

Abstract
Understanding patterns of biodiversity in microbial communities is severely constrained by the difficulty of adequately sampling these complex systems. We illustrate the problem with empirical data from small surveys (200-member 16S rRNA gene clone libraries) of four bacterial soil communities from two locations in Arizona. Among the four surveys, nearly 500 species-level groups (Dunbar et al., Appl. Environ. Microbiol. 65:662-1669, 1999) and 21 bacterial divisions were documented, including four new candidate divisions provisionally designated SC1, SC2, SC3, and SC4. We devised a simple approach to constructing theoretical null models of bacterial species abundance. These null models provide, for the first time, detailed descriptions of soil bacterial community structure that can be used to guide experimental design. Models based on a lognormal distribution were consistent with the observed sizes of the four communities and the richness of the clone surveys. Predictions from the models showed that the species richness of small surveys from complex communities is reproducible, whereas the species composition is not. By using the models, we can now estimate the required survey scale to document specified fractions of community diversity. For example, documentation of half the species in each model community would require surveys of 16,284 to 44,000 individuals. However, quantitative comparisons of half the species in two communities would require surveys at least 10-fold larger for each community.
Determining the composition of biological communities (or guilds within communities) is an essential step in deciphering the role of demographic, evolutionary, and ecological factors on ecosystem processes. For soil bacterial communities, such an undertaking is daunting due to the community size (typically 109 bacterial cells per g) and magnitude of species-level diversity. Based on DNA reassociation kinetics, the estimated number of distinct genomes present in a gram of soil ranges from 2,000 to 18,000 (29, 35, 36). The survey size must therefore be large for adequate representation of diversity.
The exact survey size needed for community representation depends on the frequency distribution of species in situ and the degree of representation that is desired in the sample. For bacterial communities, the frequency distribution of species has never been measured or even approximated due to the difficulty of obtaining sufficiently large and representative samples of community diversity. In contrast, abundance distributions of plant, animal, and insect species in samples from a wide variety of communities have been intensively studied during the past 80 years (12, 34). Numerous models, both mechanistic and statistical, have been proposed to describe the observed distributions of plant, animal, and insect species (34). Mechanistic models such as the geometric distribution are typically limited to explaining the abundance distribution of species that compete for a common niche or resource base. Statistical models, on the other hand, are more appropriate for large species assemblages that are functionally and phylogenetically diverse, such as soil bacterial communities.
The most frequently used statistical model for species abundance distributions is the lognormal distribution, applied first in ecology by Preston in 1948 (24). Lognormal distributions can arise solely from the multiplicative effects of biotic and abiotic factors on the abundance of individual species (19, 21). In other words, the distribution can be a statistical phenomenon of large numbers and does not depend upon specific biological or ecological mechanisms. A lognormal distribution is therefore appealing as a null model for the distribution of bacterial species abundance. Such null models can guide experimental studies of community structure and composition. In particular, a model distribution can be used to estimate the survey size required for confident documentation of a specified fraction of community diversity.
The most common way to comprehensively survey the phylogenetic diversity present in soil bacterial communities is by PCR amplification, cloning, and sequencing of 16S rRNA genes (16S rDNA) from extracted soil DNA. However, due to the expense of this method, the survey sizes have typically been quite small, consisting of only 100 to 300 clones per soil sample (1, 15, 17, 27) (see reference 40 for a recent exception). The surveys conducted by Kuske et al. (15) for arid soil bacterial communities in Arizona, United States, are of typical size (200 clones per library). The libraries were derived from pinyon pine rhizosphere and interspace (between-tree) soil communities at a cinder field created by volcanic eruption over 900 years ago (Sunset Crater) and at a site 20 km away (Cosnino) that has sandy loam soil typical of the arid northern Arizona region. The libraries were created in 1994 to identify bacterial populations specifically involved in assisting plant colonization and growth in the hot, dry, volcanic cinder soil. Unfortunately, the data exhibited characteristic limitations of small phylogenetic surveys of large, complex communities. That is, most (93%) of the species-level groups in each library were represented by only one or two clones each, suggesting inadequate sampling and a high probability of sampling error (7).
In the present study, we analyzed the four libraries at the division level and used the species richness of the libraries and the observed sizes of the Arizona soil communities to guide the construction of theoretical lognormal models of bacterial species abundance. The models provide an important baseline for understanding the general structure of soil bacterial communities, the limitations of phylogenetic surveys as currently practiced, and the requirements for improving future surveys of bacterial species diversity. We emphasize that the theoretical models are null models. That is, the models provide the best possible description of community structure at present based on currently available data, but their accuracy on a fine scale requires experimental validation.
MATERIALS AND METHODS
Soil samples.
Soil samples were collected from two field sites (Sunset Crater and Cosnino) 20 km apart in northern Arizona that have similar plant communities (pinyon-juniper woodlands), elevation, and general weather patterns but differ dramatically in soil type (4, 15). The sites and collection of soil samples were described in detail previously (15).
Bacterial biomass measurement.
A 50-cm3 portion of each of the four soil samples was shipped on ice to the Soil Microbial Biomass Service, Oregon State University, Corvallis, Oreg. (now Soil Food Web Inc., Corvallis, Oreg.; www.soilfoodweb.com) for measurement of bacterial biomass. Metabolically active bacteria and total bacteria were stained with fluorescein diacetate and fluorescein isothiocyanate, respectively, and counted by using epifluorescence microscopy. Active and total cell counts were obtained for soil samples from the April 1994, September 1994, and September 1995 collections.
DNA extraction and clone libraries from soil and cinders.
Extraction of DNA and construction of 16S rRNA gene clone libraries were described previously (15). Each library contained approximately 200 clones. All clones were characterized by restriction fragment length polymorphism (RFLP) analysis (7). Clones with identical RsaI-BstUI RFLP patterns were counted as a species-level group. To evaluate the extent to which this approach underestimated the number of species-level groups in the libraries, the number of RFLP groups among a set of 221 clones from the Cosnino interspace (C0) and Sunset Crater interspace (S0) clone libraries was compared to the number of groups delineated by a criterion of ≥97% sequence similarity. Sequence similarity was assessed over the 16S rRNA gene region corresponding to Escherichia coli positions 270 to 768, excluding the variable loop region between positions 451 and 460. The 270 to 768 region was the longest sequenced region common to all 221 clones used in this comparison.
