

Abstract
Background
Branchio-oto-renal syndrome is a rare autosomal dominant disorder characterized by branchial arch anomalies, hearing loss, and renal dysplasia. Its diagnosis remains challenging due to clinical heterogeneity and overlapping features with other syndromes. This case report aims to enhance awareness of branchio-oto-renal syndrome and highlight multidisciplinary management strategies.
Case presentation
A 20-year-old Han Chinese female presented with bilateral preauricular and lateral neck fistulas since birth, accompanied by intermittent discharge. Physical examination revealed cup-shaped right ear deformity, bilateral preauricular fistulas, and branchial fistulas. Audiometry showed bilateral sensorineural hearing loss. Imaging studies identified an enlarged vestibular aqueduct and a hypoplastic left kidney. The patient underwent bilateral preauricular fistulotomy and bilateral branchial fistulotomy. Half a year after surgery, the patient’s incision healed well with no discharge or signs of recurrence. Unfortunately, the patient did not undergo genetic testing.
Conclusion
The diagnosis of branchio-oto-renal syndrome requires a high degree of clinical suspicion. Multidisciplinary collaboration is crucial for comprehensive management, including surgical intervention, audiological support, and long-term kidney monitoring.
Keywords: Branchio-oto-renal syndrome, Preauricular fistula, Branchial fistula, Renal dysplasia, Hearing loss
Introduction
Branchio-oto-renal syndrome (BORS) is a rare autosomal dominant disorder with an estimated prevalence of 1/40,000 in the general population and 2% among children with profound hearing loss [1]. It is characterized by branchial cleft anomalies (fistulas or cysts), hearing loss, preauricular pits, and renal malformations [2]. Owing to its clinical manifestations involving multiple systems and high heterogeneity, the diagnosis of BORS is relatively difficult. The currently recognized clinical diagnosis requires the following conditions to be met: no family history, meeting at least three primary criteria, or meeting two primary criteria and two secondary criteria to confirm BORS. Having a family history and meeting one of the main criteria is sufficient for diagnosis. The major criteria included second branchial anomaly, hearing loss, preauricular pits, and renal anomaly. The minor criteria included external ear anomaly, middle ear anomaly, inner ear anomaly, preauricular tags, facial asymmetry, and palate abnormality [3]. This case introduces a patient with BORS, and by documenting this case, our goal is to contribute to the limited BORS knowledge system and provide insights that will aid in the diagnosis and management of similar cases in the future.
Case presentation
A 20-year-old Han Chinese female presented to the Department of Otorhinolaryngology Head and Neck Surgery with bilateral preauricular and lateral neck fistulas noted since birth. The fistulas intermittently discharged foul-smelling fluid. The patient consciously had no significant hearing loss and normal speech development.
Specialized physical examination: physical examination shows that the patient has normal intelligence, cup-shaped deformity in the right ear, normal appearance in the left auricle, small fistulas in front of the two ears, unobstructed external auditory canals on both sides, intact eardrums on both sides, and clear markings. A fistula was observed at the anterior edge of the middle segment of the bilateral sternocleidomastoid muscles, and scar tissue was observed around the fistula on the right side. A strip-like tissue was palpated under the skin, and a small amount of mucous-like secretion was squeezed out of the surrounding skin (Fig. 1).
Fig. 1.

A shows the right ear as a cup-shaped ear, with arrows indicating the openings of the right preauricular fistula and branchial cleft fistula. B shows the normal shape of the left ear auricle, with arrows indicating the openings of the left preauricular fistula and branchial cleft fistula
Auxiliary examination: pure tone audiometry showed bilateral sensorineural hearing loss. The average hearing threshold of the right ear airway was 35 dBnHL, and the average hearing threshold of the left ear airway was 45 dBnHL (Fig. 2). Temporal bone computed tomography (CT)/magnetic resonance imaging (MRI) showed enlargement of the left vestibular aqueduct (Fig. 3). MRI showed bilateral branchial fistulas (Fig. 4). Abdominal ultrasound examination indicated a small volume of the left kidney (Fig. 5). Normal renal function (creatinine 72.0 μmol/L, eGFR 103.48 mL/min). The patient was born into a nonconsanguineous marriage and neither of their parents had a similar medical history.
Fig. 2.

Pure tone audiometry shows bilateral sensorineural hearing loss, with air conduction hearing thresholds of 35 dBnHL and 45 dBnHL for the right and left ears, respectively
Fig. 3.

