100 mM sodium citrate (pH 6.0). The column was washed with 200 ml of 100 mM sodium citrate (pH 6.0) and then with a 600-ml linear gradient that began with 100 mM sodium citrate (pH 6.0) and ended with 70% ethylene glycol in 10 mM sodium citrate (pH 6.0). The NADH:quinone reductase activity eluted in a single peak at an ethylene glycol concentration of about 45%. The active fractions were concentrated by ultrafiltration and dialyzed under a vacuum in a collodion bag apparatus (10-kDa cutoff) against 20 mM sodium citrate (pH 6.0) that contained 20% (vol/vol) ethylene glycol. The final volume was less than 100 μl.
This sample was brought to a volume of 10 ml with 20 mM sodium phosphate (pH 6.0) and was immediately applied to a preequilibrated 2-ml column of hydroxyapatite (Macroprep Ceramic type 1; particle size, 8 μm; Bio-Rad) that was then washed with the same phosphate buffer. The quinone reductase activity eluted in this passthrough fraction, which was immediately dialyzed against 20 mM citrate buffer-20% ethylene glycol and concentrated as described above. Any delay in the removal of phosphate resulted in an increased loss of enzyme activity.
The enzyme preparation was brought to a volume of about 7 ml in 10 mM sodium phosphate buffer (pH 6.0) and was immediately applied to a column of DEAE Sepharose (9.0 by 1.0 cm; Pharmacia) that had been equilibrated beforehand with the same buffer. The column was washed with 20 ml of this buffer and then with a 100-ml linear gradient from 10 to 100 mM sodium phosphate (pH 6.0). The NADH:quinone reductase activity eluted in a single peak at a phosphate concentration of about 40 mM. The recovered enzyme was immediately concentrated and dialyzed against 20 mM citrate buffer-20% ethylene glycol in the collodion bag apparatus. Use of phosphate as the eluant salt in this step resulted in some loss of activity but was unavoidable because the reductase did not adhere to the column reproducibly when other salts, such as chloride or acetate, were used.
The enzyme sample (approximately 300 μl) was further purified by gel permeation chromatography on a column of Superdex 200 (30.0 by 1.0 cm; Pharmacia), which was operated on a Pharmacia fast-performance liquid chromatography apparatus in 100 mM sodium citrate (pH 6.0) at room temperature and was calibrated with a protein molecular weight standard kit (Pharmacia). The NADH:quinone reductase eluted in a single peak. It was concentrated and dialyzed against 20 mM citrate (pH 6.0) that contained 20% ethylene glycol and was stored at −20°C.
Enzyme and protein assays.
For routine assays of quinone reduction, it was most convenient to use 2,6-DMBQ as the acceptor instead of 2,5-DMBQ or 4,5-DMBQ. 2,5-DMBQ dissolves slowly in water, which makes stock solutions difficult to prepare, and 4,5-DMBQ is not commercially available.
NAD(P)H:quinone reductase activity was assayed spectrophotometrically by monitoring the decrease in absorbance at 340 nm due to NAD(P)H oxidation (ɛ340 = 6.2 mM−1 cm−1). The amount of enzyme that catalyzed the oxidation of 1 μmol of NADH/min was defined as 1 U of activity. The standard 2.0-ml assay mixture contained enzyme, sodium citrate (50 mM, pH 6.0), NAD(P)H (200 μM), and quinone (100 μM). The reactions were conducted in magnetically stirred 1-cm-path-length cuvettes that were maintained at 25°C. For Michaelis-Menten experiments, the substrate concentrations were varied as required and each assay was done in quadruplicate. The standard deviations for the rates in replicate assays were ±5% or less.
The same assay was used to look for sugar oxidases that use quinones as alternate electron acceptors, except that glucose (200 μM) replaced NAD(P)H, and the reduction of 2,6-DMBQ was monitored directly at 394 nm. Glucose oxidase activity was also assayed with O2 as the acceptor by using a coupled colorimetric assay with horseradish peroxidase and 2,2′-azino-bis-(3-ethylbenzthiazoline-6-sulfonic acid) (15).
To determine the stoichiometry of quinone reduction by the G. trabeum quinone reductase, the amount of hydroquinone produced was determined by HPLC as described previously (12). Protein was assayed with a Coomassie blue dye binding assay (Bio-Rad), which was standardized with bovine serum albumin. Purified quinone reductase was also quantitated from its absorbance at 450 nm due to flavin mononucleotide (FMN) (ɛ = 12.2 mM−1 cm−1), assuming two FMN molecules per enzyme molecule and a subunit molecular weight of 22,000. With purified G. trabeum quinone reductase, the two methods gave values that agreed within less than 10%.
