

Abstract
The rubredoxin from Clostridium pasteurianum (CpRd) provides an excellent system for investigating how the protein sequence modulates the reduction potential of the active site in an iron–sulfur protein. 15N NMR spectroscopy has allowed us to determine with unprecedented accuracy the strengths of all six key hydrogen bonds between protein backbone amides and the sulfur atoms of the four cysteine residues that ligate the iron in the oxidized (FeIII) and reduced (FeII) forms of wild-type CpRd and nine mutants (V44G, V44A, V44I, V44L, V8G, V8A, V8I, V8L, and V8G/V44G). The length (or strength) of each hydrogen bond was inferred from the magnitude of electron spin delocalized across the hydrogen bond from the iron atom onto the nitrogen. The aggregate lengths of these six hydrogen bonds are shorter in both oxidation states in variants with higher reduction potential than in those with lower reduction potential. Differences in aggregate hydrogen bonding upon reduction correlate linearly with the published reduction potentials for the 10 CpRd variants, which span 126 mV. Sequence effects on the reduction potential can be explained fully by their influence on hydrogen-bond strengths.
Keywords: iron–sulfur protein, reduction potential tuning, 15N NMR, paramagnetic NMR
Members of electron transfer chains, such as those involved in photosynthesis and respiration, have evolved finely tuned reduction potentials that facilitate electron transfer with exceptional specificity and efficiency (1–3). Just how these proteins achieve the proper setting of the reduction potential remains a fundamental question. An important category of electron transfer proteins are the iron–sulfur proteins, whose reduction potentials range from -700 to +400 mV (4–6). Different types of iron–sulfur clusters have varying intrinsic reduction potentials. Within a single cluster type, three major factors have been advanced in explaining reduction potential variations: the electrostatic environment of the redox center provided by surrounding charged residues, hydrogen-bonds (H-bonds) to the cluster, and the solvent exposure of the redox center (7–11).
Rubredoxin, whose redox active site consists of a single iron ligated by four cysteinyl sulfurs, is a member of the simplest type of iron–sulfur protein. Wild-type rubredoxins can be divided into two categories on the basis of their reduction potentials. Those with lower reduction potentials, such as Clostridium pasteurianum rubredoxin (CpRd), contain Val at residue 44, whereas those with reduction potentials ≈50 mV higher, such as Pyrococcus furiosus rubredoxin (PfRd), have Ala-44 (12, 13). The covalent character of the iron–sulfur bonds in CpRd and PfRd has been determined quantitatively by K-edge x-ray absorption spectroscopy; however, these studies revealed no direct correlation between iron–sulfur covalency and the reduction potential (14). A further detailed comparison of the crystal structures ascribed the reduction potential difference to different orientations of the peptide dipole at residue 44, along with a change in the distance between the Xaa44 N and Cys-42 Sγ (15, 16).
NMR offers a sensitive approach for examining redox centers in iron–sulfur proteins (17, 18). Interactions between the unpaired electrons of the iron and the surrounding nuclei are manifested in NMR spectra by hyperfine shifts. In the case of CpRd, the 15N hyperfine shifts of the backbone nitrogens of 12 residues are dominated by Fermi contact coupling, which arises from delocalization of unpaired electron spin density from the iron onto the protein through chemical bonds (including H-bonds) (19, 20). In CpRd, six backbone amides are involved in H-bonds with Cys Sγ atoms: residues Val-8, Cys-9, Tyr-11, Leu-41, Cys-42, and Val-44 (Fig. 1a), and their hyperfine 15N chemical shifts are dominated by electron delocalization through the Fe-S·····H-15N H-bond (19, 20). The 15N chemical shifts can resolve changes in H-bond distances on the order of 0.01 Å (19). Recently, an NMR study of a set of CpRd mutants revealed that electron delocalization onto the nitrogen of residue 44 varied with side-chain substitutions at this residue in a way that tracked changes in the reduction potential of the protein (21). This result has prompted us to carry out a comprehensive analysis of all six Fe-S·····HN H-bonds of CpRd in a series of 10 sequence variants in their oxidized (FeIII) and reduced (FeII) states. The question investigated was whether aggregate changes in H-bond strengths as reported by trans-H-bond electron delocalization between the oxidized and reduced states could explain the changes in reduction potential. We report here that the 126-mV span of reduction potentials of these 10 rubredoxin variants can be explained fully by observed changes in the strengths of H-bonds resulting from the mutations. We show that aggregate changes in multiple H-bonds, rather than changes in a single H-bond, must be taken into consideration to explain the full set of data.