showing the enlarged left vestibular aqueduct. A: computed tomography shows an inner diameter of 0.6 cm in the left vestibular aqueduct. B: computed tomography shows an enlarged middle diameter of 0.3 cm in the vestibular aqueduct. C: magnetic resonance T2-weighted image shows an enlarged vestibular aqueduct
Fig. 4.
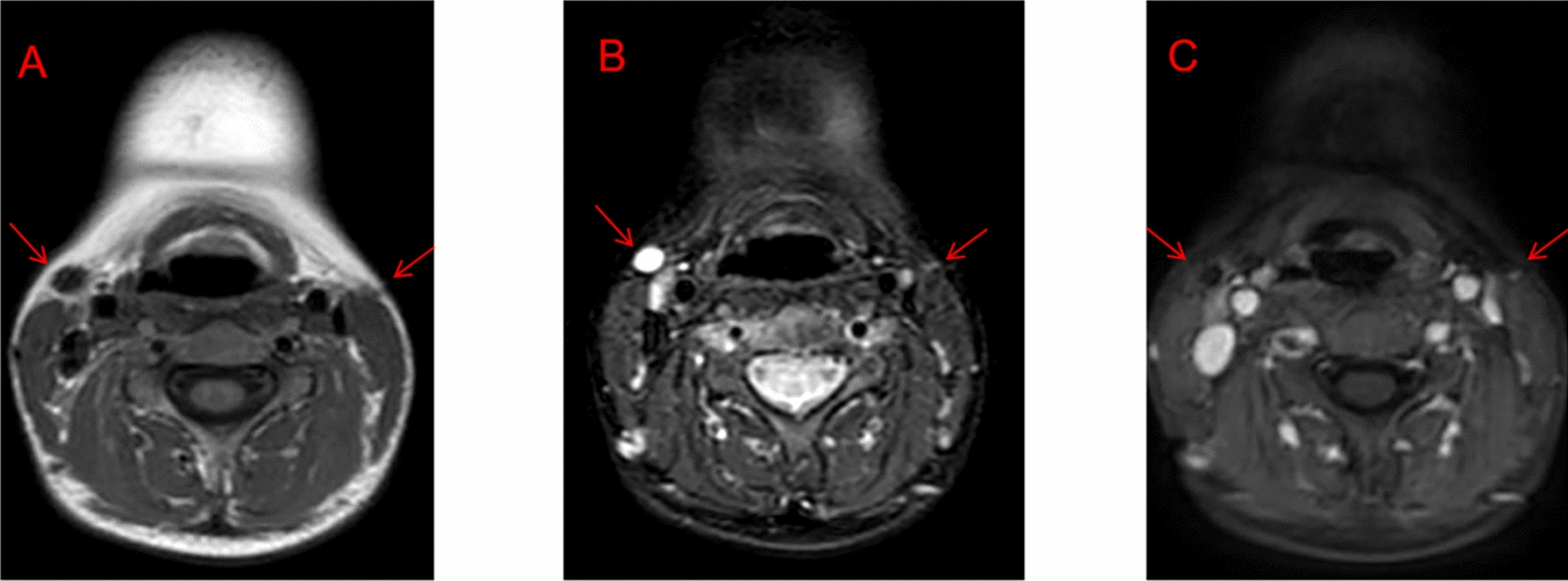
A: magnetic resonance T1-weighted images with bilateral arrows show bilateral branchial fistulas, B: magnetic resonance T2-weighted images with bilateral arrows show bilateral branchial fistulas, C: magnetic resonance-enhanced images with bilateral arrows show bilateral branchial fistulas
Fig. 5.

Renal ultrasound shows a reduction in the size of the left kidney, approximately 76 × 25 mm
The admission diagnosis was BORS. After completing preoperative examinations, bilateral preauricular fistulotomy and bilateral branchial fistulotomy were performed under general anesthesia. Intraoperative staining of the fistula with methylene blue (Fig. 6) was performed. Half a year after surgery, the patient’s incision healed well with no discharge.
Fig. 6.