DNA sequencing.
The 16S rRNA gene (rDNA) templates for DNA sequencing reactions were amplified directly from glycerol stocks of 16S rRNA gene clones. Primers M13-20 (5′-GTAAAACGACGGCCAGT) and M13-24 (5′-AACAGCTATGACCATG) were used for PCR amplification. Amplified DNA was purified by using a Qiaquick PCR cleanup kit (Qiagen, Inc., Chatsworth, Calif.), and DNA concentrations were estimated by gel electrophoresis and ethidium bromide staining. Approximately 100 ng of 16S rDNA was used as the template in dye terminator cycle sequencing reactions (ABI Prism dye terminator cycle sequencing kit; Perkin-Elmer, Foster City, Calif.).
Primer p3MODrc (5′-GGACTACHAGGGTATCTAAT, E. coli positions 806 to 787) was used in sequencing reactions to obtain partial DNA sequences. Full-length sequences were obtained from 45 clones (see section on phylogenetic analysis, below) by using primers M13-20, M13-24, P3MOD (5′-ATTAGATACCCTDGTAGTCC, E. coli positions 787 to 806) (38), P3MODrc, and 533 forward (5′-CCAGCSGCCGCGGTAA, E. coli positions 519 to 533) (16) in sequencing reactions. Electrophoresis was performed with 4.0% polyacrylamide gels on a 373A Stretch DNA sequencer (Applied Biosystems, Inc., Foster City, Calif.). The nucleotide sequences determined in this study have been deposited in the NCBI database under accession numbers AF507374 to AF507801.
Phylogenetic analysis.
16S rDNA sequences were compared with sequences from the Ribosomal Database Project (RDP; version 7.0) (20) by using the Similarity Rank program to obtain Sab values to database sequences. RDP sequences with less than 307 nucleotides for comparison were excluded from the analysis. Clone sequences were assigned to recognized bacterial divisions (or “uncertain” status) based on the affiliation of nearest-neighbor sequences from the RDP. Full-length sequences were obtained from all clones with uncertain affiliation based on partial sequence comparisons (Sab values < 0.50) and from all clones that appeared to represent new candidate divisions. Full-length sequences were checked for chimeric artifacts by using the Chimera-Check program (20) and secondary-structure analyses. Full-length sequences were then used in bootstrapped phylogenetic analyses and either assigned to recognized divisions based on reliable branching order or assigned as “uncertain” if branching order was inconsistent and unreliable.
Lognormal model of bacterial species abundance.
The general lognormal species abundance distribution is as follows:
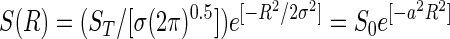 |
(1) |
where R is a log2 species abundance interval, or octave (the modal octave is 0), S(R) is the number of species in the Rth octave with average population size NR = N02R, N0 is the population size of the modal species, ST is the total number of species in the community, σ2 is the variance of the distribution, a = (0.5/σ2)0.5 is the dispersion constant, and S0 is ST a/π0.5, the number of species in the modal octave.
Assuming that one species occupies each tail of the Gaussian distribution of log2 abundance values, it follows that 1 = S(Rmax). By using this substitution in equation 1, a set of lognormal distributions was created by solving reiteratively for σ when ST ranged from 2,000 to 20,000 and Rmax ranged from 10 to 12 (i.e., the population size of the most abundant species ranged from 1 × 106 to 1.7 × 107 cells [g of soil]−1). The range of values for ST was obtained from studies of the renaturation kinetics of soil bacterial DNA (29, 35, 36). The range of Rmax values was chosen so that the calculated community size, NT, from a given lognormal distribution would be consistent with the range of observed NT values (epifluorescence direct counts of total cells) from the Arizona soils used in this study.
Estimation of survey size.
Species richness sampling curves were constructed by rarefaction (i.e., simulated sampling without replacement) (13, 30). Theoretical values of species abundance were used for calculating species-level sampling curves. The theoretical values (i.e., the abundance of each theoretical species in a model community) were obtained from lognormal distribution models of communities containing approximately 108 individuals total and 2,000 to 10,000 species. For each sample size calculation, 1,000 simulations of sampling without replacement were performed by using R software (a public-domain data analysis, graphics, and programming environment, available at www.r-project.org).
Estimates of the sample sizes required for sampling a specified set of j species with 95% confidence were calculated by using the following equation:
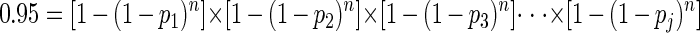 |
(2) |
where p1, p2, p3,…pj are the relative proportions of the 1st, 2nd, 3rd,… jth species in the community and n is the survey size (number of individuals). For each set of j species, the equation was solved reiteratively for n.
RESULTS
Observed bacterial community sizes.
Bacterial biomass, measured as total fluorescent cell counts by microscopy, varied in the four Arizona soils over time and between sites. For the April 1994 soil samples from which clone libraries were constructed, the Sunset Crater soils had an average of 5.3 × 107 bacterial cells (g of soil)−1 (n = 6), whereas the Cosnino samples had an average of 1.4 × 108 cells (g of soil)−1 (n = 6). The averages for Sunset Crater and Cosnino soils over a 2-year period (measurements from April 1994, September 1994, and September 1995) were 1.0 × 108 and 1.6 × 108 cells (g of soil)−1, respectively.
Division-level diversity of Arizona soil surveys.
Each Arizona soil was surveyed by constructing a 16S rRNA gene clone library. A total of 21 bacterial divisions were found among the four surveys, based on analysis of 766 clones. The affiliation of 16 of the 766 clones could not be reliably determined. The 16 full-length sequences clustered inconsistently in different divisions from one analysis to the next (data not shown) and were therefore assigned to the uncertain category, as shown in Fig. 1. Most of the clones (722 total) were affiliated with nine well-established bacterial divisions. Twenty sequences were affiliated with recently proposed candidate bacterial divisions OP3, OP4, OP10, OP11, TM6, TM7, OS-K, and WS-2. Eight clones failed to cluster closely with any previously identified bacterial division and are represented in Fig. 1 as four distinct groups provisionally named SC1, SC2, SC3, and SC4. Full-length sequences from the eight clones showed no evidence of being chimeric. Instead, the sequences appeared to represent four deeply branching bacterial lineages that have not been described previously. The depth of branching of the four lineages and the dissimilarity of the sequences to all known 16S rRNA gene sequences are consistent with criteria that have been used previously to delineate bacterial divisions (10). Thus, the sequences appear to represent four new candidate divisions.