Physical characterization of the quinone reductase.
The UV-visible absorption spectrum of the purified enzyme (0.2 mg/ml) was recorded in 20 mM sodium citrate (pH 6.0) that contained 20% ethylene glycol.
Flavin was extracted from G. trabeum quinone reductase by boiling a sample for 10 min. Because flavin adenine dinucleotide (FAD) can be hydrolyzed to FMN, a control sample of the FAD-containing glutathione reductase from Saccharomyces cerevisiae was also boiled, as were authentic standards of FAD and FMN. These controls established that FAD was not hydrolyzed under our extraction conditions and that the recovery of both flavins for analysis was nearly quantitative. The samples were analyzed by HPLC using external FAD and FMN standards. The column (Phenylhexyl Luna; 150 by 4.6 mm; particle size, 5 μm; Phenomenex) was operated in water-acetonitrile-formic acid (87.5:12.5:0.1) at a rate of 1.0 ml/min and at the ambient temperature. FAD eluted at 6 min, and FMN eluted at 10 min.
Sodium dodecyl sulfate (SDS) gel electrophoresis and analytical isoelectric focusing of the purified quinone reductase were done with precast polyacrylamide gels in a Pharmacia Phast System apparatus. The gels were calibrated with protein standard kits from Pharmacia and were stained with Coomassie blue R250.
Tryptic digestion, mass spectral analysis, and internal Edman sequencing of the reductase were done at the Keck Foundation Biotechnology Resource Laboratory at Yale University. Samples were prepared as outlined on the Keck website (http://info.med.yale.edu/wmkeck).
Isolation of cDNA and genomic clones.
To obtain RNA, mycelial pellets were snap frozen in liquid nitrogen, and the poly(A) RNA was purified by magnetic capture with oligo(dT)15 Dynabeads (Dynal, Great Neck, N.Y.) used according to the manufacturer's recommendations. RNA was stored as an ethanol precipitate at −20°C. To obtain total genomic DNA, pellets from log-phase cultures were extracted as described previously (20).
Experimentally determined peptide sequences were used to design two degenerate primers, 5′-AAXGTXCCXGCXCCCCAXGG-3′ and 5′-AAYTAYCAYCGXTTYYTXTT-3′. Poly(A) RNA was reverse transcribed with oligo(dT) and used as a template in 50-μl PCR mixtures with 50 to 150 pmol of degenerate primer. The reaction cycles were as follows: 94°C for 6 min, 58°C for 2 min, and 72°C for 40 min for one cycle, followed by 94°C for 1 min, 58°C for 2 min, and 72°C for 5 min for 35 cycles and finally an extension step consisting of 72°C for 15 min. Several reaction mixtures were pooled, and a 300-bp fragment was subcloned into pGEM-T (Promega Biotech, Madison, Wis.). Nucleotide sequences were determined by using an ABI Prism Big Dye terminator cycle sequencing kit (PE Applied Biosystems, Foster City, Calif.) with an ABI377 DNA sequencer.
From the partial cDNA sequence, cloning was extended into the flanking genomic sequence by using a Universal genome walking kit (Clonetech Laboratories, Palo Alto, Calif.) with the following primers: 5′-CGTTCTGGGACTCCACTGGTCCCCTCT-3′ and 5′-AAACGAAGAGACCCGCGTACTTGCCG-3′. Several extension products were subcloned and sequenced. Based on the extended genomic sequences, primers 5′-AGACGCAGCGGGCGTAATG-3′ and 5′-GAGACGCCACGCAGTTTACC-3′ were designed to PCR amplify the full-length cDNA and corresponding genomic clones with a high-fidelity polymerase, Pfu (Stratagene, La Jolla, Calif.). Sequences were analyzed and compared by using DNASTAR software (DNASTAR, Madison, Wis.).
Nucleotide sequence accession numbers.
The G. trabeum genomic and cDNA sequences have been deposited in the GenBank database under accession numbers AF465405 and AF465406, respectively.
RESULTS
Quinone reductase activity of G. trabeum in vivo.