Intraoperative resection of the branchial cleft fistula, with the arrow indicating the fistula stained with methylene blue
Discussion
BORS is a rare autosomal dominant genetic disorder with significant heterogeneity in clinical practice, and the penetrance rate is highly variable even within the same family. Research has shown that BORS exhibits hearing loss (noticed in 98.5% of cases), gill abnormalities (49–73%), preauricular depression (53–83%), and renal abnormalities (38–70%) [4, 5]. In addition to the typical clinical manifestations of ears, gills, and kidneys mentioned above, many studies have also reported other accompanying symptoms of BORS, such as developmental delay, intellectual disability, hypospadias, and bone defects. [6–8]. Hearing loss is the most common symptom of BORS, with 64–100% of patients experiencing some degree of hearing loss [3, 6, 9–11]. Mixed hearing loss is the most common type of hearing loss in patients with BORS (33.7–50%), followed by sensorineural (10.98–29.8%) and conductive (7.84–30%) hearing loss [6, 11–13]. Imaging studies have shown that structural abnormalities in the inner/middle ear, such as underdeveloped cochlea and enlarged vestibular aqueduct, are closely related to the progression of hearing impairment. Imaging studies can detect abnormalities in the inner ear (18–92%) and/or middle ear (15–100%) of BORS [9, 14]. Cochlear hypoplasia is the most common inner ear abnormality (33–100%), followed by vestibular aqueduct enlargement (24–50%) and inner ear canal abnormalities (17–86%) [13, 15, 16]. Kemperman analyzed long-term series audiometry data of patients with BORS, and the results showed that patients with BORS with large endolymphatic vessels and/or cysts on MRI seemed more likely to develop more severe hearing impairment. Furthermore, in clinical practice, about 70% of patients with hearing loss do not progress, and 30% of patients with progressive hearing loss often suffer from vestibular aqueduct enlargement [17].
The patient presented with bilateral branchial fistula, preauricular fistula, auricular malformation, large vestibular aqueduct, and left renal dysplasia, which meet the diagnostic criteria of BORS. For this patient, we removed both fistulas separately, ensuring the integrity of the anatomical structure while reducing the risk of fistula recurrence. There is no cure for BORS in patients with sensorineural hearing loss, and wearing hearing aids can be used as an adjuvant treatment. For patients with BORS who cannot obtain good hearing assistance from hearing aids, cochlear implantation is expected to serve as an alternative approach, enabling patients with advanced hearing impairment to achieve a good hearing prognosis. In this case report, the patient suffered from bilateral sensorineural hearing loss, but the patient consciously did not have significant hearing loss, which may be related to the fact that the patient has adapted to hearing loss. Patients may need to use hearing aids or cochlear implants to further improve hearing. In addition, the patient also has an enlarged vestibular aqueduct, which predisposes them to progressive hearing loss. Therefore, hearing should be checked at least every 6 months, and ototoxic medications should be avoided to prevent further hearing deterioration. BORS exhibits high heterogeneity in renal malformations, with clinical manifestations ranging from mild structural abnormalities to end-stage renal disease. The latest research shows that about 10–20% of patients with BORS have renal abnormalities, including renal dysplasia (such as volume reduction or structural disorder), unilateral/bilateral kidney absence, polycystic kidney disease, ureteral dilation, or hydronephrosis [18]. Among them, 6% of cases present with severe renal malformations (such as bilateral renal hypoplasia), which may progress to chronic renal failure within 20 years after birth and require dialysis or kidney transplantation [19]. Severe kidney deformities can also lead to Potter’s facial features, including wide eye distance, low ear position, mandibular retraction, and flat nose [20]. In this case, the patient had unilateral renal dysplasia and required regular monitoring and management of renal abnormalities, including at least biannual ultrasound examinations and estimated glomerular filtration rate (eGFR) monitoring, to prevent the progression of end-stage renal disease. In recent years, significant progress has been made in the genetic research of BORS. Research has found that mutations in the EYA1, SIX1, and SIX5 genes are closely related to BORS, with the EYA1 gene being the most common pathogenic gene [3]. In the European and American populations, about 40% of patients carry this gene mutation [3, 21]. The protein encoded by the EYA1 gene plays an important role in embryonic development, especially in the development of gill arches, ears, and kidneys [9]. Research has shown that the EYA1 gene regulates organogenesis. Mutations in human EYA1 can lead to BORS, while targeted inactivation of mouse EYA1 can impair early development of multiple organs.. In addition, EYA1 is also involved in controlling key early induction events involved in thymus, parathyroid, and thyroid morphogenesis [22]. In addition to BORS, the EYA1 gene is also closely related to the occurrence and development of stem cell carcinoma, melanoma, breast cancer, and other tumors [23–25].