FIG. 1.

Relative abundance of bacterial divisions found in 16S rRNA gene clone libraries four Arizona soils. Absence of a visible bar indicates that no representative clones were found in the library.
The relative abundance of the 21 bacterial divisions among the four clone libraries is shown in Fig. 1. Nine divisions (Acidobacterium, Proteobacteria, Verrucomicrobiales, gram positives, Cytophaga-Flexibacter-Bacteroides, Nitrospira, green nonsulfur bacteria, Planctomyces, and OP11) were identified previously from analysis of 168 sequences (7, 15). The relative abundance of these nine divisions has not changed substantially with the inclusion of data from additional sequences. The Acidobacterium and Proteobacteria divisions are the most abundant, accounting for 327 and 127 of the 766 clones, respectively.
In the first report of division-level diversity among the clones (15), the abundance of the Acidobacterium division was listed as 54% of 60 analyzed sequences and was revised later to 51% of 356 clones (7). Analysis of the full data set indicates that the division accounts for 49% of 766 clones. The abundance of the Proteobacteria has increased slightly, from 12% of 60 sequences (15), to 17% of 766 clones. Similar small changes in relative abundance of the other seven previously identified divisions occurred after including data from the recently sequenced clones.
Nearly half of the 21 divisions were found in all four libraries. The average abundance of the nine common divisions ranged from 2 to 82 clones per library. The divisions occurring in only one library were represented by one or two clones each. The low representation of the rare divisions made it impossible to interpret the unique occurrence of these divisions. For the nine common divisions, three qualitative points of interest were noted. First, at each site, the relative abundance of Proteobacteria was lower in the interspace soil survey than in the rhizosphere soil survey (Fig. 1). Second, the relative abundance of gram-positive bacteria was higher in the interspace soil survey than in the rhizosphere soil survey at each site. For both the Proteobacteria and gram-positive divisions, the differences in abundance between the Cosnino interspace and rhizosphere soils were small, whereas the differences between the Sunset Crater interspace and rhizosphere soils were larger. Third, the abundance of the Cytophaga-Flexibacter-Bacteroides group was higher in the Cosnino soil surveys than in the Sunset Crater soil surveys.
Abundance distribution of bacterial divisions.
Analysis of the abundance distribution of bacterial divisions in the clone libraries demonstrated significant differences between the Sunset Crater soil communities and the Cosnino soil communities (Fig. 2). More bacterial divisions were found in the volcanic cinder soil libraries (average, 14; combined total, 18 for rhizosphere and interspace libraries) than in the libraries from the sandy loam soil at Cosnino (average, 12; combined total, 14), suggesting a more skewed distribution of division abundance in the Cosnino soils.
FIG. 2.

Division-level sampling curves. Curves were calculated by rarefaction (13, 30). Data from the Yellowstone hot spring were obtained from reference 10.
Rarefaction analysis of the combined data indicated that the differences in distribution of division-level diversity in the Sunset Crater soils and the Cosnino soils were significant at the 95% confidence level (results not shown). For comparison, a data set describing division-level diversity in a Yellowstone hot spring sample (10) was also analyzed. The abundance distribution of bacterial divisions in the hot spring community exhibited significantly less skew than the Arizona soil samples. These data show that the abundance distribution of bacterial divisions can be used at least in some cases to identify differences in the structure of bacterial communities from different locations.
Extrapolation of division-level diversity in environmental samples.
The total number of bacterial divisions in environmental samples is completely unknown but could potentially be estimated from partial survey data. Since the efficacy of different extrapolation methods is dependent on the data set, trial-and-error application of different methods is often necessary (26). We applied three different statistical methods to data from the Arizona soil surveys and a hot spring survey (10) in an attempt to estimate the total number of divisions in each environment.
Asymptotes of the division-level sampling curves in Fig. 2 were estimated by fitting the data to a two-parameter linear model of the form S(n) = Smax − BS(n)/n, where n is the number of individuals sampled, S is division-level diversity, and B is a fitted constant (26). This model is the Eadie-Hofstee transformation routinely used for estimating Vmax in enzyme kinetics. The parameters Smax and B were estimated by using a maximum-likelihood technique (26). For the Yellowstone hot spring data, the two-parameter model provided an estimate of 30 divisions maximum versus the 26 actually observed. The model fit the data from the Arizona soil samples poorly, providing estimates of Smax for the Cosnino samples that were slightly lower than the actual observed values and equal to the observed values from the Sunset Crater samples.
A ranked abundance distribution of the bacterial divisions detected in the four Arizona soils was plotted (Fig. 3) to evaluate the possibility of fitting the division data to a lognormal distribution (a parametric extrapolation method). Each set of data was best fit by a power function. However, the plotted data from each environment did not exhibit clear evidence of a mode. If the data were lognormally distributed and if over half of the divisions in each environment were represented in the sample, transformation of the ranked division abundance data to a log2 abundance scale would yield a normal distribution truncated to the left of the modal octave. The division abundance data from the four Arizona soils displayed no evidence of having a normal distribution with a mode (data not shown). The hot spring data displayed the possible beginning of a normal distribution but lacked a defined mode (data not shown). If the abundance distribution of bacterial divisions in the soils is in fact lognormal, the data suggest that less than half of the divisions in each soil have been documented.
FIG. 3.

Ranked abundance plot of division diversity. For each environmental sample, the divisions documented in clone libraries were ranked in order of decreasing abundance. Curves shown in the plot are best-fit power functions. The spread of data points was examined visually for evidence of a lognormal distribution.