Fig. 1.

The Fe–S center in CpRd. (a) Schematic representation of the H-bond network (blue arrows). The Sγ atoms of the four Cys residues are denoted by S; the other letters are conventional single-letter amino acid designations. (b) Close-up view of the 3D structure showing the positions of the mutated residues Val-8 and -44.
Materials and Methods
Site-Specific Mutagenesis. The V44G, V44I, V44L, V8G, V8A, and V8I mutants were constructed by the overlap extension method in which gene halves are synthesized with overlapping ends (22). Two internal complementary primers were synthesized to contain the site to be mutated. Each of these mutagenic primers was combined with the corresponding end primer to amplify the half gene fragment in two separate PCR reactions. The two products were annealed to create a full-length gene fragment, which was amplified by PCR, digested by restriction enzyme NdeI and BamH I, and then ligated into a pET3a vector (Novagen). The ligation plasmid, encoding the mutation, was subsequently transformed into Escherichia coli strain XL1-Blue (Stratagene) for amplification of the plasmid. The construct was verified by DNA sequencing carried out at the University of Wisconsin Biotechnology Center. E. coli strain BL21(DE53)/pLysS (Novagen) was used for general protein expression.
The Stratagene QuikChange II site-directed mutagenesis method was used to generate the V44A, V8L, and V8G/V44G mutants. Two synthetic oligonucleotide primers were designed to contain the desired mutation and to be complementary to the plasmid template. These mutagenic primers were extended during thermal cycling by Pfu DNA polymerase (Stratagene). After the synthesis of the mutation-containing plasmid, DpnI endonuclease (Stratagene) was added to digest the methylated, nonmutated DNA plasmid to remove the original DNA template. The final product was transformed into E. coli strains XL1-Blue (Stratagene) and BL21(DE53)/pLysS (Novagen) for DNA sequence identification and protein production.
Production of Labeled CpRd Protein Variants. The CpRd mutants were overexpressed by following procedures similar to those described in ref. 23. Cells of E. coli BL21(DE53)/pLysS (Novagen), containing mutated CpRd/pET3a, were grown in LB media with 100 mg·liter-1 ampicillin (Sigma-Aldrich) and 34 mg·liter-1 chloramphenicol (Sigma-Aldrich) at 37°C with vigorous shaking. As the A600 of the culture reached 1.3, IPTG was added to a concentration of 100 mg·liter-1 to induce protein expression. After continuous incubation for 2.5 h, the culture was harvested by centrifugation, and the cell pellets were resuspended in 50 mM Tris·Cl buffer (pH 7.4).
Selective labeling patterns were achieved by residue-specific and inverse residue-specific labeling (24). By manipulating the composition of the growth medium, protein can be either uniformly labeled with 15N or selectively labeled with a specific amino acid containing 15N. The medium used for uniform 15N-labeling was M9 containing 15NH4Cl. Residue-specific labeling was accomplished with standard M9 plus a mixture of all amino acids at natural abundance with the exception of that to be labeled. The 15N-labeled amino acid was added to the culture at the time of induction. For inverse residue-specific labeling, the protein was grown in the uniform 15N-labeling medium. At the time of induction, a natural abundance (14N) amino acid was added, which resulted in reduction of the 15N NMR signal intensity from that residue type.
To enhance the yield of holoprotein, the iron center was reconstituted in vitro. Cells were lysed by a freeze–thaw cycle followed by sonication. After adding urea to a final concentration of 8 M, the cell lysate was stirred for ≈2 h at room temperature to allow protein denaturation. The mixture was then degassed by alternately applying vacuum and purging with argon gas. Degassed DTT (Sigma-Aldrich) was added to a final concentration of 100 mM to ensure a reducing environment. Under flushing with argon gas, FeCl3 was added until a black precipitate formed. The mixture then was stirred for another 10 min under purging argon gas. Next, it was diluted eightfold into degassed 50 mM Tris·Cl buffer (pH 7.4), and finally it was diluted twofold into undegassed buffer to allow protein refolding and iron center reconstitution.