The specific activity of exogenous quinone reduction by intact mycelium remained roughly constant for the first 6 days of culture and then increased (Fig. 1). A t test of the data indicated 98% confidence that the specific reduction rates on days 7 to 10 were significantly greater than those on days 1 to 6. The extracellular concentrations of 2,5-DMBQ and 4,5-DMBQ, the natural substrates of the reductase which we were looking for, declined slightly during the growth phase and then peaked on days 6 to 7 after growth ceased. These results led us to surmise that day 7 cultures would provide the best chance to identify the enzyme that drives Fenton chemistry. This assessment was consistent with our previous observations that G. trabeum mycelium from day 7 supports rapid redox cycling of fungal quinones and produces extracellular Fenton reagent (12, 14).
FIG. 1.

Production and reduction of quinones by G. trabeum. The top panel shows the average extracellular concentration of 2,5-DMBQ in each culture (○), the average extracellular concentration of 4,5-DMBQ in each culture (□), and the average dry weight of mycelium in each culture (▿). The error bars show the standard deviations for triplicate determinations. Quinones were detected on day zero in this experiment because they were already present in the agar plate inoculum (see Materials and Methods). The bottom panel shows the average rate of 2,6-DMBQ reduction per milligram (dry weight) of mycelium by three harvested and pooled mycelial mats (see Materials and Methods). The error bars show the standard deviations for triplicate determinations. In an additional experiment, we found that the specific rate of 2,5-DMBQ reduction on day 7 was 0.0027 U/mg (dry weight), the same as the rate of 2,6-DMBQ reduction.
Subcellular location of the principal quinone reductase.
We fractionated a day 7 mycelial extract by differential centrifugation and assayed the fractions for 2,6-DMBQ reduction (Table 1). The extract contained a high level of NADH-dependent quinone reductase activity, more than 95% of which was soluble rather than membrane associated. The rate of NADH-dependent quinone reduction by the soluble fraction (0.19 U/mg [dry weight]) was more than sufficient to account for the rate of quinone reduction by intact G. trabeum mycelium on day 7 (0.0027 U/mg [dry weight]). A lower level of NADPH-dependent activity, comprising less than 10% of the total, was also detected. Glucose oxidase activity was undetectable, and no quinone reduction occurred when glucose rather than NADH was used as the electron donor; that is, the extract lacked sugar oxidases that can use quinones as alternative electron acceptors (15). Repetitions of these assays with 2,5-DMBQ or 4,5-DMBQ as the electron acceptor gave results little different from those obtained with 2,6-DMBQ. We concluded that the enzyme most likely to drive the G. trabeum quinone redox cycle is a soluble NADH:quinone oxidoreductase that exhibits low specificity for benzoquinones.
TABLE 1.
Subcellular fractionation of G. trabeum quinone reductase activitya
| Fraction | Total protein (mg) | Reductase activity
|
|||
|---|---|---|---|---|---|
| With NADH
|
With NADPH
|
||||
| Total activity (U) | Sp act (U/mg) | Total activity (U) | Sp act (U/mg) | ||
| 10,000-x-g supernatant | 54 | 233 | 4.3 | 22 | 0.41 |
| 100,000-x-g supernatant | 40 | 233 | 5.8 | 14 | 0.35 |
| 100,000-x-g pellet | 13 | 12 | 0.92 | 2 | 0.15 |
Data for a quantity of mycelium equivalent to 1,285 mg (dry weight).
Purification and physical characterization of the reductase.
We purified the soluble activity by conventional chromatographic methods (Table 2). A single peak of activity appeared in all chromatographic steps, and the results of the final gel permeation chromatography step indicated that the holoenzyme's molecular weight was about 50,000. Typical preparations were purified 250- to 330-fold, had specific activities between 2,500 and 3,000 U/mg, and produced a single 22-kDa band during SDS-polyacrylamide gel electrophoresis (Fig. 2). Analytical isoelectric focusing of the native protein confirmed its purity and gave an isoelectric point of 3.3. The UV-visible absorption spectrum of the purified native enzyme (Fig. 3) was typical of a flavoprotein, with an A275/A450 ratio of 7.3. HPLC analysis of boiled enzyme samples showed that the holoenzyme contained 0.9 mol of FMN per mol of subunit. In summary, the soluble G. trabeum quinone reductase is a dimer of two identical 22-kDa subunits, each with one molecule of FMN.
TABLE 2.
