

Abstract
In vitro cell cultures were compared to neonatal mice for measuring the infectivity of five genotype 2 isolates of Cryptosporidium parvum. Oocyst doses were enumerated by flow cytometry and delivered to animals and cell monolayers by using standardized procedures. Each dose of oocysts was inoculated into up to nine replicates of 9 to 12 mice or 6 to 10 cell culture wells. Infections were detected by hematoxylin and eosin staining in CD-1 mice, by reverse transcriptase PCR in HCT-8 and Caco-2 cells, and by immunofluorescence microscopy in Madin-Darby canine kidney (MDCK) cells. Infectivity was expressed as a logistic transformation of the proportion of animals or cell culture wells that developed infection at each dose. In most instances, the slopes of the dose-response curves were not significantly different when we compared the infectivity models for each isolate. The 50% infective doses for the different isolates varied depending on the method of calculation but were in the range from 16 to 347 oocysts for CD-1 mice and in the ranges from 27 to 106, 31 to 629, and 13 to 18 oocysts for HCT-8, Caco-2, and MDCK cells, respectively. The average standard deviations for the percentages of infectivity for all replicates of all isolates were 13.9, 11.5, 13.2, and 10.7% for CD-1 mice, HCT-8 cells, Caco-2 cells, and MDCK cells, respectively, demonstrating that the levels of variability were similar in all assays. There was a good correlation between the average infectivity for HCT-8 cells and the results for CD-1 mice across all isolates for untreated oocysts (r = 0.85, n = 25) and for oocysts exposed to ozone and UV light (r = 0.89, n = 29). This study demonstrated that in vitro cell culture was equivalent to the “gold standard,” mouse infectivity, for measuring the infectivity of C. parvum and should therefore be considered a practical and accurate alternative for assessing oocyst infectivity and inactivation. However, the high levels of variability displayed by all assays indicated that infectivity and disinfection experiments should be limited to discerning relatively large differences.
Cryptosporidium parvum is an enteric coccidian parasite (phylum Apicomplexa) that causes acute, normally self-limiting diarrhea in immunocompetent humans who become infected and chronic cryptosporidiosis, with potentially fatal consequences, in immunocompromised individuals. The environmentally resistant thick-walled oocyst stage of the organism is a common contaminant in surface waters (rivers and lakes) worldwide and is resistant to disinfection with chlorine at the concentrations typically applied in drinking water treatment plants (2 to 6 mg/liter). There have been more than 50 documented outbreaks of human cryptosporidiosis associated with drinking water (surface water and groundwater), swimming pools, water parks, and fountains. In the most notorious outbreak (Milwaukee, 1993), the estimates of the numbers of infected individuals ranged from approximately 15,000 to more than 400,000 people, and up to 100 deaths were attributed to the incident (13, 18). Correctly operating water treatment plants usually remove oocysts from source water with high efficiency. However, oocysts have been detected in 3.8 to 40% of treated drinking water samples at concentrations of up to 48 oocysts/100 liters (27). To fully assess the public health significance of C. parvum oocysts in drinking water sources, it is necessary to know whether oocysts in the environment are infectious. Also, due to the resistance of C. parvum to most chemotherapeutic agents and the chlorine-based disinfectants used in many water treatment plants, an increasingly important application of infectivity assays is to determine the anticryptosporidial efficacy of alternative drugs and disinfectants.
The three methods used to assess C. parvum infectivity are human volunteer studies, animal models, and in vitro cell culture. In one study, the 50% infective dose (ID50) of C. parvum in healthy human volunteers was 132 oocysts, and some individuals became infected with 34 ± 3 oocysts, the lowest challenge dose tested (10). In a second study, the ID50 for humans ranged from 9 to 1,042 oocysts for three different genotype 2 isolates (21). However, human infectivity assays are not practical for use on a routine basis due to the difficulty of obtaining a sufficiently large number of study subjects, ethical concerns about human testing, and the potentially serious and long-term adverse health effects for volunteers. A wide variety of animals, including hamsters, macaques, pigs, lambs, and opossums, have been used for C. parvum infectivity assays, but the most commonly used animal models are various strains of adult and suckling mice. Although infectivity in mice is generally considered to be the “gold standard” for measuring C. parvum infectivity, there are drawbacks to this approach. The use of animals in scientific research raises ethical concerns, and animal-based assays are expensive and time-consuming and have significant hidden costs, such as the maintenance of accredited facilities and license fees. Also, there is considerable variation in the C. parvum disinfection data generated with mouse infectivity models, although much of the variation may have been due to experimental design rather than a failing of the actual mouse models. A further disadvantage is the inability of mouse infectivity models, including gamma knockout mice, and other standard animal models to support infection with genotype 1 isolates of C. parvum (22, 30). Consequently, infectivity assays based on in vitro cell culture have been developed to circumvent many of the disadvantages of human and animal-based assays.