The rubredoxin-containing mixture was centrifuged and filtered to remove precipitates. The supernatant was loaded onto a DE53 anion exchange column, and the refolded holoprotein was subsequently eluted with an NaCl gradient in 50 mM Tris·Cl buffer (pH 7.4). The eluate was subjected to further purification by gel filtration.
NMR Spectroscopy. All 1D 15N NMR spectra were acquired at 20°C on a DMX500 spectrometer (Bruker, Billerica, MA) with a 5-mm broadband probe. The buffer was 50 mM potassium phosphate (pH 6.0) and 90% 1H2O/10% 2H2O for all mutants and wild-type CpRd. To suppress diamagnetic signals, the SuperWEFT pulse sequence (25) was used for samples in the oxidized state, and the one-pulse sequence with short repetition time (≈20 ms) was used for samples in the reduced state. Reduced protein was prepared by adding small amounts of solid sodium dithionite to the protein solution under anaerobic conditions. The chemical shift of 15N was indirectly referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonate by using the 15N/1H ratio = 0.101329118 (data source: www.bmrb.wisc.edu).
Theoretical Calculations. A tetrahedral computer model of iron tetramethylthiolate was constructed by using the Fe–S bond lengths from the oxidized and reduced bis(o-xylyl-aa′-dithiolato)ferrate(II,III) anion crystal structures (26). To model an H-bonded amide, one methyl formamide was added to the structure. With the Fe–S cluster held rigidly, the geometry was optimized. For each of the oxidation states (FeII and FeIII), an additional four models were created by increasing the distance between the amide H and the sulfur atom by 0.1-Å increments away from the optimized H-bond distance. In each model, the methyl formamide(s) was reoptimized with the Fe–S cluster geometry and H–S distance held rigidly. This procedure maintained the difference in H-bond length between each pair of models for the oxidized and reduced state. The level of theory was B3LYP/6-31g**. The gaussian 2003 program (revision B.04; ref. 27) was used in performing all calculations.
The lengths of four of the six HN·····Sγ H-bonds were estimated (20) from the paramagnetic component (largely Fermi-contact) of the observed 15N chemical shift. Covalent bond effects on the 15N chemical shifts of the two H-bonded Cys residues (Cys-9 and -42) prevented estimates of their H-bond lengths. The HN·····Sγ H-bond distance for oxidized dox and reduced dred CpRd were estimated from theory as
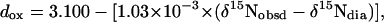 |
[1] |
and
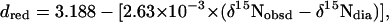 |
[2] |
where 15Nobsd is the experimental shift and 15Ndia is the diamagnetic 15N chemical shift for the residue type from Biological Magnetic Resonance Data Bank (www.bmrb.wisc.edu).
Results
Measurement of 15N NMR Hyperfine Chemical Shifts. Hyperfine 15N NMR spectra were collected for wild-type CpRd and mutants of CpRd at two residues (Val-8 and -44) that participate in Fe–S·····HN H-bonds (Fig. 1b): V44G, V44A, V44I, V44L, V8G, V8A, V8I, V8L, and V8G/V44G. Spectra were obtained for all 10 proteins in both their oxidized and reduced states. The hyperfine 15N spectral regions of these oxidized and reduced CpRd variants, ordered according the residue(s) mutated and within classes by their reduction potentials (16), (Fig. 2) exhibit striking chemical shift changes. For each variant, all 15N hyperfine-shifted signals were assigned by residue-specific 15N-labeling or inverse residue-specific 15N-labeling (I.-J.L., B. Xia, D. S. King, T.E.M., W.M.W., and J.L.M., unpublished data). All signals were assigned to residue type, and most were given sequence-specific assignments (see Table 1, which is published as supporting information on the PNAS web site). The predicted HN·····Sγ bond lengths are tabulated in Table 2, which is published as supporting information on the PNAS web site.
