

Abstract
Multicomponent supramolecular polymer gels are a class of soft matter materials which form via the assembly of two or more small molecules. Different structures can be generated with interesting potential functions and applications. Insight into the assembly mechanism is key in the design of these systems for specific applications. Herein, a series of hydrogels with diverse structures and functionalities were synthesised. Using dynamic covalent chemistry as a key tool we show that it is possible to control the monomer assembly, forming both self-sorted and co-assembled polymers and gels from the same initial components. The hierarchical structure of the gels is difficult to elucidate. We emphasise the significance of small-angle neutron scattering (SANS) and spin-echo SANS (SESANS) measurements in characterising these intricate assemblies and demonstrate that these techniques are able to differentiate among self-sorted and co-assembled structures even when using chemically similar components.
Subject terms: Supramolecular polymers, Self-assembly
Multicomponent supramolecular polymer gels can possess useful functions and applications, but the mechanisms of their assembly are not fully understood. Here, the authors use dynamic covalent chemistry as a tool to demonstrate that it is possible to control the monomer assembly, forming both self-sorted and co-assembled polymers and gels from the same initial components.
Introduction
Supramolecular gels can form via the assembly of low molecular weight gelators (LMWGs), acting as monomers, through reversible non-covalent interactions into supramolecular polymers that form a self-assembled fibrillar network (SAFiN)1,2. The reversibility of the non-covalent interactions instils an enhanced tunability and, as a result, these systems have a large range of current and future applications spanning diverse fields such as organic electronics, drug delivery, and cell culture media, to name but a few2–6.
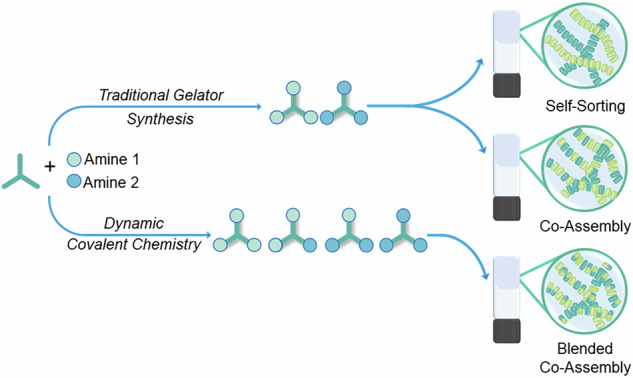
Supramolecular polymerisation of two or more different components can result in the formation of a multicomponent gel or polymer and allows fine-tuning of network properties such as the mechanical strength, spatio-temporal stability and electronic properties2,7,8. However, controlling the supramolecular co-polymerisation is not trivial. Different outcomes are possible, including narcissistic self-sorting, where each LMWG will form separate fibres; or at the opposite extreme, random co-assembly, where the LMWGs are distributed among the fibres in a non-ordered and unpredictable manner. There are, of course, several intermediate situations between these two extremes, such as block co-assembly or interweaved fibrils. (Fig. 1)9,10. Although predicting the assembly mechanism is difficult, by considering interaction energies between the building blocks, thermodynamic control over the assembly process can be established6. Self-sorted systems have been designed by selecting monomers with structural9,11–15/chiral mismatch16 and thermal annealing17–19 whereas co-assembly can be promoted by exploiting complementary electrostatics or introducing charge transfer interactions9,20–28. Many of these tools and ideas have been and will need to be utilised to develop out-of-equilibrium systems27–34.
Fig. 1. Assembly mechanisms in supramolecular gels.
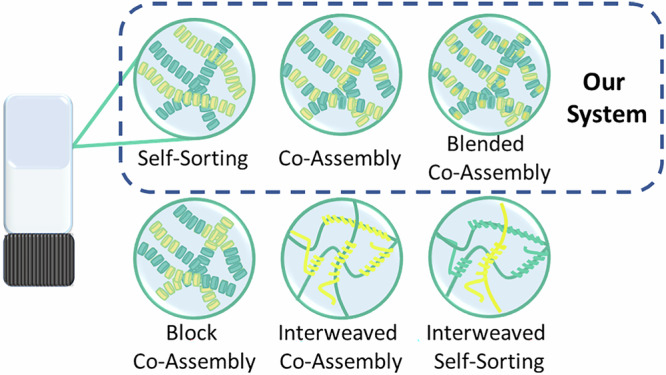
Schematic representation of some of the different supramolecular systems and gel networks formed from multicomponent assembly.
One of the great challenges is the formation of both self-sorted and co-assembled supramolecular entities from the same building blocks, with very few examples previously reported9,28,29. Since assemblies are typically formed under thermodynamic control, by considering alternative kinetic pathways, it is possible to activate sequence control during assembly. A recent report on the use of chemical fuels and cycles by Singh, Hermans and co-workers shows how selecting monomers with different chemical reactivity can push a normally co-assembled system into a self-sorted set of assemblies28. Sequence-controlled supramolecular structures were also obtained by Sarkar et al. using kinetic pathways and seeding to direct their thermodynamically controlled systems to self-sort or form block co-polymers9. By astute selection of differing triggers for the induction of assembly, Adams and co-workers have shown that it is possible to switch between co-assembled (by anti-solvent) to self-sorted (kinetic pH change) systems29.
Here, we show that blending of the chemical functionality in multi-component systems makes it possible to switch between either a self-sorted pair of supramolecular gel networks or a co-assembled network. We utilise dynamic covalent chemistry to make multifunctional LMWGs and lock that dynamic functionality into specific patterns using a non-reversible tautomerisation, as previously demonstrated by our group27. This additional complexity then adds to the statistical assembly mechanism associated with the gel formation. The simple hypothesis for this work is that the more diverse and disordered the functional groups, the more likely the co-assembly of the monomers into a multicomponent supramolecular polymer. By contrast, a binary, less disordered, multicomponent mixture prefers to self-sort by sticking to familiar functionality, and it is thermodynamically less likely to co-associate.
Distinguishing between the different assembly mechanisms within gels is particularly challenging. Gels are constituted primarily of solvent (>95% w/w) and, therefore, common laboratory techniques, such as wide-angle X-ray diffraction (XRD) and microscopy, can provide useful insight but have limitations. This is because artefacts and/or structural changes may arise during sample preparation (e.g. drying)35,36. To circumvent these issues, non-destructive techniques such as small-angle neutron scattering (SANS) can be employed to probe the structure of the as-prepared, solvated samples35,37.
SANS measurements of the scattered intensity as a function of the scattering angle provide unique information on the shape and size of the scattering object37,38. SANS can also be used to investigate the sol-to-gel transition and/or to study the finer features of the gel networks of various systems39–48. However, as SANS is a less readily available technique, typically only feasible at large-scale facilities, it is unsurprising that it has not found widespread use in characterising supramolecular gels49.