About 4% of patients with BORS carry the SIX1 gene mutation [26]. The SIX1 gene is regulated by the EYA1 gene, and the SIX1–EYA1 protein complex affects organ development during embryonic development by regulating cell proliferation and differentiation [27]. Recent studies have found that the SIX1 protein is involved in the regulation of expression patterns during early sensory development, and mutations can affect embryonic craniofacial gene expression and auditory sac development [28, 29]. In clinical practice, patients with gill kidney lineage diseases carrying SIX1 pathogenic mutations are more prone to severe sensorineural hearing loss [30]. About 5% of patients with BORS carry a pathogenic mutation in the SIX5 gene. Hoskins et al. first discovered mutations in SIX5 in patients in 2007 and demonstrated that mutations affect the transcriptional activation of the SIX5 protein and SIX5–EYA1 protein complex, indicating that SIX5 is a BORS-related gene [31]. At present, it is believed that the pathogenic mechanism of SIX5 mutation is similar to that of the SIX1 mutation. However, no SIX5 mutation was found in the subsequent large-scale screening, and some cases were re attributed to EYA1 mutation, therefore its pathogenicity is questionable.
In addition to the above three genes, nearly half of the patients still need to further explore the genetic basis of their pathogenic factors. Currently, new genes related to BORS are constantly being discovered. In 2014, Morisada reported a sporadic case of BORS in Japanese patients and found through array comparative genomic hybridization that a 6 Mb microdeletion on chromosome 16 of the SALL1 gene was associated with the BORS phenotype [32]. SALL1 mutations are usually associated with Townes Brocks syndrome (manifested as anal atresia, ear deformities), partially overlapping with the clinical phenotype of BORS, but whether the pathogenic mechanism is directly related still needs to be verified [33]. In 2013, Brophy analyzed the whole genome copy number variations of 35 patients with BORS who did not have EYA1, SIX1, or SIX5 gene mutations and found that 17 of them had significant copy number variations (11 chromosomal microdeletions and 6 microduplications), thus identifying the chromosomal hotspots of BORS and discovering that the SHARPIN, FGF3, and HOXA gene clusters were potential pathogenic genes [21].
Genetic testing, such as multiplex ligation-dependent probe amplification (MLPA) and next-generation sequencing (NGS), has significant value in the diagnosis of BORS. MLPA can detect copy number variations in the EYA1 gene, while NGS can comprehensively screen for mutations in the EYA1, SIX1, and SIX5 genes. Despite the high cost of genetic testing, its potential application prospects in the diagnosis and genetic counseling of BORS are broad. Future research should further explore the application value of genetic testing in BORS diagnosis, especially in situations where resources are limited. Unfortunately, since the patient’s family refused to proceed with further genetic testing, the type of gene mutation in this patient could not be verified. The lack of genetic testing not only confines the diagnostic basis of this case to clinical manifestations and imaging assessments but also significantly restricts the precise evaluation of genetic risks for the patient and their family. Confirmation of pathogenic variants in EYA1 or related genes would not only strengthen the diagnostic basis for BORS but also enable targeted genetic screening for other potential carriers in the family (such as parents and siblings). This would facilitate the early identification of recessive carriers and allow for risk communication, prenatal diagnosis, or genetic screening during their family planning, thereby significantly altering the family’s reproductive decision-making and risk management strategies. Meanwhile, genetic testing can also guide the multidisciplinary team in developing more personalized follow-up and intervention plans. Since genetic testing was not performed in this case, it has, to some extent, limited the long-term management of the case and the provision of precise genetic counseling to the family members.
Conclusion
BORS, as a rare genetic disorder, requires multidisciplinary collaboration for management to address otologic, renal, and genetic issues. Genetic testing is of significant importance for the early diagnosis and prognosis of BORS. Clinicians should be highly vigilant for the possibility of BORS when encountering patients with branchial arch anomalies, hearing loss, or renal malformations.
Acknowledgements
Not applicable.
Abbreviations
- BORS
Branchio-oto-renal syndrome
- CT
Computed tomography
- MRI
Magnetic resonance imaging
Author contributions
SCF, XXH, and YHW performed surgical procedures on the patient. JQY and HBY participated in writing the manuscript and obtained the patient’s consent. JQY and XXH participated in the development of the patient’s surgical plan. All authors have read and approved the final manuscript.