Fig. 2.

Shown are 1D 15N NMR spectra of CpRd variants. (a) Series of variants at position 44. (b) Series of variants at position 8. (c) Double mutant V8G/V44G. The reduction potentials indicated are those determined for these proteins by square wave voltammetry by Wedd and coworkers (16).
Correlation of 15N Shifts with Reduction Potentials. In the Val-44 series of variants, in both their oxidized and reduced states, the 15N NMR signal exhibiting the largest dependence on amino acid sequence was that of the mutated residue (Fig. 2a). All variants at position 44, with the exception of V44L, exhibited a progressive increase in the 15N hyperfine shift as a function of increased reduction potential of the protein. On the basis of the 15N hyperfine shift data, mutation of residue Val-44 of CpRd to Ile, Ala, or Gly decreases the length of the H-bond in the oxidized protein by 0.11, 0.29, and 0.47 Å, respectively (Table 2). Changes observed in other residues indicated changes in H-bonding, but they did not track the reduction potential. Although the hyperfine shift at residue 44 for V44L failed to follow the trend with reduction potential, the 15N signal from residue Val-8 in that mutant showed a large upfield shift (Fig. 2a). This result indicates that, although the H-bond from residue 44 to the iron–sulfur cluster becomes shorter in oxidized V44L, the H-bond from residue Val-8 becomes longer.
In the series of Val-8 variants, both V8I and V8L exhibited 15N hyperfine shifts and reduction potentials similar to those of wild-type CpRd in oxidized and reduced states (Fig. 2b). Substitution of Val-8 by Ala or Gly resulted in large spectral changes (Fig. 2b) as well as large changes in the reduction potential. With increasing reduction potential the 15N hyperfine shifts of residues Xaa8, Tyr-11, and Leu-41 moved to higher frequencies in the oxidized state, whereas the 15N hyperfine signals from residues 8 and 41 moved to higher frequencies in the reduced state. In the oxidized Xaa8 variants, the 8HN·····Sγ6 (HN of residue 8 H-bonded to the Sγ of residue 6), 11HN-Sγ9, and 41HN·····Sγ39 bonds tend to be shorter in mutants featuring higher reduction potentials. In the reduced state, both H-bonds (8HN·····Sγ6 and 41HN·····Sγ39) tend to be shorter in variants with higher reduction potential.
Spectra of the double mutant (V8G/V44G) displayed clear differences from the wild-type spectra in both oxidation states (Fig. 2c). Replacement of both Val-8 and -44 with Gly shifts the reduction potential from -77 to +39 mV (a change of +116 mV), the highest reduction potential among all CpRd variants (16).
Aggregate 15N Hyperfine Shift Differences. Because the data show that changes can occur in all six H-bonds, to investigate the influence of H-bonds on the reduction potential in each variant we have subtracted the sum of the hyperfine shifts in the reduced state from the sum of the hyperfine shifts in the oxidized state. We use the symbol ΔΣδ to denote this value
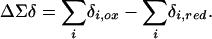 |
[3] |
Because this procedure takes into account signals from all Fe–S·····HN H-bonds, the approach provides a rigorous analysis even given unambiguous sequence-specific assignments for the signals from Cys-9/Cys-42 in all variants and Gly-8/Gly-44 in the double mutant (Table 1). Given the 15N hyperfine shift has an approximate linear relationship with the H–S distance in H-bonds to the iron–sulfur cluster in CpRd (20), and that shorter H-bonds are stronger than longer H-bonds, changes in the hyperfine shifts of 15N atoms involved in H-bonds can be interpreted in terms of changes in strengths of the H-bonds. As discussed below, for small changes in the H-bond length, the energy changes in a roughly linear fashion.