The small LMWG molecules self-assemble into long fibres, which entangle and physically cross-link to form SAFiNs. Understanding the mechanism by which LMWGs pack into highly hierarchical structures requires the use of complementary techniques able to probe a very wide range of length scales. Recently, Adams and co-workers have championed the use of SANS to study the assembly mechanism of their multicomponent gels50,51. To look at longer length scales, ultra-small-angle neutron scattering (USANS) has been used52,53. Spin-echo small-angle neutron scattering (SESANS) can also be employed for this purpose54,55 but the technique has yet to be used to study supramolecular gel networks in detail.
Due to the structural similarity of the LMWGs used in this study, the elucidation of our assemblies was only possible by conducting SANS with complementary SESANS measurements. The work presented here can be used to elucidate design principles for controlling the supramolecular polymerisation, which can lead to programmable and information-rich materials.
Results and discussion
Gelator synthesis and selection
Three different building blocks were used to prepare LMWGs. Compounds 1 and 2 (Fig. 2a) are positional isomers of each other. Compound 3 contains the additional electron-withdrawing CF3 unit (Fig. 2a). Multicomponent tripodal ketoenamine-based hydrogels with a triformylphloroglucinol core can be synthesised by two distinct routes27,56. Route A involves the mixing of two ‘pre-formed’ LMWGs (e.g. G111 with G222), whereas route B describes the reaction of two different aminobenzoic acids with 2,4,6-triformylphloroglucinol (Fig. 2a) to generate a four-component mixture (e.g. a gelator mixture G12 would be G111, G112, G122, and G222 in equal quantities). The aminobenzoic acid components were sourced commercially, while the synthesis of 2,4,6-triformylphloroglucinol has been widely published57–61. Syntheses and characterisation data are available in the “Synthesis and Characterisation” section of the SI. In route B (Fig. 2b) all starting materials (2,4,6-triformylphloroglucinol and two aminobenzoic acids) are reacted together simultaneously prior to gelation. The first step of the reaction consists of the formation of imine groups. Dynamic covalent chemistry62 operates during the formation of the imine intermediates and, provided sufficient time is allowed for equilibrium to be reached, an equal quantity of four gelators (Fig. 2b) is formed. As shown elsewhere27 the dynamic covalent chemical distribution for the reaction is 1 : 1 : 1 : 1 from HPLC analysis. This equal ratio of the LMWGs is subsequently locked in place by the tautomerisation27,56.
Fig. 2. Chemical structure and characteristics of LMWGs and gels.
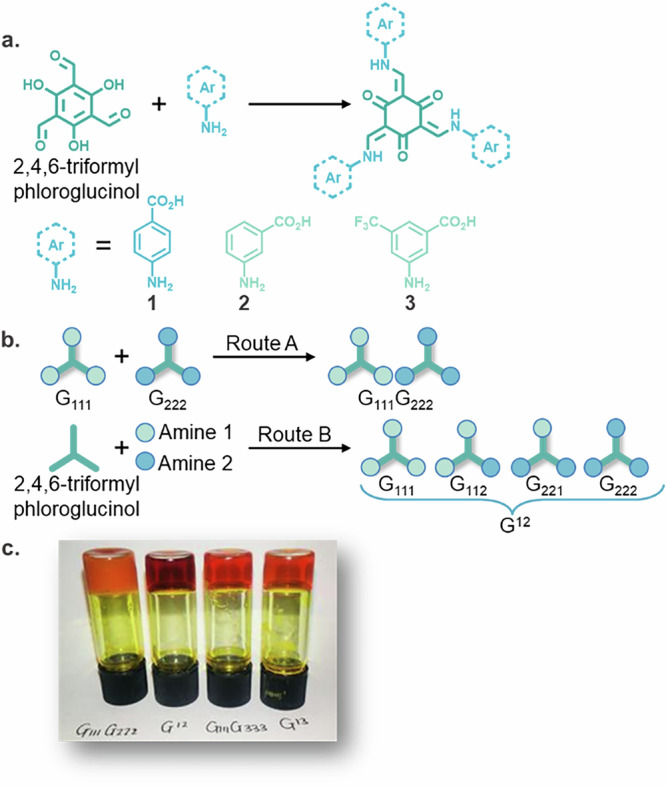
a Synthesis of ketoenamine-based hydrogels. b Pictorial representation of the LWMGs formed by different gelation routes. c Multicomponent gels showing differences in transparency.
Gelation is triggered by dissolving the LMWGs at high pH (NaOH/H2O, above the apparent pKa value of all starting materials and products) followed by the addition of glucono-δ-lactone (GdL). GdL acts as a slow release of protons and thus lowers the overall pH of the system below the apparent pKa of the LMWGs, reducing their solubility and resulting in the formation of a solid network (Table S1)63. In our previous work27,56, we reported the synthesis of the two gelator mixes G111G333 and G13, which, as shown in that work, are effectively 1:1 and 1:1:1:1 in terms of components, respectively. These gels are visually indistinguishable (Fig. 2c), and using literature-supported characterisation in the form of XRD, pH titrations to determine variation in the apparent pKa values, and solution 1H NMR, the two gelator mixes appeared to form a co-assembled fibrous network. This co-assembly occurs even though the apparent pKa values of G111 and G333 are different enough to expect self-sorting during the gradual lowering of the pH, as has been seen in the work by Adams and co-workers7,12,13. Dissimilar to the G111G333 and G13 gelator mixes, the new multicomponent gels G111G222 and G12 are visually different, as shown in Fig. 2c where G111G222 is opaque, while G12 is transparent. We thus aimed to determine if different assembly mechanisms were occurring and, consequently, if two distinct gel networks were formed. Compounds 1 and 2 are positional isomers of each other; thus, the apparent pKa values of G111 and G222 are nearly identical (6.1–5.8 and 6.5–5.9, respectively). Due to this similarity, any analytical technique following the kinetics of formation would not be able to resolve what type of assembly was occurring, self-sorted or co-assembled. As expected, the use of NMR for following the kinetics of assembly provided no notable variation (Fig. S7) while the XRD for single-component G111, G222 and multicomponent G111G222 and G12 are very similar. (Figs. S8 to S10) In previous work and as part of this study27,56 we viewed similar gels using microscopy (SEM, AFM, TEM, and super-resolution microscopy), but the resolution offered by these techniques was found to be insufficient for comparing the different gel networks. We have attempted to re-image the new gels with no further improvements in image quality; thus, we have not presented images for the materials in this paper. To overcome these limitations, we turned to neutron scattering techniques. As discussed below, SANS and SESANS offer unique tools able to distinguish between different assemblies and networks, including co-assembly or self-sorting processes.