Typical purification of the G. trabeum quinone reductasea
| Step | Total protein (mg) | Total activity (U) | Sp act (U/mg) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|
| 14,000-x-g supernatant | 92 | 826 | 9 | 100 | 1 |
| Phenyl Sepharose | 25 | 832 | 33 | 101 | 4 |
| Hydroxyapatite | 0.73 | 505 | 692 | 61 | 77 |
| DEAE Sepharose | 0.23 | 230 | 1,000 | 28 | 111 |
| Superdex 200 | 0.045 | 135 | 3,000 | 16 | 334 |
Data for a quantity of mycelium equivalent to 2,675 mg (dry weight).
FIG. 2.
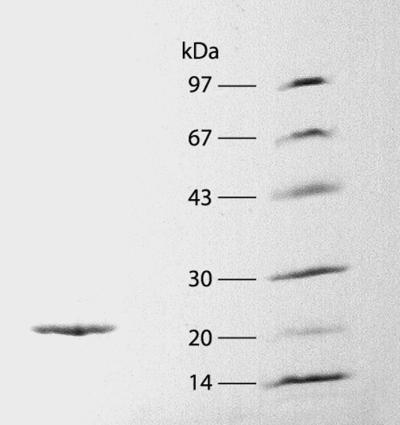
SDS gel electrophoresis of the purified G. trabeum NADH:quinone reductase (left lane) and of molecular mass standards (right lane), including phosphorylase b (97 kDa), serum albumin (67 kDa), carbonic anhydrase (30 kDa), trypsin inhibitor (20 kDa), and α-lactalbumin (14 kDa).
FIG. 3.

UV-visible absorption spectrum of the purified G. trabeum NADH:quinone reductase (0.2 mg/ml).
The purified quinone reductase was stable at −20°C in sodium citrate buffer (pH 6.0) that contained 20% (vol/vol) ethylene glycol. It lost all activity within several days in the absence of ethylene glycol or in the presence of phosphate buffer. It was about twofold more active in pH 3.0 citrate buffer than in pH 6.0 citrate buffer, but we routinely assayed it at the higher pH because it lost all activity within a few hours at pH 3. These assays showed that the reductase exhibited low specificity for quinone electron acceptors; 1,4-benzoquinone, 2-methoxy-1,4-benzoquinone, 2,6-DMBQ, 2,5-DMBQ, and 4,5-DMBQ were all similarly reactive. The enzyme was active with NADH as the donor but showed no activity with NADPH. HPLC analysis of the hydroquinone reaction products showed that the enzyme reduced 1.1 mol of quinone per mol of NADH supplied; i.e., the reaction proceeds with a 1:1 stoichiometry between reductant and oxidant.
Kinetic parameters of the reductase.
We did Michaelis-Menten experiments at pH 6.0 with the two physiological quinones, 2,5-DMBQ and 4,5-DMBQ. Each experiment was done two or three times, and all of the results yielded linear Lineweaver-Burk plots, which showed that the Km for both 2,5-DMBQ and 4,5-DMBQ was 5 to 7 μM, whereas the Km for NADH was 85 to 90 μM. Estimation of the enzyme's kcat gave values between 1,100 and 1,600 s−1. These Km and kcat values are subject to considerable error, because the enzyme's extremely high affinity for quinones required us to measure small absorbance changes during the kinetics experiments. Nevertheless, the results show that the catalytic efficiency (kcat/Km) of the enzyme for 2,5-DMBQ and 4,5-DMBQ is very high, greater than 108 M−1 s−1.
Inferred amino acid sequence of the reductase.
Attempted N-terminal analyses of the reductase yielded no data, and therefore the N terminus of the mature 22-kDa protein is probably blocked. However, tryptic digestion followed by HPLC separation and Edman degradation of selected peptides yielded three internal sequences: NYDGFLFGIPTR, GGSPWGAGTFANSDGSR, and SFYEYVAR.
We used degenerate primers based on portions of these peptide sequences to amplify cDNA from G. trabeum cultures that expressed high levels of quinone reductase activity. We sequenced the resulting cDNA clone and found that it contained nucleotide sequences that exactly matched the three experimentally determined peptide sequences. A comparison of the G. trabeum cDNA sequence with related sequences in the GenBank database showed that the SFYEYVAR peptide, in particular, is located in a highly variable region near the C terminus of the quinone reductase. Therefore, the probability is very high that the cDNA which we isolated corresponds to the enzyme which we purified.