Funding
Jiujiang Science and Technology Plan Project (S2023ZDYFN683).
Data availability
All data used during the present study are available from the corresponding author upon reasonable request.
Declarations
Ethics approval and consent to participate
The handling of this case was carried out in accordance with the Declaration of Helsinki. All programs were conducted in accordance with the ethical standards of the Clinical Research Ethics Committee of Jiujiang University Affiliated Hospital. Patient’s informed consent has been obtained.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests. The authors have no association with financial or nonfinancial organizations.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fraser FC, Sproule JR, Halal F. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss. Am J Med Genet. 1980;7(3):341–9. [DOI] [PubMed] [Google Scholar]
- 2.Li HX, Zhou P, Tong M, Zheng Y. Branchial cleft fistula to branchio-oto-renal syndrome: a case report and literature review. J Int Med Res. 2020;48(7):300060520926363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang EH, Menezes M, Meyer NC, Cucci RA, Vervoort VS, Schwartz CE, et al. Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat. 2004;23(6):582–9. [DOI] [PubMed] [Google Scholar]
- 4.Stinckens C, Standaert L, Casselman JW, Huygen PL, Kumar S, Van de Wallen J, et al. The presence of a widened vestibular aqueduct and progressive sensorineural hearing loss in the branchio-oto-renal syndrome. A family study. Int J Pediatr Otorhinolaryngol. 2001;59(3):163–72. [DOI] [PubMed]
- 5.Masuda M, Kanno A, Nara K, Mutai H, Morisada N, Iijima K, et al. Phenotype-genotype correlation in patients with typical and atypical branchio-oto-renal syndrome. Sci Rep. 2022;12(1):969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morisada N, Nozu K, Iijima K. Branchio-oto-renal syndrome: comprehensive review based on nationwide surveillance in Japan. Pediatr Int. 2014;56(3):309–14. [DOI] [PubMed] [Google Scholar]
- 7.Salinas-Torres VM, Rivera H. Branchiootorenal syndrome with skeletal defects: a novel association in a Mexican child. Clin Dysmorphol. 2015;24(1):21–3. [DOI] [PubMed] [Google Scholar]
- 8.Dutta M, Chatterjee I. Hypospadias as a new entity to define the branchio-oto-renal spectrum disorders. Ear Nose Throat J. 2019;98(1):20–2. [DOI] [PubMed] [Google Scholar]
- 9.Unzaki A, Morisada N, Nozu K, Ye MJ, Ito S, Matsunaga T, et al. Clinically diverse phenotypes and genotypes of patients with branchio-oto-renal syndrome. J Hum Genet. 2018;63(5):647–56. [DOI] [PubMed] [Google Scholar]
- 10.Krug P, Morinière V, Marlin S, Koubi V, Gabriel HD, Colin E, et al. Mutation screening of the EYA1, SIX1, and SIX5 genes in a large cohort of patients harboring branchio-oto-renal syndrome calls into question the pathogenic role of SIX5 mutations. Hum Mutat. 2011;32(2):183–90. [DOI] [PubMed] [Google Scholar]
- 11.Smith RJ, Schwartz C. Branchio-oto-renal syndrome. J Commun Disord. 1998;31 (5):411–20; (quiz 21). [DOI] [PubMed]
- 12.Lindau TA, Cardoso AC, Rossi NF, Giacheti CM. Anatomical changes and audiological profile in branchio-oto-renal syndrome: a literature review. Int Arch Otorhinolaryngol. 2014;18(1):68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen A, Francis M, Ni L, Cremers CW, Kimberling WJ, Sato Y, et al. Phenotypic manifestations of branchio-oto-renal syndrome. Am J Med Genet. 1995;58(4):365–70. [DOI] [PubMed] [Google Scholar]
- 14.Ideura M, Nishio SY, Moteki H, Takumi Y, Miyagawa M, Sato T, et al. Comprehensive analysis of syndromic hearing loss patients in Japan. Sci Rep. 2019;9(1):11976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Propst EJ, Blaser S, Gordon KA, Harrison RV, Papsin BC. Temporal bone findings on computed tomography imaging in branchio-oto-renal syndrome. Laryngoscope. 2005;115(10):1855–62. [DOI] [PubMed] [Google Scholar]