Single-component gels: determination of assembly model
G111, G222 and G333 all form single-component hydrogels. We have described previously the chemical and gel properties of G111 and G333 and full characterisation of G222 is reported in the supplementary information27,56.
The LMWG monomers are discotic molecules. Molecular dynamic simulations (see “Molecular Dynamics Simulations” in SI for detail) show that they form one-dimensional face-to-face stacks stabilised by π-π interactions and intramolecular hydrogen bonding allowing for a rigid structure, reminiscent of their liquid crystal and covalent organic framework counterparts64–67.
Despite their chemical similarities, single-component gels of G111 and G222 are strikingly different in appearance, with the latter forming a fully transparent gel. A difference in transparency is typically a result of the width of gel fibres and the extent of aggregation within the system1–5,7,8. As discussed later, through model fitting, SANS provides a measure of a range of structural parameters of the fibres within the gel networks and therefore allows us to probe the underlying reasons for the differences in optical transparency without drying the gel samples35,41.
The SANS data of the pure gels are plotted in Fig. 3a. These samples were measured at a fixed concentration (1 wt%) in D2O to provide contrast between the solid network and aqueous solution. The notable difference between the scattering profiles indicates a difference in the structure of the primary fibres (Fig. 3).
Fig. 3. Comparison between SANS data of pure gels.
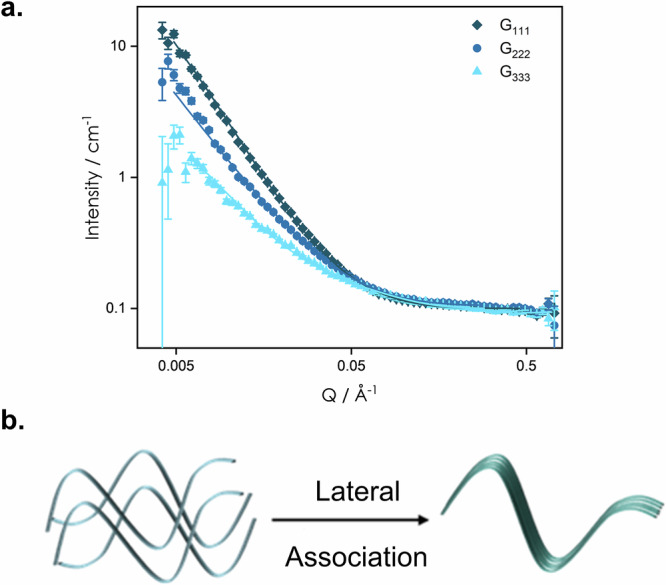
a Scattering curves for pure gels G111, G222 and G333 (symbols) with a fit to a flexible elliptical cylinder model (lines). b Schematic showing lateral association of fibres resulting in an elliptical cross-section and decreased flexibility of the fibres.
The scattering vector, Q, is inversely proportional to the size of the scattering objects. Thus, the absence of a plateau at the lowest experimental Q values (i.e. the absence of a detectable, shape-independent Guinier region) suggests that the length of our fibres lies outside the size accessible (d = 2π/Q < 151 nm) with the probed Q range. As a result, the scattering patterns will be most representative of changes to the fibril diameters.
A Q dependency of -1 is expected for cylindrical rod-like scatterers and is seen frequently in the literature for supramolecular gels44,68 and benzene-1,3,5-tricarboxamide-based supramolecular polymers, which have a similar C3 symmetric, discotic shape as our LMWGs69–71. However, fitting our data to a cylinder or a flexible cylinder model proved unsuccessful. To determine the Q dependence, the SANS data were fitted to a power law:
| 1 |
where I(0) is the scale, α characterises the Q dependence and B represents the background. Using Eq. (1), Q dependencies of -2.1, -1.8 and -1.6 were determined for gels G111 to G333, respectively. Table 1 presents a selection of parameters determined from fits of all the SANS samples measured. Full sets of fitting parameters are listed within the SI (Tables S3 and S4). A Q dependency closer to −2 (α = 2) suggests the presence of ribbon-like fibres, giving a lamellar-type structure (Fig. 3b). This is in line with the XRD data collected from dried samples (Figs. S8 to S10) in which peak positions of Bragg-like reflections have ratios of 1,2,3,4, which is consistent with lamellar-type packing72. From the Q dependency and comparison with similar systems in literature, a flexible elliptical cylinder model71–74 was chosen and fitted to the SANS data of the gels to extract dimensional information (Fig. 3, a detailed description of this process is provided in the “Small-Angle Neutron Scattering (SANS)” section of the SI)48,75,76. The elliptical cross-section is likely to arise from the lateral association of fibres (Fig. 3b)48,75,76.
Table 1.
Selected parameters from fits using the power law (Eq. 1) and the flexible elliptical cylinder77 models for all measured gels
| Gel | αa | Radius /Å | Axis ratio | Kuhn length /Å |
|---|---|---|---|---|
| G111 | 2.125 ± 0.006 | 8.5 ± 0.1 | 14 ± 2 | 281 ± 41 |
| G222 | 1.788 ± 0.008 | 7.4 ± 0.4 | 2.80 ± 0.01 | 40 ± 5 |
| G333 | 1.38 ± 0.01 | 8.50 ± 0.03 | 2.4 ± 0.1 | 146 ± 6 |
| G111G222 | 2.241 ± 0.006 | 7.9 ± 0.5 | 12 ± 2 | 144 ± 8 |
| G12 | 1.752 ± 0.009 | 7.60 ± 0.02 | 2.5 ± 0.2 | 45 ± 6 |
| G111G333 | 1.544 ± 0.008 | 8.5 ± 0.3 | 3.1 ± 0.1 | 97 ± 4 |
| G13 | 1.64 ± 0.01 | 8.47 ± 0.03 | 2.5 ± 0.1 | 88 ± 3 |
aPower law exponent, α, was determined by fitting the SANS data to Eq. (1). Fits and the full set of fitting parameters are given in the SI.
The flexible cylinder elliptical model in SasView77, used to fit the SANS data, is a function of the following structural parameters: (1) the radius of the cylinder, (2) the axis ratio, i.e. the major radius divided by the minor radius, (3) the Kuhn length characterising the flexibility of the cylinder and (4) the contour length. The minor radii for the three gels were found to vary between 7.4 and 8.5 Å, which is approximately the size of one molecule, as confirmed through molecular dynamics simulations (see “Molecular Dynamics Simulations” in SI for details). Hence, the larger the axis ratio, the greater the degree of lateral association and the more elliptical the cross-section. Values of axis ratios in Table 1 follow the same trend as α from Eq. 1, and so the two models (Eq. 1 and the elliptical model) indicate that the greatest extent of lateral association occurs for G111, which is in agreement with the opaque appearance of the gel.