The G. trabeum quinone reductase (accession number AAL67860; see the GenBank database) is similar to a protein expressed by Paracoccidioides brasiliensis (AAL50803; 66% identical) (5), to a protein encoded by the S. cerevisiae PST2 gene (CAA98854; 64% identical), and to a previously reported quinone reductase from the white-rot basidiomycete Phanerochaete chrysosporium (AAD21025; 56% identical) (1). The G. trabeum reductase also exhibited lower levels of similarity with some other known and putative quinone reductases from fungal and plant sources (16), including one encoded by a minor allergen gene of Arabidopsis thaliana (CAB16805; 42% identical).
Although we were unable to identify the N terminus of the G. trabeum reductase, the data suggest two possibilities. One is that translation of the mRNA starts with Met-55 of the sequence which we submitted (AAL67860), thus yielding a 21.9-kDa mature protein without posttranslational processing. However, it seems more likely that translation starts with Met-1 to give a 27.7-kDa precursor and that a leader sequence is then cleaved from the mature protein at an unknown location near Met-55. We draw this conclusion because putative leader sequences also occur in the mRNAs for the P. chrysosporium quinone reductase (1) and the A. thaliana allergen. The function of these leaders is unclear, but all three of them are hydrophilic, and the inferred N termini of the two fungal sequences exhibit substantial similarity.
DISCUSSION
Most of the G. trabeum Fenton system involves extracellular, nonenzymatic reactions in which electrons are transferred from hydroquinones or semiquinones to Fe3+ or O2. The fungus itself does not participate directly in these reactions, but it drives them by continuously reducing quinones back to hydroquinones so that the redox cycle can continue (12, 14). The quinone reductase that catalyzes this step is, accordingly, a critical component of the G. trabeum biodegradative arsenal. We found one major quinone reductase in cultures of G. trabeum grown under conditions that elicit high levels of extracellular quinone redox cycling. This result and the high catalytic efficiency of the reductase led us to hypothesize that G. trabeum uses this enzyme to drive Fenton chemistry.
However, an alternative (or perhaps additional) function for the G. trabeum reductase cannot be ruled out yet. This enzyme belongs to a widely distributed family of flavoprotein quinone reductases that are generally thought to detoxify intracellular quinones by maintaining them in the reduced form (16). Quinones are cytotoxic in part because they readily undergo intracellular one-electron reduction to semiquinones, which react rapidly with O2 to produce superoxide. By contrast, hydroquinones are relatively nontoxic because they undergo rapid one-electron oxidation to semiquinones only in the presence of transition metal oxidants, such as Fe3+, which are generally unavailable inside cells because they are sequestered in redox-inactive complexes (10). Since G. trabeum produces large amounts of quinones as natural metabolites (12, 14, 18), it may have an unusually high requirement for a quinone detoxification system.
Much of the current impetus for a better understanding of brown-rot mechanisms is aimed at devising better ways to inhibit wood decay. Approximately 10% of all trees cut in the United States go to replace decayed wood, and to minimize the losses, wood rot fungi are currently controlled with toxic, environmentally deleterious biocides, such as creosote, pentachlorophenol, and chromated copper arsenate (22). It would be advantageous to target these fungi with compounds that interfere more specifically with wood decay mechanisms. Given the importance of quinone metabolism in brown rot (8, 12, 14, 18) and white rot (1, 3, 4, 9), fungal quinone reductases that drive Fenton chemistry or prevent toxicity from quinones are potential targets that merit further investigation.
Acknowledgments
We are grateful to C. J. Houtman for a statistical analysis of the Fig. 1 data.
This work was supported by U.S. Department of Energy grant DE-FG02-94ER20140 to K.E.H.
REFERENCES
- 1.Akileswaran, L., B. J. Brock, J. L. Cereghino, and M. H. Gold. 1999. 1,4-Benzoquinone reductase from Phanerochaete chrysosporium: cDNA cloning and regulation of expression. Appl. Environ. Microbiol. 65:415-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backa, S., J. Gierer, T. Reitberger, and T. Nilsson. 1992. Hydroxyl radical activity in brown-rot fungi studied by a new chemiluminescence method. Holzforschung 46:61-67. [Google Scholar]
- 3.Brock, B. J., and M. H. Gold. 1996. 1,4-Benzoquinone reductase from the basidiomycete Phanerochaete chrysosporium: spectral and kinetic analysis. Arch. Biochem. Biophys. 331:31-40. [DOI] [PubMed] [Google Scholar]
- 4.Brock, B. J., S. Rieble, and M. H. Gold. 1995. Purification and characterization of a 1,4-benzoquinone reductase from the basidiomycete Phanerochaete chrysosporium. Appl. Environ. Microbiol. 61:3076-3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunha, A. F., M. V. Sousa, S. P. Silva, R. S. Jesuino, C. M. Soares, and M. S. Felipe. 1999. Identification, N-terminal region sequencing and similarity analysis of differentially expressed proteins in Paracoccidioides brasiliensis. Med. Mycol. 37:115-121. [PubMed] [Google Scholar]
- 6.Ek, M., J. Gierer, and K. Jansbo. 1989. Study on the selectivity of bleaching with oxygen-containing species. Holzforschung 43:391-396. [Google Scholar]
- 7.Gilbertson, R. L., and L. Ryvarden. 1986. North American polypores. Fungiflora, Oslo, Norway.