The reduction potential is related to the energy differences between the reduced and oxidized states of the molecule. Because the sum of the 15N hyperfine shifts of the H-bonded residues in a given oxidation state reflects the total strengths of the H-bonds, the difference between the sums of the hyperfine shifts in the reduced and oxidized states represents the contribution of changes in H-bonding to the energy difference or reduction potential. The correlation between the difference of the sums of hyperfine shifts in two oxidation states and the reduction potential is linear for the series of 10 variants investigated (Fig. 3). This finding suggests that the mechanism for transmitting the effect of the side-chain substitution(s) to the iron center is mediated largely by changes in H-bond strengths. The slope of the line is 0.20 mV·ppm-1 (r2 = 0.86). Better linear correlations are achieved by fitting the data from the Val-44 and -8 series individually (excluding the double mutant) to lines with different slopes: Val-44 series (0.24 mV·ppm-1, r2 = 0.97) and Val-8 series (0.16 mV·ppm-1, r2 = 0.98). The double mutant falls on the line for the Val-44 series.
Fig. 3.

Plot of ΔΣδ, the difference (oxidized - reduced) of the summed 15N chemical shifts of each H-bonded nitrogen (15N signals from residues 8, 9, 11, 41, 42, and 44) for each of the 10 CpRd variants vs. the reduction potential of the variant. The circles represent the Val-44 series of variants; the triangles represent the Val-8 series of variants; and the diamond represents the double mutant (V8G/V44G). The solid line is the best linear fit to the full set of data. The results indicate that the reduction potential within a series can be predicted from the ΔΣδ value, which reflects the contribution of H-bonding energy. As noted in the text, significantly better linear fits can be obtained by considering the Val-44 and -8 series separately, as shown by the dashed lines.
Theoretical Calculations. We carried out quantum chemical calculations to investigate the theoretical basis for the observed results. A harmonic oscillator model of the energy as a function of the H–S distance yielded a quadratic term for the reduced state ≈15% larger than that for the oxidized state. This result indicates that as the H-bond becomes shorter, the energy of the reduced state decreases faster than that of the oxidized state and results in a shift of the reduction potential to more positive values, as observed in the experimental data.
The fact that the sum of the 15N hyperfine shifts can be used to estimate the total H-bond strengths in a given mutant suggests the redox center of CpRd can be modeled in terms of a single amide moiety with a H-bond to a sulfur atom in a cluster model such as iron tetramethylthiolate. To examine the relationship between the reduction potentials and the H-bond strengths, we calculated the gas-phase reduction potential and the ΔΣδ for a series of two such models (FeII and FeIII) with increasing N-H·····S H-bond lengths. The crude theoretical model yielded a linear slope of 0.08 mV·ppm-1 (Fig. 4).
Fig. 4.

Plot of the theoretical (oxidized - reduced) hyperfine shifts (ΔΣδ in analogy with Fig. 3; however, here the sum is over a single H-bond) vs. the theoretical reduction potential (gas phase) for iron tetramethylthiolate with an H-bonded methyl formamide as a model of the rubredoxin redox center. The solid line is a linear regression analysis (r2 = 0.89). The values are relative, with the largest set equal to 0 mV.
Discussion
Detailed comparison of available x-ray crystal structures of CpRd variants has revealed that slight changes in the structure surrounding the iron-sulfur cluster are responsible for the range of observed reduction potentials (15, 16, 28). In our study, 15N hyperfine shifts were used to detect subtle changes in HN·····Sγ bond lengths in CpRd variants. The 15N hyperfine shifts report changes in HN·····Sγ bond lengths at the iron center with remarkable sensitivity: a change in HN·····Sγ bond length of only 0.1 Å changes the 15N hyperfine shift by nearly 90 ppm (20). Each hyperfine shift characterizes the interplay of a specific nucleus with the delocalized unpaired electrons of the iron–sulfur cluster. By using a variety of labeling strategies, we have assigned the well-resolved hyperfine shifts unambiguously to specific amino acid residues. The hyperfine shifts arising from this interaction serve as “fingerprints” of the protein in the vicinity of iron–sulfur cluster.