The Kuhn length parameter gives an indication of the flexibility of the fibres. The larger the difference between the Kuhn and the cylinder lengths, the more flexible the fibres. For pure and multicomponent gels of G111 and G222 the contour length was fixed at the same value and therefore the smaller Kuhn lengths of G222 and G12 indicate more flexible fibres.
Time, frequency, and amplitude sweep rheological measurements were carried out under the same conditions as the SANS data to confirm the smaller mesh size (which normally arises in a more robust gelatinous material)1–5,7,8. As SANS measurements were carried out in D2O, the effect of switching from H2O to D2O was also investigated, and it was found that the rate of gelation decreased. This is unsurprising as the rate of GdL hydrolysis is reduced in a deuterated solvent78. Regardless, overall trends are consistent and therefore complete rheological measurements were carried out in H2O. The time sweeps (Fig. 4) show that G222 and G333 initially form mechanically stronger gels than G111, confirming the hypothesis that they form more compact, interlocked network structures. However, once the maximum strength is reached, a decrease in G′ values is observed, followed by a lower plateau at longer time sweeps. This behaviour is indicative of syneresis, i.e. shrinking of the gel as water is expelled. Lateral association of fibres is known to affect the mechanical strength of a gel and its stability. We believe that the absence of syneresis observed for G111 (Fig. 4) is consistent with the larger extent of lateral association and greater stiffness of this gel, as indicated by the SANS measurements (Table 1).
Fig. 4. Time sweep measurements for pure gels.
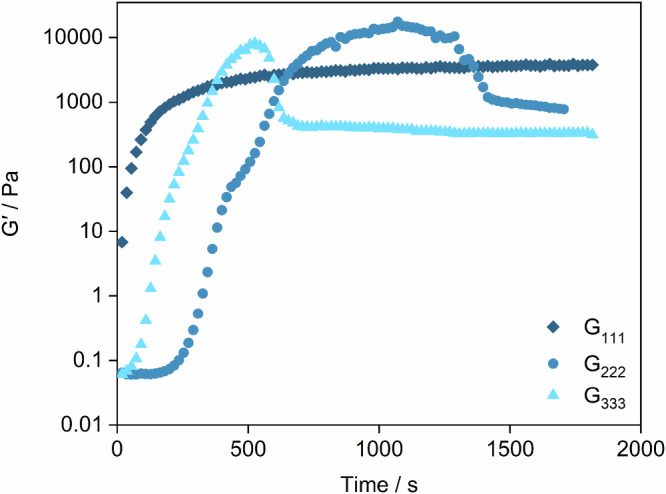
Time sweeps for G111, G222 and G333 showing the initial robustness of the G222 and G333 before syneresis occurs.
Due to the syneresis, limited information can be extracted from the frequency or amplitude sweeps; however, all are consistent with the expected behaviour of gel materials (Figs. S24 to S26).
SESANS was used to further investigate the network structures of G111, G222 and their multicomponent gels. SESANS is an emerging technique that, in contrast to the SANS measurements obtained in reciprocal (Q) space, measures the extent of the depolarisation of a spin-polarised beam as a function of a real-space parameter, the spin-echo length. The spin-echo length can be defined as the correlation distance probed in the sample. For a detailed explanation of the SESANS technique, we refer readers to the referenced articles54,79.
The SESANS data in Fig. 5 display the normalised SESANS signal, ln(P/P0)/(tλ2), where P is the polarisation of the beam through the sample, P0 is the polarisation of the reference beam going through the empty instrument, t is the sample thickness, and λ is the wavelength. The depolarisation depends on t and λ, so this normalised parameter removes instrumental dependence. As shown in Fig. 5c, e the normalised SESANS signal from our samples is constant as a function of spin-echo length, which indicates that there are no structural features at the length scales probed (between approximately 0.9 and 13.8 μm) and that the samples appear homogeneous. We note that the depolarisation effect varies from sample to sample (Fig. 5).
Fig. 5. Structural and rheological properties of pure and multicomponent gels.

a Time sweeps for pure gels G111, G222 and their G111G222 and G12 multicomponent gels. b SESANS data for G111, G222 and their G111G222 and G12 multicomponent gels. The lines are guides to the eye. c SESANS data for the 1:3 and 3:1 G111G222 and G12 multicomponent gels showing the general shift towards more G111 or G222 characteristics. d Scattering curves for pure gels G111, G222 and their G111G222 and G12 multicomponent gels with fit to an elliptical cylinder model. e Scattering curves for pure gels G111, G333 and their G111G333 and G13 multicomponent gels with fit to an elliptical cylinder model. f Image of the mixed ratios gels of G111, G222 and their G111G222 and G12 multicomponent versions, with the transparency indicating how the gels are altering.
The normalised SESANS signal, ln(P/P0)/(tλ2), is related to the contrast, the volume fraction and the correlation length (see “Spin-Echo Small-Angle Neutron Scattering (SESANS)” section in SI for details). Due to the similar structures of our LMWGs, the contrast is the same for all samples. By doing measurements at the same concentration, we can assume a constant volume fraction. Thus, all parameters related to the loss in polarisation are identical, except for the correlation length54. Therefore, by measuring the extent of depolarisation, we can directly compare the correlation lengths of our samples.
We note that to increase the SESANS signal, the SESANS samples were five times more concentrated (5 wt%) than those used for the SANS experiments (1 wt%). Additional SANS measurements were run on the pure gels G111 and G222 at the higher concentration, in a thicker cell, to mimic the SESANS conditions (Fig. S18). In the absence of structural changes, the scattered intensity should be directly proportional to the concentration of the scattering objects. As shown in Figs. S18 to S20, this is not the case for our gels. A network structure forms, with increasing concentration and therefore the scattering data provide a measure of the network mesh size.
G111 was found to depolarise the beam to a greater extent than G222, indicating a larger mesh size (Fig. 5c). The Kuhn length extracted from the SANS data (Table 1) indicates that G222 is much more flexible than G111. Therefore, the shorter correlation length associated with the SESANS depolarisation of G222 is unsurprising given that the thinner and more flexible fibres of these gels would have a greater propensity to form entanglements1–5,7,8. This is fully supported by SANS measurements at 5 wt% (Fig. S20), which are well described by a correlation length model (see section 6.4 of the SI).
Overall, it can be hypothesised that the greater propensity for lateral association, as seen for G111, is related to the substitution pattern on the aromatic amines, with the para-substituted amines producing the most elliptical and least flexible fibres which in turn produce mechanically weaker gel networks.