Division-level diversity was also estimated by using a nonparametric extrapolation method. The Chao 1 estimator (3), like other equations for nonparametric extrapolation of diversity (5), uses the distribution of rare taxa observed in a sample to extrapolate the total number of taxa present in the environment. The Chao 1 equation has the form Smax = Sobs + (a2/2b), where a is the number of singletons observed in a sample and b is the number of doubletons. Chao 1 estimates of the division-level diversity in each Arizona soil environment are listed in Table 1. The Cosnino interspace, Sunset Crater interspace, and Sunset Crater rhizosphere soil samples were estimated to contain two additional divisions each, whereas six additional divisions were estimated to occur in the Cosnino interspace soil sample. The estimate from the Cosnino interspace sample was substantially higher than the other three due to the fact that only one division in the Cosnino library was represented by two individuals, a circumstance that maximizes estimates from the Chao 1 equation. Estimates were also derived from pooled survey data in order to extrapolate the total number of divisions in soil from the Cosnino area, the Sunset Crater area, and from the region in general. As shown in Table 1, estimates of division-level diversity were only slightly larger than the observed totals from the four small clone library surveys. The large variance associated with each estimate demonstrates that reliable extrapolations of division-level diversity will depend upon surveys that are either much larger in size or well replicated.
TABLE 1.
Extrapolation of division-level diversity in four Arizona soils
| Divisions | No. of divisions (variance)a
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Cosnino
|
Sunset Crater
|
All soils | Hot spring | |||||
| Interspace soil | Rhizosphere soil | Interspace + rhizosphere | Interspace soil | Rhizosphere soil | Interspace + rhizosphere | |||
| Observed | 12 | 12 | 14 | 15 | 14 | 18 | 21 | 18 |
| Estimated | 20 (69.0) | 14 (7.9) | 16 (7.9) | 16 (2.3) | 17 (8.5) | 20 (5.0) | 24 (9.4) | 38 (95.0) |
Species-level diversity of Arizona soil surveys.
Species-level diversity in the 200-member clone libraries from the four Arizona soils was estimated previously by determining the number of RsaI-BstUI RFLP patterns among the clones (6). To assess whether the RFLP analysis underestimated the species-level richness of the libraries, we compared the previous estimates to new estimates (obtained for a subset of clones from the Sunset Crater and Cosnino interspace libraries) based on sequence similarity. Clone sequences with 97% or greater sequence similarity (22, 28, 31) were counted as a species-level group. For the Sunset Crater interspace library, 73 sequence similarity groups were found in a set of 114 sequences, versus 80 RFLP groups among the same set. In a set of 107 clones from the Cosnino interspace library, 81 sequence similarity groups and 81 RFLP groups occurred. These data indicate that the previous RFLP analysis provided reasonable approximations of species-level richness in the four libraries.
Modeling bacterial species abundance.
To obtain a null model of bacterial species abundance for the Arizona soil communities, a series of lognormal distributions were constructed. The observed bacterial community size and observed sample diversity from each Arizona soil community were used to constrain the set of theoretical distribution models. For each theoretical distribution, the community size, NT, was calculated mathematically and compared with empirical data. Likewise, the species richness in a sample size of 200 individuals was estimated from each theoretical model by simulated sampling and compared with the observed values from the Arizona clone libraries. With this approach, we identified a reasonable set of models for the Arizona soil communities.
As shown in Fig. 4, lognormal models with about 3,000 to 8,000 species and an Rmax value of 11 produced results most consistent with the observed data from the four Arizona communities. The calculated community sizes for models with an Rmax value of 10 ranged from 2.4 × 107 to 7.8 × 107 cells (g of soil)−1 (for communities with 2,000 to 10,000 species) and from 2.5 × 108 to 6.7 × 108 cells (g of soil)−1 for models with an Rmax value of 12. These community sizes were generally either too low or too high to be consistent with the observed community sizes (total cell counts) from the Arizona soils. The calculated community size from models with an Rmax value of 11 (and 3,000 to 8,000 species) ranged from 9.8 × 107 to 1.9 × 108 individuals, compared with averages of 1.0 × 108 and 1.6 × 108 cells (g of soil)−1 observed in the Arizona soils. For the Rmax = 11 (Rmax11) models, the predicted species richness for a sample size of 200 individuals ranged from 124 ± 12 to 161 ± 11, compared with the 134 to 161 RFLP groups found previously (7) in the Arizona soil clone libraries. Given the consistency of these observations, the Rmax11 models were the most reasonable lognormal null models for the distribution of species abundance in the Arizona soils.
FIG. 4.

Comparison of theoretical lognormal models with observed data from four Arizona soils. Fifteen model communities were constructed by using three different values for Rmax (10, 11, and 12) and five different values for ST (2,000, 4,000, 6,000, 8,000, and 10,000 species). Data from 12 of the models are shown in the figure. The solid square and diamond represent observed data from Sunset Crater and Cosnino interspace soils, respectively. The semifilled square and diamond represent observed data from Sunset Crater and Cosnino rhizosphere soils, respectively. The plotted symbols for the Arizona communities would be positioned artificially high (by 5 to 20%) if the surveys contained PCR artifacts.
Lognormal model details.
Five lognormal models (Rmax11) are illustrated in Fig. 5A. The models represent theoretical communities containing 2,000, 4,000, 6,000, 8,000, or 10,000 species. In each model community, the most abundant species has a population size of approximately 6.3 × 106 cells (g of soil)−1, corresponding to an Rmax value of 11 (the interval containing species with population sizes of 4.2 × 106 to 8.4 × 106 cells [g of soil]−1). The community sizes, NT, calculated from the five distributions ranged from 7.6 × 107 to 2.2 × 108 cells (g of soil)−1. The dispersion constant, a, ranged from 0.213 to 0.244, close to the value of 0.2 observed by Preston (23, 24) for lognormal distributions from plant, animal, and insect communities. The modal octave for all five model communities contained species with population sizes of 2,048 to 4,095 cells (g of soil)−1 and included 12 to 14% of the total number of species in each model community. Most of the species (71 to 78%) in each model community had population sizes within approximately ±1 standard deviation of the mode (i.e., population sizes of 3 × 102 to 6 × 104 cells [g of soil]−1). These species collectively accounted for 12 to 22% of the biomass (total cell counts; Fig. 5B). The number of dominant species (population sizes of 7 × 104 to 6 × 106 cells [g of soil]−1, about 10- to 1,000-fold more abundant than the modal species) in the model communities ranged from 185 to 544. The dominant species accounted for a disproportionate fraction (45 to 26%) of community biomass.