For each of the 10 variants, the difference (oxidized - reduced) between the sums of hyperfine 15N chemical shifts of the six amides involved in Fe–S·····H–N H-bonds provided a measure of the changes in H-bonding. The high degree of correlation between these ΔΣδ values and with the reduction potentials of the CpRd variants (Fig. 3) indicated that H-bonding can account fully for the modulation of reduction potential. The experimental slopes (Fig. 3) were 0.20 mV·ppm-1 for the CpRd Val-44 series of variants and 0.17 mV·ppm-1 for the CpRd Val-8 series. The calculated differences between the reduced and oxidized spin densities and energies (Fig. 4) provide a theoretical analogy with the experimental data shown in Fig. 3. Although the sophistication and accuracy of the model might be increased by the use of continuum solvent methods, it is clear that this simple model captures the essence of the modulation of the reduction potential by the change in the length/strength of the H-bond. The experimental data (Fig. 3) may appear more linear than the theoretical curve (Fig. 4) because they represent the summed effects of multiple H-bonds of varying strength.
In wild-type CpRd, V44I, V44A, and V44G, the length of a single H-bond, 44HN·····Sγ42, was found to shorten in proportion to more positive reduction potential values. Because changes in the other H-bonds are minimal, the fine tuning of the reduction potential in this limited number of variants can be attributed to variations in this one H-bond. In other CpRd variants investigated, changes were observed in the lengths of more than one H-bond. In mutant V44L, the effect of lengthening of the 44HN·····Sγ42 bond is compensated by the effect of shortening of the 8HN·····Sγ6 bond. These H-bonds cooperatively regulate the reduction potential in the V44L mutant. In the case of Val-8 series of variants, a correlation was observed between the shortening of multiple H-bonds and higher reduction potential. The slopes of the plots of ΔΣδ vs. the reduction potential (Fig. 3) are slightly different in the two series of mutants. One possible interpretation of this result is that the H-bond effect in the Val-8 series (represented by ΔΣδ) is offset by an opposing energetic factor that alters the relative stabilities of the oxidized and reduced states in a manner that scales with the aggregate strengths of the H-bonds. Steric strain induced by tightening of the iron-center H-bonds offers a possible mechanism with the required properties. Because residue 8 is located in the Cys-X-X-Cys loop, its motion presumably is more restricted than that of residue 44, which is located outside the two Cys-X-X-Cys loops (Fig. 1a). Thus, a decrease in the length of the residue 8 H-bond may lead to an increase in steric strain, whereas the residue 44 H-bond length may have little or no steric coupling. The finding that the double mutant (V44G/V8G) falls on the line with the V44 series can be explained by its lack of strain as the result of the V44G substitution.
Although modulation of the reduction potential by H-bonding was observed in both the Val-44 and -8 series of variants, the Val-44 series offers the simpler paradigm for the manipulation of the reduction potential. By varying the length of a single H-bond supplied by residue 44, the reduction potential in CpRd can be varied by nearly 80 mV. By contrast, mutations at position 8 affect H-bonds at other sites in addition to 8. Unlike residue Val-44, which forms the only backbone amide H-bond to the Sγ atom of residue Cys-42, the backbone amides of both residues Val-8 and Cys-9 form H-bonds to the Sγ atom of residue Cys-6 (Fig. 1b). Residue Val-8 is thus coupled more tightly to the H-bonding network of the iron–sulfur cluster than residue Val-44. Hence, mutations at residue 8 lead to more extensive effects around the iron–sulfur cluster. In nature, whereas position 8 is conserved in different species as Val, Ile, or Leu (substitutions that change the reduction potential by ≤5 mV), position 44 is generally Val or Ala (a substitution that changes the reduction potential by 53 mV). It may be too energetically expensive and destabilizing to modulate the reduction potential by substitutions at the Val-8 site. By contrast, the overall structural insensitivity to mutations at position 44, except for V44L, allows for efficient manipulation of the reduction potential by changing the amino acid residue, without accompanying structural changes. In the Val-44 series, V44L is the only mutation that leads to appreciable changes in multiple H-bonds.
In the case of double mutation, neither the reduction potential nor the H-bonding change can be predicted from adding the effects of the individual mutations. The change in the reduction potential for the double mutant (116 mV) is less than the sum of the reduction potential changes for the single mutants (70 mV + 77 mV = +147 mV). Parallel to this finding, the H-bond contribution to the reduction potential as measured by ΔΣδ is much less for the double mutant (ΔΣδ = 817 ppm) than for the sum of the two single mutants (ΔΣδ = 1,472 ppm for the sum of ΔΣδ = 846 ppm for V8G plus ΔΣδ = 626 ppm for V44G).