Thus, the combined SANS and SESANS data provide insight into the hierarchical structure of the gel networks. The initial columnar aggregates, confirmed by XRD analysis and molecular dynamics simulations, undergo lateral association into elliptical fibres, as shown by fitting the SANS data to a flexible elliptical cylinder model. From the SANS fitting parameters, we can estimate the degree of association (see “Determination of the Degree of Association” section in the SI). A large degree of association (12–15 strands) limits the flexibility of the fibres, evident from the larger Kuhn length, and consequently, the SAFiN mesh size, resulting in opaque gels. Conversely, transparent gels have a limited aggregation of approximately 2-3 strands forming a larger number of fibre,s which are more flexible (smaller Kuhn length) and thus a denser and mechanically stronger gel network is formed. It can be hypothesised that the greater propensity for lateral association, as seen for G111, is related to the substitution pattern on the aromatic amines, with para-substituted amines exhibiting the most elliptical fibril cross sections.
With these clear assembly models for the pure gels in hand, we can now turn to the data and models for the multicomponent gels.
Multicomponent gels: self-sorted and/or co-assembly
Compounds 1 and 2 are regioisomers and consequently G111, G222, G111G222 and G12 exhibit near identical apparent pKa values. As a result, pH titrations and 1H NMR could not be used to distinguish between different assembly mechanisms, as was the case for G111G333 and G1327,56. As the apparent pKa values are nearly identical, it is highly unlikely that the kinetic behaviour supplied by the judicious use of GdL, as seen by others29, can contribute to the self-sorting of the different gelators. So this study aims to answer the question: Do compounds 1 and 2 match the assembly of 1 and 3? The XRD patterns for the xerogels (Figs. S8 to S10) exhibit only minor differences, and no conclusion can be drawn with regard to the assembly mechanism.
In multicomponent gels, the two LMWGs may work synergistically to improve the mechanical properties or antagonistically, producing a weaker gel than the individual LMWGs35. Time sweeps during gel formation showed that the maximum value for the elastic modulus (G′) is higher for the multicomponent gels than for G111 (Fig. 5a). It can therefore be concluded that the presence of G222 strengthens both multicomponent gels. Similar to G222, the G12 gel exhibits syneresis, albeit delayed with respect to the pure gel (Fig. 5a). Overall, different gel properties are formed via the different synthesis routes, but it is unclear if this is the product of different assembly mechanisms or the chemical differences arising from the dynamic covalent chemistry giving mechanically different but structurally similar networks.
To fully characterise our gels, SANS measurements were conducted, and prior to a detailed analysis, it is immediately obvious that the scattering curves of the G111G222 and G12 multicomponent gels are significantly different (Fig. 5d). Again, power law and flexible elliptical cylinder models were fitted to the data, and values of fitting parameters are reported in Table 1.
Further details regarding the fitting procedure can be found in sections 6.2 and 6.3 of the SI. As shown in Table 1, all fitting parameters for the G111G222 gel are closer to those of G111 compared to G222, with a significant amount of lateral association represented by the relatively large axis ratio and the Q-2.2 dependence. Conversely, the values extracted for the G12 gel are closer to those of G222. The same trend is observed for SESANS measurements (Fig. 5c), with both the G111 and G111G222 gels depolarising the beam to a greater extent than both the G12 and G222 gels. This trend substantiates longer correlation lengths for the G111 and G111G222 gels, resulting from limited entanglements due to the thicker, less flexible (based on the Kuhn length values) and less abundance of the fibres.
In a self-sorting system, the scattering would be expected to be more representative of G111 as its fibres are larger and would therefore dominate the scattering, as is the case for the G111G222 gels. However, the scattering profile for the G12 gel resembles closer that of G222 which suggests a co-assembly mechanism is in play and with interdigitating LMWGs, the lateral association typically seen for G111 is limited.
As mentioned previously, a common approach for designing self-sorting supramolecular polymers and gels consists of combining substantially different compounds9,11–15. Therefore, with LMWGs 1 and 2 being regioisomers, it is unsurprising that G12 would form a co-assembled gel network. However, by changing the synthesis route, unprecedented behaviour arises and a self-sorting mechanism is accessed from the same starting materials for G111G222. It is believed that this propensity for homo-recognition despite similar molecular structures is a result of the differing substitution pattern on the peripheral leg units, resulting in the formation of LMWGs with slightly different shapes10.
For comparison purposes and to further corroborate the co-assembling mechanism previously suggested for both G111G333 and G13 gels27, SANS experiments were carried out. As G111 and G333 have distinctly different apparent pKa values, we can highlight that the kinetics of the pH change during the gel setting appear not to influence the selection to co-assembly and indicate that the intermittency of the components is likely occurring before gelation27. A different scenario than for G111G222 and G12 emerges, where the scattering curves for the G111G333 and G13 gels are similar (Fig. 5e). This is consistent with both gels co-assembling. As was the case for the G12 gel, which is also thought to co-assemble, the lateral association is limited for both gels, with the elliptical cylinder model returning a small axis ratio and the power law model returning a lower Q dependency in comparison to the G111 and G111G222 gels (Table 1).
Therefore, counterintuitively, despite the substantial structural mismatch and differing apparent pKa values of the LMWGs, G111G333 and G13 form co-assembling SAFiNs via both synthetic mixing methods. However, co-assembly can be explained and triggered by the formation of donor-acceptor charge transfer-like π-π interactions20–25. MD simulations and literature highlight the π-π propensity of the monomers64–67. Due to the highly electron-withdrawing nature of the CF3 group, the aromatic rings of the 3 components will be electron-deficient with respect to the 1 components, and it is likely that ”aromatic donor-acceptor” interactions encourage co-assembly80. For 1 and 2-based compounds such charge transfer-like π-π interactions are not possible, and despite the similar structures, the different substitution pattern is sufficient to encourage self-recognition.