FIG. 5.

Theoretical lognormal model series most consistent with observed data from four Arizona soils. (A) Lognormal species rank abundance plots. Each curve shows the population size of each species (ranked in order of decreasing abundance) from the theoretical community. The sum of population sizes for all the species in a community is the calculated community size, NT. The inset panel illustrates the normal distribution achieved by plotting species population sizes on a geometric scale, log2. The first interval (the −11 octave) contains a single species represented by one individual (1 cell [g of soil]−1). The second octave contains species represented by two or three individuals, etc. (B) Cumulative biomass distribution for species from lognormal models shown in panel A. The species are ranked in order of decreasing abundance.
Sample size estimation for documenting species richness.
To predict the magnitude of sampling required for more comprehensive surveys of species diversity in the Arizona communities, sampling curves were constructed (Fig. 6A) for each lognormal distribution from Fig. 5A. The curves can be used to predict the return of a specified sampling effort or to predict the sampling effort required for a specified return. For example, a specified sampling effort of 2,000 individuals (10-fold larger than the size of the present surveys) would be expected to document 442 to 1,019 species, depending on the model. To document 50% of the species in each model community, a sample size of approximately 16,000 to 50,000 individuals would be required, depending on the model. The data show that theoretical models combined with simulated sampling can guide the design of surveys intended to achieve specific sampling goals. It is important to note, however, that the prediction of species richness in a simulated sample does not specify the identities of the species that comprise the particular sample.
FIG. 6.

Sampling curves for lognormal model communities. (A) Species richness sampling curves. The curves were calculated by rarefaction. The number at the apex of each curve shows the percentage of the total number of species, ST, from each community expected in a sample size of 50,000 individuals. The triangles indicate the sample size required to document half the species richness of each model community. (B) Species composition sampling curves based on a 95% confidence interval. Each curve shows the sample size required for confident sampling of a specified set of species (each set includes the given species number on the x axis and all the preceding species on the x axis). Species are ranked in order of decreasing population size. Sampling curves from model communities that differ 2.5-fold in species number (Rmax11 curves) and 8-fold in community size (Rmax10 and Rmax12 curves) are shown.
Sample size estimation for species detection.
If the sampling goal is to document particular species instead of a particular number of random species, sample sizes are significantly affected. Sample size estimates for reproducibly documenting specified sets of species are illustrated in Fig. 6B. Comparison of data from Fig. 6A and 6B shows that the sample size required for detection (at the 95% confidence level) of a specified set of species is significantly larger than the sample size required for documenting a specified number of random species. For example, a sample size of only one individual would be sufficient to document a single species, but documentation of species no. 1 (the most abundant species) with 95% confidence from each of the five model communities in Fig. 5A would require sample sizes ranging from 35 to 106 individuals.
For the 4,000-species-community model (shown in Fig. 5A), the minimum sample size for documentation of species no. 1 with 95% confidence is 55 individuals (Fig. 6B). A sample of this size would be expected to include species no. 1 and 46 (±5) additional species. If a second sample of 55 individuals were taken, only species no. 1 of the 47 (±5) species in the first sample would be expected to recur at the 95% confidence level. There is progressively less chance that the other 46 species (ranked in order of decreasing population size) from the first sample would co-occur with species no. 1 in a second sample.
Applying this analysis to the Arizona soil clone libraries, only 6 to 8 of the 134 to 161 species-level groups in each library are predicted to be reproducible at the 95% confidence level. On a larger scale, confident documentation of the most abundant 2,000 species (the top 50%) from the 4,000-species model community would require a sample size of 285,400 individuals. This sample size is 11.4-fold larger than the sample size of 25,000 individuals needed for documenting a random set of 2,000 species. These data demonstrate that if 16S rDNA surveys are used (as currently practiced) for comparing the composition of complex soil bacterial communities, the sample sizes must be dramatically larger than the sizes commonly used at present. Furthermore, only a fraction of the species present in a survey will be reproducible. Given a suitable model of the abundance distribution of species in a community, the reproducible fraction of species in a sample can be estimated.
Model inaccuracy.
The models we constructed depend on observed community sizes (total cell counts) and observed sample diversity. To determine the impact of model inaccuracies (over- or underestimates of community size or diversity) on estimates of sample scale, sampling curves were calculated for lognormal models based on Rmax values of 10, 11, and 12 (Fig. 6B). By using these values, theoretical communities were constructed that contained the same number of species (4,000) but varied in size by a factor of 8 (NT = 4 × 107, 1.2 × 108, and 4 × 108 cells, respectively). The required sample size for detection of the most abundant 50 species in each community was 1,639, 1,481, and 1,472 individuals, respectively. Varying the total number of species (ST) also had a minor effect on sample scale estimates. For example, for model communities containing 4,000, 6,000, or 10,000 species (Rmax = 11), the required sample sizes were 1,481, 1,759, and 2,140 individuals, respectively. The data show that for sampling the top fraction of the community, sample size increases as species diversity (ST) increases or as community size (Rmax) decreases. These relationships change, however, as progressively larger fractions of community diversity are sampled (Fig. 6B). Nonetheless, the magnitude of calculated sample sizes for surveying a specified set of species is generally similar despite small changes (potential inaccuracies) in model parameters.
DISCUSSION
Phylogenetic surveys of complex bacterial communities have historically been small, constrained by the technical difficulty and expense of characterizing a large number of individuals to the species level. We used four standard-sized surveys (approximately 200 clones each) for comparison of the composition of four soil communities from two sites in Arizona. The surveys were intended to assess unusual features of bacterial communities in the hot, dry, cinder soil of 900-year-old Sunset Crater volcano. After the eruption and cooling of the volcano, the area was recolonized by a few plant species and also, presumably, by the bacteria from the surrounding sandy loam soil that is typical of the region. Previously, we compared the species-level richness and structure of the libraries (7). In the present study, we examined patterns of diversity at the division level and constructed species abundance distributions to determine requirements for adequate sampling.