- 16.Kemperman MH, Koch SM, Joosten FB, Kumar S, Huygen PL, Cremers CW. Inner ear anomalies are frequent but nonobligatory features of the branchio-oto-renal syndrome. Arch Otolaryngol Head Neck Surg. 2002;128(9):1033–8. [DOI] [PubMed]
- 17.Kemperman MH, Koch SM, Kumar S, Huygen PL, Joosten FB, Cremers CW. Evidence of progression and fluctuation of hearing impairment in branchio-oto-renal syndrome. Int J Audiol. 2004;43(9):523–32. [DOI] [PubMed] [Google Scholar]
- 18.Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006;17(10):2864–70. [DOI] [PubMed] [Google Scholar]
- 19.Izzedine H, Tankere F, Launay-Vacher V, Deray G. Ear and kidney syndromes: molecular versus clinical approach. Kidney Int. 2004;65(2):369–85. [DOI] [PubMed] [Google Scholar]
- 20.Carmi R, Binshtock M, Abeliovich D, Bar-Ziv J. The branchio-oto-renal (BOR) syndrome: report of bilateral renal agenesis in three sibs. Am J Med Genet. 1983;14(4):625–7. [DOI] [PubMed] [Google Scholar]
- 21.Brophy PD, Alasti F, Darbro BW, Clarke J, Nishimura C, Cobb B, et al. Genome-wide copy number variation analysis of a Branchio-oto-renal syndrome cohort identifies a recombination hotspot and implicates new candidate genes. Hum Genet. 2013;132(12):1339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu PX, Zheng W, Laclef C, Maire P, Maas RL, Peters H, et al. Eya1 is required for the morphogenesis of mammalian thymus, parathyroid and thyroid. Development. 2002;129(13):3033–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou JJ, Huang Y, Zhang X, Cheng Y, Tang L, Ma X. Eyes absent gene (EYA1) is a pathogenic driver and a therapeutic target for melanoma. Oncotarget. 2017;8(62):105081–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong D, Ma W, Zhang D, Cui Q, Wang K, Tang J, et al. EYA1 promotes cell migration and tumor metastasis in hepatocellular carcinoma. Am J Transl Res. 2019;11(4):2328–38. [PMC free article] [PubMed] [Google Scholar]
- 25.Guan H, Dai Z, Ma Y, Wang Z, Liu X, Wang X. Microrna-101 inhibits cell proliferation and induces apoptosis by targeting EYA1 in breast cancer. Int J Mol Med. 2016;37(6):1643–51. [DOI] [PubMed] [Google Scholar]
- 26.Kochhar A, Orten DJ, Sorensen JL, Fischer SM, Cremers CW, Kimberling WJ, et al. SIX1 mutation screening in 247 branchio-oto-renal syndrome families: a recurrent missense mutation associated with BOR. Hum Mutat. 2008;29(4):565. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426(6964):247–54. [DOI] [PubMed] [Google Scholar]
- 28.Shah AM, Krohn P, Baxi AB, Tavares ALP, Sullivan CH, Chillakuru YR, et al. Six1 proteins with human branchio-oto-renal mutations differentially affect cranial gene expression and otic development. Dis Model Mech. 2020. 10.1242/dmm.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato S, Furuta Y, Kawakami K. Regulation of continuous but complex expression pattern of Six1 during early sensory development. Dev Dyn. 2018;247(1):250–61. [DOI] [PubMed] [Google Scholar]
- 30.Chen A, Song J, Acke FRE, Mei L, Cai X, Feng Y, et al. Otological manifestations in branchiootorenal spectrum disorder: a systematic review and meta-analysis. Clin Genet. 2021;100(1):3–13. [DOI] [PubMed] [Google Scholar]
- 31.Hoskins BE, Cramer CH, Silvius D, Zou D, Raymond RM, Orten DJ, et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet. 2007;80(4):800–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morisada N, Sekine T, Ishimori S, Tsuda M, Adachi M, Nozu K, et al. 16q12 microdeletion syndrome in two Japanese boys. Pediatr Int. 2014;56(5):e75–8. [DOI] [PubMed] [Google Scholar]
- 33.Engels S, Kohlhase J, McGaughran J. A SALL1 mutation causes a branchio-oto-renal syndrome-like phenotype. J Med Genet. 2000;37(6):458–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used during the present study are available from the corresponding author upon reasonable request.