The switch to the co-assembly mechanism during G12 gelation (in contrast to the self-sorting of G111G222) is therefore a consequence of the presence of asymmetric LMWGs formed via the dynamic covalent chemistry and the blending of the monomers, which encourages diversity and discourages self-recognition
Having confirmed the switch in the assembly mechanism by changing the synthetic route for a 1:1 ratio mixture for G111G222 and G12, we used SESANS (Fig. 5c) and rheology (Fig. S30) to investigate the effect of varying the ratio of the components. As shown in Fig. 5f, the three G111G222 gels are visually opaque compared to their G12 counterparts, suggesting similar assembly mechanisms. For a 3:1 ratio, the difference between G111G222 and G12 gels is still evident (Fig. 5c). In the case of a co-assembling mechanism, mixed fibres are produced. and the presence of G222 prevents the greater degree of lateral association and longer correlation length seen for pure fibres of G111. Therefore, the lower depolarisation for the G12 gel is indicative of possible co-assembly and even with a higher ratio of G111 in the gel, the small amount of G222 in the mixed fibres still limits the association and thus, the correlation length. The G111G222 gel depolarises the beam to a greater extent, suggesting that a self-sorting mechanism is occurring and that the larger fibres and longer correlation lengths of G111 dominate the scattering as seen with the 1:1 ratio. From the rheology, it was found that the syneresis, previously observed for the G12 (1:1) gel, does not occur for the higher ratio of 1 (G12 3:1), and the gels are still different, with the G111G222 3:1 gel forming a tighter gel network. (Fig. S30) When the ratio is reversed, the situation is less clear. The extent of depolarisation for the G111G222 1:3 gel is no longer much greater than for G222 and the G12 1:3 gel. (Fig. 5c) This is to be expected due to the slightly more transparent appearance of the gel and could be indicative of a change in assembly from self-sorting to co-assembly. However, with only 25% G111 present, even in the case of self-sorting, there would be a reduced number of the larger fibres of G111, and this could account for the smaller correlation length, decreasing the depolarisation.
Conclusion
In this work, we have shown how a blending method can be used to overcome self-recognition of monomers within a supramolecular polymer, allowing for a switch from a self-sorted set of networks into a co-assembled network. A four-component mixture was synthesised using dynamic covalent chemistry, and the properties of the resulting gel (G12) were compared to the two-component gel mixture (G111G222). By using SANS (primarily) with complementary SESANS, MD, XRD, and rheological data, we have been able to establish the assembly mechanism responsible for forming the gel fibres. Properties of G12 and G111G222 are compared to those of previously reported gels (G111 and G333)27. We have shown that, depending on the strength of the π−π interactions between the different LMWGs, it is possible to form either self-sorted or co-assembled gels. The addition of an electron-withdrawing CF3 moiety leads to an electron-deficient π-system, which tends to co-assemble. This effect appears to dominate even though the differing apparent pKa values of the LMWGs should result in kinetic separation of the assembly through the use of the GdL. On the contrary, when using two structurally similar LMWGs with almost identical apparent pKas, 1 and 2, we find that homo-recognition is the preferred route, allowing self-sorted gels to be formed. By exploiting dynamic covalent chemistry, asymmetric LMWGs were formed. In this case, a co-assembled gel is obtained, thus providing a facile route for producing different gels for specific applications from the same components (1 and 2). We aim to use this technology to develop active components embedded into the gel’s fibres either in their self-sorted or co-assembled network, and we hope that the concept of blending supramolecular monomers can be applied to select between self-sorted or co-assembled supramolecular polymers with other potentially multifunctional monomers.
Methods
Materials
See Section 1.1 of Supplementary Information (SI).
Experimental techniques and MD simulations
See Sections 1.2 to 1.10 of SI.
Synthetic procedures
Characterisation of LMWGs
Sample preparation
See Sections 3 and 4 of SI.
Supplementary Information
The Supplementary Information file provides details on the methods used in this work and data analysis.
Supplementary information
Acknowledgements
M.J. would like to thank HWU and EPSRC for PhD funding from the EPSRC DTP. G.O.L. thanks the Royal Society of Edinburgh for a Scottish Government Personal Research fellowship while at HWU. We gratefully acknowledge ISIS for the SANS2D and SESANS beam time allocated for the RB2010573 and RB2010583 experiments. This work benefited from the use of the SasView application, originally developed under NSF Award DMR-0520547. SasView contains code developed with funding from the EU Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement No 654000.
Author contributions
All authors contributed to the project and approved the final version of the manuscript. Overall conceptualisation: G.O.L.: planning of neutron scattering experiments, data acquisition and interpretation: M.C.J., V.A., G.N.S., R.D., L.P.C., G.O.L. and F.V.; Computational work: S.R.E.; Experimental work, analysis and first drafting of manuscript, M.C.J.; writing, review and editing, G.O.L., F.V., S.R.E. and V.A.; supervision G.O.L., F.V. and V.A.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
The SANS2D experiment at ISIS was allocated beam time under experiment number RB2010573 and the SESANS on LARMOR was assigned the experiment number RB2010583.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Filipe Vilela, Email: f.vilela@hw.ac.uk.
Valeria. Arrighi, Email: v.arrighi@hw.ac.uk
Gareth O. Lloyd, Email: glloyd@lincoln.ac.uk
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-025-01657-1.
References
- 1.Steed, J. W. Supramolecular gel chemistry: developments over the last decade. Chem. Commun.47, 1379–1383 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Draper, E. R. & Adams, D. J. Low-molecular-weight gels: the state of the art. Chem3, 390–410 (2017). [Google Scholar]