Interpreting division-level community composition and structure.
The structure and composition of biological communities are indicators of ecological complexity, evolutionary history, and community boundaries. At the species level, community structure can reveal differences in resource availability or in resource partitioning and succession status. However, at higher taxonomic levels such as the division level, the ecological significance of community composition and structure is ambiguous. The ecological relevance of differences in division abundance is impossible to interpret unless the change can be ascribed to natural selection acting upon a phenotype shared by most or all members of a division. While this situation is true for a few bacterial divisions, many divisions (e.g., the Proteobacteria) are known for the remarkable metabolic and ecological diversity of member species. It is therefore difficult to imagine that patterns of diversity at higher taxonomic levels are strongly shaped by primary ecological mechanisms.
The division-level structure and composition of bacterial communities may simply be a signature of communities with shared colonization history (i.e., who arrived and adapted first). As shown in Fig. 2, bacterial communities from different environments (rhizosphere versus interspace) that had shared history exhibited similar patterns of division-level structure. Environments with a common geologic history and geographic proximity would be expected to experience similar demographic processes, resulting in shared patterns of diversity at the coarsest levels of phylogenetic resolution. Such patterns may be maintained, despite environmental changes, as a result of differential population responses (declines in some populations can be counteracted by increases in the others) that buffer division-level abundance.
Determining the extent of local division diversity.
One of the primary uses of bacterial community surveys is to document the scope of phylogenetic diversity in natural environments. Every survey conducted to date has documented novel lineages at the species level or at higher taxonomic levels. The Arizona soil bacteria surveys included four deeply branching lineages (provisionally named SC1, SC2, SC3, and SC4) that appear to represent novel candidate divisions. Members of the SC1 and SC4 lineages were documented previously in surveys of a marine sample (unpublished NCBI sequence AF007732) and a Wisconsin soil sample (1), respectively. The independent collection of sequences representing these lineages supports the contention that the sequences represent legitimate, deeply branching bacterial groups. Sequences closely related to the SC2 and SC3 lineages have not yet been reported in other surveys. If the division-level status of these lineages is substantiated by additional studies, these putative divisions would raise the number of confirmed and candidate divisions in the bacterial domain from about 35 (9) to 39.
The total number of bacterial divisions that exist on a local or global scale is unknown. Since surveys of terrestrial bacterial communities are typically dominated by members of the Acidobacterium, Proteobacteria, and gram-positive divisions, determining the global extent of division-level diversity within the bacterial domain will depend upon sampling rare divisions that occur in local environments. The scale of surveys required for complete documentation of division-level diversity in a local environment could be estimated if the total number and abundance distribution of divisions were known. However, our unsuccessful attempts to extrapolate the total number of bacterial divisions in each Arizona soil community based on the observed survey data demonstrated that extrapolation of division-level diversity on a local scale will require empirical data from surveys that are either significantly larger in size, well replicated, or both.
Theoretical sampling of species diversity.
Larger surveys are also required to document the extent of bacterial species diversity in nature, but we can now predict the magnitude of these surveys by use of theoretical species abundance models. Modeling the distribution of species abundance in biological communities typically involves two distinct problems: fitting the curvature of the upper portion and fitting the curvature of the lower portion of the true distribution. The upper portion of the distribution represents the most abundant, easily sampled species, while the lower portion (the most uncertain portion, prone to greatest modeling error) represents the rare species that are difficult or impossible to sample. A single model may provide a good fit for only one portion of the distribution. For example, a community may generally fit a lognormal distribution but have a long lower tail in the distribution due to an overabundance of rare species (especially true for communities with high immigration rates) (8).
Addressing the two problems depends on a researcher's needs. If parametric extrapolation of species diversity (based on partial survey data) is the goal, selecting a model that well describes both portions of the true distribution is essential. However, in our case, error in the lower tail of the distribution is tolerable because we are most concerned with predicting sampling requirements for the dominant bacterial species (presumably, the species that contribute most to ecosystem processes). Therefore, we require a reasonable model describing at least the upper portion of the bacterial species abundance distribution.
We used a lognormal distribution as the basic model for the distribution of bacterial species abundance. At present the lognormal is the best choice as a null model of nonuniform bacterial species abundance because it is a purely statistical model requiring no assumptions about demographic, ecological, or evolutionary mechanisms that might shape bacterial community structure. A uniform distribution was recently suggested as the most appropriate distribution for bacterial communities in surface soils based on data from 16S rDNA surveys (40). The Arizona soil communities were in some regards similar to several of the communities described by Zhou et al. (40). For example, the arid Arizona soils were low-carbon (0.3% organic matter), unsaturated environments. Low clone dominance was observed in the Arizona bacterial surveys, with only six to eight clones comprising the most abundant species-level group in three of the libraries. The Arizona community surveys also yielded index values (1/D = 52, 100, 104, and 107) that were intermediate in the range of index values reported by Zhou et al. (40). Nonetheless, application of uniform distribution models to the Arizona community data produced implausible results. Most importantly, sampling simulations from uniform distribution models yielded surveys in which the abundance distribution of species-level groups differed markedly from those observed in the Arizona surveys (data not shown). In contrast, lognormal distribution models (Fig. 5) produced results consistent with the Arizona soil community surveys.
The lognormal model generates a distribution that is concave downward in the upper portion of the curve (when plotted as in Fig. 5A), a feature characteristic of the observed abundance distributions of species from plant, animal, and insect surveys (34). The traditional approach to constructing lognormal models for biological communities has been to derive the models directly from survey data. Survey data are plotted to identify the modal octave of the lognormal distribution and to measure the standard deviation or dispersion constant. This type of approach has not been possible for bacterial communities due to the extremely small size of the surveys conducted to date. Therefore, we devised a new approach that enables the construction and partial validation of lognormal null models for bacterial communities without extensive survey data.
Lognormal models of species abundance in bacterial communities can be constructed by using only two critical parameters: an estimate of the population size of the most abundant species (this defines Rmax) and an estimate of species richness. Both estimates can be selected by trial and error. The estimates create a model community of a fixed size, NT, that can be compared to an observed community size to partially validate the model. Models with community sizes that differ greatly from observed data can be quickly rejected. To further validate a model, the species richness in a simulated sample from a model can be compared to the species richness observed in a sample of identical size from a natural community. Combined, the size of a natural community and the observed species richness in a sample from the community impose severe restrictions on the modeling space and circumscribe a small set of feasible models, as shown in Fig. 4.