It is clear that H-bonds can play an important role in the modulation of reduction potentials in iron–sulfur proteins. In all cases the average HN·····Sγ bond length was observed to be shorter in variants that have higher reduction potentials. This finding is intuitively satisfying in that shorter H-bonds allow for more efficient delocalization (and stabilization) of the negative charge in the reduced state. Observed changes in H-bond lengths are sufficient to explain the reduction potentials in this series of 10 CpRd variants investigated here. The results show that the reduction potential of CpRd can be modulated by as much as 126 mV by altering one or more H-bond lengths. The simple linear correlation between difference of sums of 15N hyperfine shifts, and the reduction potential observed in this study offers the basis for further exploitation in the bioengineering field. Furthermore, these results permit the interpretation of the structural roles played by residues 8 and 44 in determining the functional properties of rubredoxin. The present work not only provides insight into the elucidation of sequence–structure–function relationship in CpRd but also shows the versatility of NMR as a sensitive tool for the study of metal-containing biomolecules. It will be of interest to test the HN·····Sγ bond lengths predicted from the 15N chemical shifts (Table 2) as suitable diffraction methods become available.
It will be of interest to determine whether the approach used here, quantitative analysis of H-bonding from measurements of hyperfine NMR shifts, is applicable to other types of metal centers in proteins. H-bonding has been shown to affect reduction potentials in blue-copper proteins, as demonstrated by comparisons of proteins from different species (29) and from mutagenesis investigations (30, 31). Blue-copper proteins may constitute another well-studied system worthy of investigation.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Institute of General Medical Sciences. NMR data were collected at the National Magnetic Resonance Facility at Madison, which is supported by grants from the National Institutes of Health National Center for Research Resources and Institute of General Medical Sciences.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CpRd, Clostridium pasteurianum rubredoxin; 8HN·····Sγ6, H-bond between the backbone amide H of residue 8 and the side-chain S of residue 6.
Data deposition: 15N NMR chemical shifts for the oxidized and reduced CpRd variants have been deposited at the Biological Magnetic Resonance Data Bank, www.bmrb.wisc.edu (accession codes 6659–6678).
References
- 1.Marcus, R. A. & Sutin, N. (1985) Biochim. Biophys. Acta 811, 265-322. [Google Scholar]
- 2.Gray, H. B. & Winkler, J. R. (1996) Annu. Rev. Biochem. 65, 537-561. [DOI] [PubMed] [Google Scholar]
- 3.Moser, C. C., Keske, J. M., Warncke, K., Farid, R. S. & Dutton, P. L. (1992) Nature 355, 796-802. [DOI] [PubMed] [Google Scholar]
- 4.Beinert, H. (2000) J. Biol. Inorg. Chem. 5, 2-15. [DOI] [PubMed] [Google Scholar]
- 5.Holm, R. H., Kennepohl, P. & Solomon, E. I. (1996) Chem. Rev. 96, 2239-2314. [DOI] [PubMed] [Google Scholar]
- 6.Imsande, J. (1999) Plant Physiol. Biochem. 37, 87-97. [Google Scholar]
- 7.Bertrand, P., Mbarki, O., Asso, M., Blanchard, L., Guerlesquin, F. & Tegoni, M. (1995) Biochemistry 34, 11071-11079. [DOI] [PubMed] [Google Scholar]