- 8.Goodell, B., J. Jellison, J. Liu, G. Daniel, A. Paszczynski, F. Fekete, S. Krishnamurthy, L. Jun, and G. Xu. 1997. Low molecular weight chelators and phenolic compounds isolated from wood decay fungi and their role in the fungal biodegradation of wood. J. Biotechnol. 53:133-162. [Google Scholar]
- 9.Guillén, F., C. Muñoz, V. Gómez-Toribio, A. T. Martínez, and M. J. Martínez. 2000. Oxygen activation during oxidation of methoxyhydroquinones by laccase from Pleurotus eryngii. Appl. Environ. Microbiol. 66:170-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halliwell, B., and J. M. C. Gutteridge. 1999. Free radicals in biology and medicine, 3rd ed. Oxford University Press, Oxford, United Kingdom.
- 11.Hirano, T., H. Tanaka, and A. Enoki. 1997. Relationship between production of hydroxyl radicals and degradation of wood by the brown-rot fungus Tyromyces palustris. Holzforschung 51:389-395. [Google Scholar]
- 12.Jensen, K. A., Jr., C. J. Houtman, Z. C. Ryan, and K. E. Hammel. 2001. Pathways for extracellular Fenton chemistry in the brown rot basidiomycete Gloeophyllum trabeum. Appl. Environ. Microbiol. 67:2705-2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerem, Z., W. Bao, and K. E. Hammel. 1998. Rapid polyether cleavage via extracellular one-electron oxidation by a brown-rot basidiomycete. Proc. Natl. Acad. Sci. USA 95:10373-10377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerem, Z., K. A. Jensen, and K. E. Hammel. 1999. Biodegradative mechanism of the brown rot basidiomycete Gloeophyllum trabeum: evidence for an extracellular hydroquinone-driven fenton reaction. FEBS Lett. 446:49-54. [DOI] [PubMed] [Google Scholar]
- 15.Leitner, C., J. Volc, and D. Haltrich. 2001. Purification and characterization of pyranose oxidase from the white rot fungus Trametes multicolor. Appl. Environ. Microbiol. 67:3636-3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matvienko, M., A. Wojtowicz, R. Wrobel, D. Jamison, Y. Goldwasser, and J. I. Yoder. 2001. Quinone oxidoreductase message levels are differentially regulated in parasitic and non-parasitic plants exposed to allelopathic quinones. Plant J. 25:375-387. [DOI] [PubMed] [Google Scholar]
- 17.McFee, W. W., and E. L. Stone. 1966. The persistence of decaying wood in the humus layers of northern forests. Soil Sci. Soc. Am. Proc. 30:513-516. [Google Scholar]
- 18.Paszczynski, A., R. Crawford, D. Funk, and B. Goodell. 1999. De novo synthesis of 4,5-dimethoxycatechol and 2,5-dimethoxyhydroquinone by the brown rot fungus Gloeophyllum trabeum. Appl. Environ. Microbiol. 65:674-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prati, L., and M. Rossi. 1995. A simple route to 4,5-dialkoxy-o-quinones by catalytic oxidation of phenol. Gazz. Chim. Ital. 125:83-86. [Google Scholar]
- 20.Raeder, U., and P. Broda. 1985. Rapid preparation of DNA from filamentous fungi. Lett. Appl. Microbiol. 1:17-20. [Google Scholar]
- 21.Schlosser, D., K. Fahr, W. Karl, and H.-G. Wetzstein. 2000. Hydroxylated metabolites of 2,4-dichlorophenol imply a Fenton-type reaction in Gloeophyllum striatum. Appl. Environ. Microbiol. 66:2479-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zabel, R. A., and J. J. Morell. 1992. Wood microbiology: decay and its prevention. Academic Press, San Diego, Calif.