By using the approach above, we identified a set of lognormal models consistent with observed data from the Arizona soil communities. The set of feasible models describes communities in which about half of the species have population sizes of between 1 × 103 and 6 × 106 cells (g of soil)−1. These species comprise 99% of the bacterial biomass. The remaining 1% of biomass is distributed unequally between the remaining half of species with population sizes of less than 103 cells (g of soil)−1 (Fig. 5A and 5B). These models clearly create testable predictions. The population size of the dominant species can be confirmed experimentally, or additional surveys can be conducted to test predictions of sample diversity. The accuracy of the models, and therefore the true structure and composition of the natural communities, can thus be examined further by rational experimentation guided by concrete model predictions.
Equipped with a species abundance model, one can easily estimate the scale of surveys required for documentation of species diversity (as shown in Fig. 6A and 6B). For such predictions, the intended purpose of the survey has profound consequences on the required survey size. For example, one may wish to predict the sample size required to document a specified fraction (e.g., 10%) of the species diversity in a community or, alternatively, to predict the sample size needed for partial comparison of the species composition of two different communities. In the first case, a specified number of random species is required in a sample, whereas the identity of each species is irrelevant. For such a survey, sample size depends on additive sampling probabilities. In the second case, the identity or relative rank of species desired in the sample is paramount. In this case, the required survey size is a multiplicative function of the sampling probability of each specified species (or species rank) and is therefore much larger. In fact, as our results clearly showed, adequate documentation of even a modest number of soil bacterial species for partial comparison of community composition requires sample sizes orders of magnitude larger than those currently used.
To better illustrate the limitations of small surveys for comparison of species diversity in soil bacterial communities, we compared the species richness and species composition of simulated surveys. Five independent surveys (200 individuals each) were obtained by randomly selecting individuals from the 6,000-species model community. Species richness varied little between surveys (average, 159 species; range, 154 to 164 species; standard deviation [S.D.] = 3.6), in accord with the contention of Hughes et al. (11) that species richness, even in small surveys, has sufficient precision for use as a relative diversity index. In fact, rarefaction analysis of the model communities in Fig. 5A demonstrated over a broad range of sample sizes that species richness is reproducible (for a given sample size) with low sample-to-sample variance (data not shown).
Species composition, on the other hand, is highly variable. For example, based on the Arizona community models, 94 to 99% of the species occurring in a random sample of 200 individuals were predicted to vary between samples. This prediction was consistent with comparisons of the five simulated surveys. Only seven species were common to all five surveys (data not shown). In comparisons of four or three surveys, 11 (S.D. = 2) and 18 (S.D. = 3.5) common species were identified, respectively. In pairwise comparisons, an average of 38 common species (S.D. = 3) were identified, or in other words, paired surveys were 48% similar. The pairwise similarity of the existing clone libraries from the Arizona soils ranged from 11 to 22% (7). These observations demonstrate not only the limitations of small surveys but also, more importantly, the strength of theoretical species abundance models in guiding the interpretation of survey data.
A theoretical null model may not be an exact representation of a natural community. Since no sampling method is error free, species abundance models based on observed sample diversity may incorporate biases. PCR amplification and cloning of DNA sequences are known to introduce errors. Most of these errors distort the relative abundance of individual populations in a sample (18, 32, 33, 39), and some may inflate richness estimates up to 20% (14, 25, 37). However, we demonstrated (Fig. 6) that inaccurate modeling of community size (up to eightfold) or species diversity (up to 2.5-fold) has only minor effects on the estimates of sample sizes required for surveying the most abundant species. Consequently, we argue that theoretical models based on observed community sizes and sample diversity can effectively demonstrate the scope of sampling problems and the magnitude of surveys needed for adequate documentation of diversity in natural bacterial communities and can serve effectively as classical null models for hypothesis testing.
Acknowledgments
This work was supported by research grants to C.R.K. from the U.S. Department of Energy Program for Ecosystem Research and the DOE Microbial Genome Program.
We thank Joe Busch, Jody Davis, and Greg Fisher for technical assistance.
REFERENCES
- 1.Borneman, J., P. W. Skroch, K. M. O'Sullivan, J. A. Palus, N. G. Rumjanek, J. L. Jansen, J. Nienhuis, and E. R. Triplett. 1996. Molecular microbial diversity of an agricultural soil in Wisconsin. Appl. Environ. Microbiol. 62:1935-1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chao, A. 1987. Estimating the population size for capture-recapture data with unequal catchability. Biometrics 43:783-791. [PubMed] [Google Scholar]
- 3.Chao, A. 1984. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11:265-270. [Google Scholar]
- 4.Cobb, N. S., S. Mopper, C. A. Gehring, M. Caouette, K. M. Christensen, and T. G. Whitham. 1997. Increased moth herbivory associated with environmental stress of pinyon pine at local and regional levels. Oecologia 109:389-397. [DOI] [PubMed] [Google Scholar]
- 5.Colwell, R. K., and J. A. Coddington. 1994. Estimating terrestrial biodiversity through extrapolation. Phil. Trans. R. Soc. London 345:101-118. [DOI] [PubMed] [Google Scholar]
- 6.Dunbar, J., and C. R. Kuske. 2000. Assessment of microbial diversity in two southwestern U.S. soils by terminal restriction fragment (TRF) analysis. Appl. Environ. Microbiol. 66:2943-2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunbar, J., S. Takala, S. M. Barns, J. A. Davis, and C. R. Kuske. 1999. Levels of bacterial community diversity in four arid soils compared by cultivation and 16S rRNA gene cloning. Appl. Environ. Microbiol. 65:1662-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaston, K. J. 1994. Rarity. Chapman and Hall, London, England.