- 3.Sangeetha, N. M. & Maitra, U. Supramolecular gels: functions and uses. Chem. Soc. Rev.34, 821 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Dong, R., Pang, Y., Su, Y. & Zhu, X. Supramolecular hydrogels: synthesis, properties and their biomedical applications. Biomater. Sci.3, 937–954 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Babu, S. S., Prasanthkumar, S. & Ajayaghosh, A. Self-assembled gelators for organic electronics. Angew. Chem. Int. Ed.51, 1766–1776 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Das, A. et al. Supramolecular copolymers: structure and composition revealed by theoretical modeling. J. Am. Chem. Soc.139, 7036–7044 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buerkle, L. E. & Rowan, S. J. Supramolecular gels formed from multi-component low molecular weight species. Chem. Soc. Rev.41, 6089 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Raeburn, J. & Adams, D. J. Multicomponent low molecular weight gelators. Chem. Commun.51, 5170–5180 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Sarkar, A. et al. Self-sorted, random, and block supramolecular copolymers via sequence controlled, multicomponent self-assembly. J. Am. Chem. Soc.142, 7606–7617 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Safont-Sempere, M. M., Fernández, G. & Würthner, F. Self-sorting phenomena in complex supramolecular systems. Chem. Rev.111, 5784–5814 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Shigemitsu, H. et al. An adaptive supramolecular hydrogel comprising self-sorting double nanofibre networks. Nat. Nanotechnol.13, 165–172 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Morris, K. L. et al. Chemically programmed self-sorting of gelator networks. Nat. Commun.4, 1480 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Draper, E. R., Eden, E. G. B., McDonald, T. O. & Adams, D. J. Spatially resolved multicomponent gels. Nat. Chem.7, 848–852 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Cornwell, D. J., Daubney, O. J. & Smith, D. K. Photopatterned multidomain gels: multi-component self-assembled hydrogels based on partially self-sorting 1,3:2,4-dibenzylidene-D-sorbitol derivatives. J. Am. Chem. Soc.137, 15486–15492 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Prasanthkumar, S. et al. Organic donor–acceptor assemblies form coaxial p–n heterojunctions with high photoconductivity. Angew. Chem. Int. Ed.54, 946–950 (2015). [DOI] [PubMed] [Google Scholar]
- 16.He, H. et al. Chirality on dendrimers: “roll booster” of the molecule-level self-sorting assembly in two-component supramolecular gel system. Chem. Commun.56, 2983–2986 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Smith, M. M. & Smith, D. K. Self-sorting multi-gelator gels—mixing and ageing effects in thermally addressable supramolecular soft nanomaterials. Soft Matter7, 4856 (2011). [Google Scholar]
- 18.Moffat, J. R. & Smith, D. K. Controlled self-sorting in the assembly of ‘multi-gelator’ gels. Chem. Commun. 2009, 316–318 (2009). [DOI] [PubMed]
- 19.Singh, N. et al. Tandem reactions in self-sorted catalytic molecular hydrogels. Chem. Sci.7, 5568–5572 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao, K. V., Jayaramulu, K., Maji, T. K. & George, S. J. Supramolecular Hydrogels and High-Aspect-Ratio Nanofibers through Charge-Transfer-Induced Alternate Coassembly. Angew. Chem. Int. Ed.49, 4218–4222 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Frisch, H. et al. pH-switchable ampholytic supramolecular copolymers. Angew. Chem. Int. Ed.52, 10097–10101 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Xu, X.-D. et al. Coassembly of oppositely charged short peptides into well-defined supramolecular hydrogels. J. Phys. Chem. B114, 2365–2372 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Kumar, M., Venkata Rao, K. & George, S. J. Supramolecular charge transfer nanostructures. Phys. Chem. Chem. Phys.16, 1300–1313 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Wang, C. et al. Supramolecular amphiphiles based on a water-soluble charge-transfer complex: fabrication of ultralong nanofibers with tunable straightness. Angew. Chem. Int. Ed.48, 8962–8965 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Frisch, H. et al. Kinetically controlled sequential growth of surface-grafted chiral supramolecular copolymers. Angew. Chem. Int. Ed.55, 7242–7246 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Wang, F., Liao, R. & Wang, F. Pathway control of π-conjugated supramolecular polymers by incorporating donor-acceptor functionality. Angew. Chem. Int. Ed. Eng l. 62, e202305827 (2023). [DOI] [PubMed]
- 27.Foster, J. S., Prentice, A. W., Forgan, R. S., Paterson, M. J. & Lloyd, G. O. Targetable mechanical properties by switching between self-sorting and co-assembly with in situ formed tripodal ketoenamine supramolecular hydrogels. ChemNanoMat4, 853–859 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh, N., Lopez-Acosta, A., Formon, G. J. M. & Hermans, T. M. Chemically fueled self-sorted hydrogels. J. Am. Chem. Soc.144, 410–415 (2022). [DOI] [PubMed] [Google Scholar]
- 29.Ravarino, P. et al. Controlled annealing in adaptive multicomponent gels. Angew. Chem. Int. Ed.62, e202215813 (2023). [DOI] [PMC free article] [PubMed]
- 30.López-Gandul, L., Morón-Blanco, A., García, F. & Sánchez, L. L. Supramolecular block copolymers from tricarboxamides. Biasing co-assembly by the incorporation of pyridine rings. Angew. Chem. Int. Ed. 62, e202308749 (2023). [DOI] [PubMed]
- 31.Jansen, S. A. H., Su, H., Schnitzer, T., Vantomme, G. & Meijer, E. W. Temperature directs the majority-rules principle in supramolecular copolymers driven by triazine–benzene interactions. Chem. Eur. J.29, e202301726 (2023). [DOI] [PubMed]
- 32.Wang, Y. et al. Transient supramolecular hydrogels formed by aging-induced seeded self-assembly of molecular hydrogelators. Adv. Sci.7, 1902487 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura, K. et al. Phototriggered spatially controlled out-of-equilibrium patterns of peptide nanofibers in a self-sorting double network hydrogel. J. Am. Chem. Soc.143, 19532–19541 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Wang, Y. et al. Access to metastable gel states using seeded self-assembly of low-molecular-weight gelators. Angew. Chem. Int. Ed.58, 3800–3803 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Draper, E. R. & Adams, D. J. How should multicomponent supramolecular gels be characterised?. Chem. Soc. Rev.47, 3395–3405 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Weyandt, E. et al. How to determine the role of an additive on the length of supramolecular polymers? Org. Mater.02, 129–142 (2020). [Google Scholar]
- 37.Hollamby, M. J. Practical applications of small-angle neutron scattering. Phys. Chem. Chem. Phys.15, 10566 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Jackson, A. J. Introduction to Small-angle Neutron Scattering and Neutron Reflectometry. 1–24 (NIST Center for Neutron Research, 2008).