- 8.Kassner, R. J. (1972) Proc. Natl. Acad. Sci. USA 69, 2263-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodgers, K. K. & Sligar, S. G. (1991) J. Am. Chem. Soc. 113, 9419-9421. [Google Scholar]
- 10.Adman, E., Watenpaugh, K. D. & Jensen, L. H. (1975) Proc. Natl. Acad. Sci. USA 72, 4854-4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carter, C. W., Jr. (1977) J. Biol. Chem. 252, 7802-7811. [PubMed] [Google Scholar]
- 12.Swartz, P. D. & Ichiye, T. (1997) Biophys. J. 73, 2733-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swartz, P. D., Beck, B. W. & Ichiye, T. (1996) Biophys. J. 71, 2958-2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rose, K., Shadle, S. E., Eidsness, M. K., Kurtz, D. M., Scott, R. A., Hedman, B., Hodgson, K. O. & Solomon, E. I. (1998) J. Am. Chem. Soc. 120, 10743-10747. [Google Scholar]
- 15.Eidsness, M. K., Burden, A. E., Richie, K. A., Kurtz, D. M., Scott, R. A., Smith, E. T., Ichiye, T., Beard, B., Min, T. P. & Kang, C. H. (1999) Biochemistry 38, 14803-14809. [DOI] [PubMed] [Google Scholar]
- 16.Xiao, Z., Maher, M. J., Cross, M., Bond, C. S., Guss, J. M. & Wedd, A. G. (2000) J. Biol. Inorg. Chem. 5, 75-84. [DOI] [PubMed] [Google Scholar]
- 17.Bertini, I. & Luchinat, C. (1986) NMR of Paramagnetic Molecules in Biological Systems (Benjamin/Cummings, Menlo Park, CA).
- 18.Bertini, I., Turano, P. & Vila, A. J. (1993) Chem. Rev. 93, 2833-2932. [Google Scholar]
- 19.Xia, B., Wilkens, S. J., Westler, W. M. & Markley, J. L. (1998) J. Am. Chem. Soc. 120, 4893-4894. [Google Scholar]
- 20.Wilkens, S. J., Xia, B., Weinhold, F., Markley, J. L. & Westler, W. M. (1998) J. Am. Chem. Soc. 120, 4806-4814. [Google Scholar]
- 21.Lin, I. J., Gebel, E. B., Machonkin, T. E., Westler, W. M. & Markley, J. L. (2003) J. Am. Chem. Soc. 125, 1464-1465. [DOI] [PubMed] [Google Scholar]
- 22.Ho, S. N., Hunt, H. D., Horton, R. M., Pullen, J. K. & Pease, L. R. (1989) Gene 77, 51-59. [DOI] [PubMed] [Google Scholar]
- 23.Xia, B., Westler, W. M., Cheng, H., Meyer, J. P., Moulis, J.-M. & Markley, J. L. (1995) J. Am. Chem. Soc. 117, 5347-5350. [Google Scholar]
- 24.Cheng, H., Westler, W. M., Xia, B., Oh, B.-H. & Markley, J. L. (1995) Arch. Biochem. Biophys. 316, 619-634. [DOI] [PubMed] [Google Scholar]
- 25.Inubushi, T. & Becker, E. D. (1983) J. Magn. Reson. 51, 128-133. [Google Scholar]
- 26.Lane, R. W., Ibers, J. A., Frankel, R. B., Papaefthymiou, G. C. & Holm, R. H. (1977) J. Am. Chem. Soc. 99, 84-98. [DOI] [PubMed] [Google Scholar]
- 27.Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Montgomery, J. A., Jr., Vreven, T., Kudin, K. N., Burant, J. C., et al. (2003) gaussian 03 (Gaussian, Pittsburgh), Revision B.04.
- 28.Park, I. Y., Eidsness, M. K., Lin, I. J., Gebel, E. B., Youn, B., Harley, J. L., Machonkin, T. E., Frederick, R. O., Markley, J. L., Smith, E. T., et al. (2004) Proteins 57, 618-625. [DOI] [PubMed] [Google Scholar]
- 29.Li, H., Webb, S. P., Ivanic, J. & Jensen, J. H. (2004) J. Am. Chem. Soc. 126, 8010-8019. [DOI] [PubMed] [Google Scholar]
- 30.Dong, S., Ybe, J. A., Hecht, M. H. & Spiro, T. G. (1999) Biochemistry 38, 3379-3385. [DOI] [PubMed] [Google Scholar]
- 31.Machczynski, M. C., Gray, H. B. & Richards, J. H. (2002) J. Inorg. Biochem. 88, 375-380. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.