- 9.Hugenholtz, P. 2002. Exploring prokaryotic diversity in the genomic era. Genome Biol. 3:3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hugenholtz, P., C. Pitulle, K. L. Hershberger, and N. R. Pace. 1998. Novel division-level bacterial diversity in a Yellowstone hot spring. J. Bacteriol. 180:366-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes, J. B., J. J. Hellmann, T. H. Ricketts, and B. J. M. Bohannan. 2001. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl. Environ. Microbiol. 67:4399-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes, R. G. 1986. Theories and models of species abundance. Am. Nat. 128:879-899. [Google Scholar]
- 13.Hurlbert, S. H. 1971. The nonconcept of species diversity: a critique and alternative parameters. Ecology 52:577-586. [DOI] [PubMed] [Google Scholar]
- 14.Kopczynski, E. D., M. M. Bateson, and D. M. Ward. 1994. Recognition of chimeric small-subunit ribosomal DNAs composed of genes from uncultivated microorganisms. Appl. Environ. Microbiol. 60:746-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuske, C. R., S. M. Barns, and J. D. Busch. 1997. Diverse uncultivated bacterial groups from soils of the arid southwestern United States that are present in many geographic regions. Appl. Environ. Microbiol. 63:3614-3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lane, D. 1991. 16S/23S rRNA sequencing, p. 115-175. In E. Stackebrandt and M. Goodfellow (ed.), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, New York, N.Y.
- 17.Liesack, W., and E. Stackebrandt. 1992. Occurrence of novel groups of the domain Bacteria as revealed be analysis of genetic material isolated from an Australian terrestrial environment. J. Bacteriol. 174:5072-5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liesack, W., H. Weyland, and E. Stackebrandt. 1991. Potential risks of gene amplification by PCR as determined by SSU rDNA analysis of a mixed culture of strict barophilic bacteria. Microb. Ecol. 21:191-198. [DOI] [PubMed] [Google Scholar]
- 19.MacArthur, R. M. 1960. On the relative abundance of species. Am. Nat. 94:25-36. [Google Scholar]
- 20.Maidak, B. L., J. R. Cole, T. G. Lilburn, C. T. Parker Jr., P. R. Saxman, J. M. Stredwick, G. M. Garrity, B. Li, G. J. Olsen, S. Pramanik, T. M. Schmidt, and J. M. Tiedje. 2000. The RDP (Ribosomal Database Project) continues. Nucleic Acids Res. 28:173-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.May, R. M. 1975. Patterns of species abundance and diversity, p. 81-120. In M. L. Cody and J. M. Diamond (ed.), Ecology and evolution of communities. Belnap Press, Cambridge, Mass.
- 22.O'Donnell, A. G., M. Goodfellow, and D. L. Hawksworth. 1994. Theoretical and practical aspects of the quantification of biodiversity among microorganisms. Phil. Trans. R. Soc. London B 345:65-73. [DOI] [PubMed] [Google Scholar]
- 23.Preston, F. W. 1962. The canonical distribution of commonness and rarity. Ecology 43:185-215. [Google Scholar]
- 24.Preston, F. W. 1948. The commonness and rarity of species. Ecology 29:254-283. [Google Scholar]
- 25.Qiu, X., L. Wu, H. Huang, P. E. McDonal, A. V. Palumbo, J. M. Tiedje, and J. Z. Zhou. 2001. Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Appl. Environ. Microbiol. 67:880-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raaijmakers, J. G. W. 1987. Statistical analysis of the Michaelis-Menten equation. Biometrics 43:793-803. [PubMed] [Google Scholar]
- 27.Ravenschlag, K., K. Sahm, J. Pernthaler, and R. Amann. 1999. High bacterial diversity in permanently cold marine sediments. Appl. Environ. Microbiol. 65:3982-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosselló-Mora, R., and R. Amann. 2001. The species concept for prokaryotes. FEMS Microbiol. Rev. 25:39-67. [DOI] [PubMed] [Google Scholar]
- 29.Sandaa, R., V. Torsvik, Ø. Enger, F. L. Daae, T. Castberg, and D. Hahn. 1999. Analysis of bacterial communities in heavy metal-contaminated soils at different levels of resolution. FEMS Microbiol. Ecol. 30:237-251. [DOI] [PubMed] [Google Scholar]
- 30.Simberloff, D. 1978. Use of rarefaction and related methods in ecology, p. 150-165. In K. L. Dickson, J. J Cairns, and R. J. Livingston (ed.), Biological data in water pollution assessment: quantitative and statistical analyses. American Society for Testing and Materials, West Conshohocken, Pa.
- 31.Stackebrandt, E., and B. M. Goebel. 1994. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 44:846-849. [Google Scholar]
- 32.Suzuki, M., M. S. Rappe, and S. J. Giovannoni. 1998. Kinetic bias in estimate of coastal picoplankton community structure obtained by measurements of small-subunit rRNA gene PCR amplicon length heterogeneity. Appl. Environ. Microbiol. 64:4522-4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki, M. T., and S. J. Giovannoni. 1996. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62:625-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tokeshi, M. 1993. Species abundance patterns and community structure. Adv. Ecol. Res. 24:111-186. [Google Scholar]
- 35.Torsvik, V., J. Goksoyr, and F. L. Daae. 1990. High diversity in DNA of soil bacteria. Appl. Environ. Microbiol. 56:782-787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torsvik, V., R. Sorheim, and J. Goksoyr. 1996. Total bacterial diversity in soil and sediment communities—a review. J. Ind. Microbiol. 17:170-178. [Google Scholar]
- 37.Wang, G. C., and Y. Wang. 1997. Frequency of formation of chimeric molecules as a consequence of PCR coamplification of 16S rRNA genes from mixed bacterial genomes. Appl. Environ. Microbiol. 63:4645-4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson, K. H., R. B. Blitchington, and R. C. Green. 1990. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J. Clin. Microbiol. 28:1942-1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wintzingerode, F. V., U. B. Gobel, and E. Stackebrandt. 1997. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 21:213-229. [DOI] [PubMed] [Google Scholar]
- 40.Zhou, J., B. Xia, D. S. Treves, L. Y. Wu, T. L. Marsh, R. V. O'Neill, A. V. Palumbo, and J. M. Tiedje. 2002. Spatial and resource factors influencing high microbial diversity in soil. Appl. Environ. Microbiol. 68:326-334. [DOI] [PMC free article] [PubMed] [Google Scholar]