- 39.Dawn, A. et al. Investigating the effect of supramolecular gel phase crystallization on gel nucleation. Soft Matter14, 9489–9497 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Kumari, H. et al. Fluorous ‘ponytails’ lead to strong gelators showing thermally induced structure evolution. Soft Matter11, 8471–8478 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Gao, Y. et al. Supramolecular self-assembly of a model hydrogelator: characterization of fiber formation and morphology. Gels2, 27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin, A. D. et al. Controlling self-assembly of diphenylalanine peptides at high pH using heterocyclic capping groups. Sci. Rep.7, 43947 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jamieson, S. A. et al. Small angle neutron scattering (SANS) studies on the structural evolution of pyromellitamide self-assembled gels. Langmuir30, 13987–13993 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Willemen, H. M. et al. A small-angle neutron scattering study of cholic acid-based organogel systems. Langmuir20, 2075–2080 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Thomson, L., Schweins, R., Draper, E. R. & Adams, D. J. Creating transient gradients in supramolecular hydrogels. Macromol. Rapid Commun.41, 2000093 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Colquhoun, C. et al. The effect of self-sorting and co-assembly on the mechanical properties of low molecular weight hydrogels. Nanoscale6, 13719–13725 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Cardoso, A. Z. et al. The influence of the kinetics of self-assembly on the properties of dipeptide hydrogels. Faraday Discuss.166, 101 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Draper, E. R. et al. Using small-angle scattering and contrast matching to understand molecular packing in low molecular weight gels. Matter2, 764–778 (2020). [Google Scholar]
- 49.Arrighi, V. & Higgins, J. S. Structural investigation of polymers by neutron scattering. Plast., Rubber Compos.33, 313–330 (2004). [Google Scholar]
- 50.Cross, E. R., Sproules, S., Schweins, R., Draper, E. R. & Adams, D. J. Controlled tuning of the properties in optoelectronic self-sorted gels. J. Am. Chem. Soc.140, 8667–8670 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Sugiyasu, K., Kawano, S. -i, Fujita, N. & Shinkai, S. Self-sorting organogels with p−n heterojunction points. Chem. Mater.20, 2863–2865 (2008). [Google Scholar]
- 52.Nolan, M. C. et al. Optimising low molecular weight hydrogels for automated 3D printing. Soft Matter13, 8426–8432 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Hule, R. A. et al. Correlations between structure, material properties and bioproperties in self-assembled β-hairpin peptide hydrogels. Faraday Discuss.139, 251 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andersson, R., Heijkamp, L., Schepper, I. & Bouwman, W. Analysis of spin-echo small-angle neutron scattering measurements. J. Appl. Crystallogr.41, 868–885 (2008). [Google Scholar]
- 55.Rehm, C., Barker, J., Bouwman, W. G. & Pynn, R. DCD USANS and SESANS: a comparison of two neutron scattering techniques applicable for the study of large-scale structures. J. Appl. Crystallogr.46, 354–364 (2013). [Google Scholar]
- 56.Foster, J. S. et al. Gelation landscape engineering using a multi-reaction supramolecular hydrogelator system. J. Am. Chem. Soc.137, 14236–14239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chong, J. H., Sauer, M., Patrick, B. O. & MacLachlan, M. J. Highly stable keto-enamine salicylideneanilines. Org. Lett.5, 3823–3826 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Singh, A., Samanta, D. & Maji, T. K. Realization of oxygen reduction and evolution electrocatalysis by in situ stabilization of Co nanoparticles in a redox-active donor-acceptor porous organic polymer. ChemElectroChem6, 3756–3763 (2019). [Google Scholar]
- 59.Gottschling, K. et al. Molecular insights into carbon dioxide sorption in hydrazone-based covalent organic frameworks with tertiary amine moieties. Chem. Mater.31, 1946–1955 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li, X. et al. A Triformylphloroglucinol-based covalent organic polymer: synthesis, characterization and its application in visible-light-driven oxidative coupling reactions of primary amines. Chem. Res. Chin. Univ.36, 1017–1023 (2020). [Google Scholar]
- 61.Zou, L., Yang, X., Yuan, S. & Zhou, H.-C. Flexible monomer-based covalent organic frameworks: design, structure and functions. CrystEngComm19, 4868–4871 (2017). [Google Scholar]
- 62.Rowan, S. J., Cantrill, S. J., Cousins, G. R. L., Sanders, J. K. M. & Stoddart, J. F. Dynamic covalent chemistry. Angew. Chem. Int. Ed.41, 898–952 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Adams, D. J. et al. A new method for maintaining homogeneity during liquid–hydrogel transitions using low molecular weight hydrogelators. Soft Matter5, 1856 (2009). [Google Scholar]
- 64.Yelamaggad, C. V., Achalkumar, A. S., Shankar Rao, D. S. & Prasad, S. K. Self-assembly of C3h and Cs symmetric keto-enamine forms of tris(N-salicyl-ideneanilines) into columnar phases: a new family of discotic liquid crystals. J. Am. Chem. Soc.126, 6506–6507 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Achalkumar, A. S. & Yelamaggad, C. V. Light emitting, star-shaped tris(N-salicylideneaniline) discotic liquid crystals bearing trans-stilbene fluorophores: synthesis and characterization. Tetrahedron Lett.53, 7108–7112 (2012). [Google Scholar]
- 66.Yelamaggad, C. V., Achalkumar, A. S., Rao, D. S. S. & Prasad, S. K. A new class of discotic mesogens derived from Tris(N-salicylideneaniline)s existing in C3h and Cs keto-enamine forms. J. Org. Chem.72, 8308–8318 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Kandambeth, S. et al. Construction of crystalline 2D covalent organic frameworks with remarkable chemical (acid/base) stability via a combined reversible and irreversible route. J. Am. Chem. Soc.134, 19524–19527 (2012). [DOI] [PubMed] [Google Scholar]
- 68.Guilbaud, J.-B. & Saiani, A. Using small angle scattering (SAS) to structurally characterise peptide and protein self-assembled materials. Chem. Soc. Rev.40, 1200–1210 (2011). [DOI] [PubMed] [Google Scholar]
- 69.Besenius, P. et al. Controlling the growth and shape of chiral supramolecular polymers in water. Proc. Natl Acad. Sci. USA107, 17888–17893 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Basuyaux, G. et al. Extra hydrogen bonding interactions by peripheral indole groups stabilize benzene-1,3,5-tricarboxamide helical assemblies. Chem. Commun.55, 8548–8551 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Desmarchelier, A. et al. Tuning the nature and stability of self-assemblies formed by ester benzene 1,3,5-tricarboxamides: the crucial role played by the substituents. Soft Matter12, 7824–7838 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Hamley, I. & Castelletto, V. In Small-Angle Scattering of Block Copolymers. 1021–1081 (Springer, 2008).
- 73.Pedersen, J. S. & Schurtenberger, P. Scattering functions of semiflexible polymers with and without excluded volume effects. Macromolecules29, 7602–7612 (1996). [Google Scholar]
- 74.Chen, W.-R., Butler, P. D. & Magid, L. J. Incorporating intermicellar interactions in the fitting of SANS data from cationic wormlike micelles. Langmuir22, 6539–6548 (2006). [DOI] [PubMed] [Google Scholar]
- 75.Terech, P. et al. Structural variations in a family of orthodialkoxyarenes organogelators. J. Colloid Interface Sci.302, 633–642 (2006). [DOI] [PubMed] [Google Scholar]
- 76.McAulay, K. et al. Isotopic control over self-assembly in supramolecular gels. Langmuir36, 8626–8631 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Doucet, M. et al. SasView version 5.0.3. http://www.sasview.org/ (2020).
- 78.Pocker, Y. & Green, E. Hydrolysis of D-glucono-.delta.-lactone. I. General acid-base catalysis, solvent deuterium isotope effects, and transition state characterization. J. Am. Chem. Soc.95, 113–119 (1973). [DOI] [PubMed] [Google Scholar]
- 79.Rekveldt, M. T. Novel SANS instrument using neutron spin echo. Nucl. Instrum. Methods Phys. Res. Sect. B: Beam Interact. Mater. At.114, 366–370 (1996). [Google Scholar]
- 80.Martinez, C. R. & Iverson, B. L. Rethinking the term “pi-stacking. Chem. Sci.3, 2191 (2012). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The SANS2D experiment at ISIS was allocated beam time under experiment number RB2010573 and the SESANS on LARMOR was assigned the experiment number RB2010583.