A variety of cell culture-based methods have been developed to study the infectivity of C. parvum by utilizing at least 20 different cell lines, various growth conditions, and a variety of assay formats. However, cell culture is not yet recognized as an acceptable technique because the relationship between cell culture-based infectivity and animal infectivity has not been thoroughly investigated. Although many cell culture methods have been developed for C. parvum, it has not been proven that infectivity in cell culture is a good indicator of the ability of oocysts to cause infections in animals. Also, the apparent levels of oocyst inactivation by disinfectants may be affected by the method used to measure inactivation; cell culture-based procedures may not give the same results as animal infectivity assays. Therefore, the objective of this study was to assess the correlation between cell culture-based infectivity and animal-based infectivity of C. parvum to determine whether the different methods vary in their sensitivities to infection and dose-response curves. Infection in the widely used CD-1/ICR mouse model was compared with infectivity in HCT-8, Caco-2, and Madin-Darby canine kidney (MDCK) cell monolayers by using five genotype 2 isolates and a single genotype 1 isolate.
MATERIALS AND METHODS
All animals used in this research for propagation of oocysts and infectivity assays were housed, handled, and monitored in an accredited animal care facility at the University of Arizona in accordance with the Animal Welfare Act of 1985, the Public Health Service Policy of 1986, the Good Laboratory Practice Act of 1978, and the National Academy of Sciences Guide for the Care and Use of Laboratory Animals (1996).
C. parvum oocysts.
The C. parvum isolates used in this investigation are described in Table 1. Oocysts of genotype 2 isolates (zoonotic) were propagated by artificial infection in Holstein calves. The total daily output from infected calves was collected, screened through sieves, and concentrated by centrifugation. Oocysts were purified from fecal material by sequential density gradient centrifugation through sucrose and cesium chloride gradients by using previously described methods (1). Purified oocysts were suspended in phosphate-buffered saline (PBS) containing 0.01% Tween 20, 100 U of penicillin per ml, 100 μg of streptomycin per ml, and 100 μg of gentamicin per ml and stored at 4°C until they were used for infection. The excystation rate was determined for each batch of freshly purified oocysts by microscopic observation following sequential incubation at 37°C in acidified Hanks balanced salt solution for 1 h and in 0.8% trypsin-0.75% sodium taurocholate for 1 h, followed by incubation at room temperature for 30 min. Most experiments were performed with freshly purified oocysts within 10 to 18 days after shedding. Genotype 1 C. parvum isolates (anthroponotic) were obtained from human immunodeficiency virus patients in Lima, Peru, and were supplied by R. Gilman (Johns Hopkins University). Since these oocysts could not be propagated in cows, they were purified and inoculated directly onto cell monolayers 62 days after isolation from the infected individuals. C. parvum oocysts were confirmed to be either genotype 1 or 2 by PCR targeting of a 620-bp polymorphic region of the β-tubulin gene by using previously published procedures (25). For negative infection controls, oocysts were inactivated by heating at 70°C for 30 min.
TABLE 1.
C. parvum isolates used in this investigation
| Isolate | Host | Source | Oocyst age (days)a | Excystation rate (%)b |
|---|---|---|---|---|
| Iowa | Cow | H. Moon, National Animal Disease Center, Ames, Iowa | 8-15 | 95 |
| TAMU | Horse | C. L. Chappell, University of Texas Health Science Center, Houston | 12-49 | 95 |
| Moredun | Deer | S. E. Wright, Moredun Research Institute, Peniculk, Scotland | 11-37 | 98 |
| Maine | Humanc | M. J. Arrowood, Centers for Disease Control, Atlanta, Ga. | 6 | 97 |
| Glasgow | Humand | H. V. Smith, Scottish Parasite Diagnostic Laboratory, Glasgow, Scotland | 8 | 98 |
| Peru-HIV2 | Humane | R. Gilman, Johns Hopkins University, Baltimore, Md. | 62 | 81 |
Oocyst age at the time of infection.
In vitro excystation rate determined within 10 days after shedding (except Peru-HIV2 [62 days]).
Isolated during an outbreak traced to contaminated apple cider.
Isolated during an outbreak traced to contaminated drinking water.
Isolated from a human immunodeficiency virus patient attending a clinic in Lima, Peru.
Enumeration of oocyst challenge doses.
Purified oocysts were enumerated by flow cytometry, and challenge doses were prepared and enumerated by flow cytometric sorting by using an Epics Elite flow cytometer (Coulter Corporation, Miami, Fla.) at the Wisconsin State Laboratory of Hygiene (Madison, Wis.). This ensured the validity of the dose-response curve data by providing the same standardized oocyst preparations for each of the three laboratories and each infectivity model involved in this investigation.
Cell culture infectivity assays.
Monolayers of the human ileocecal adenocarcinoma cell line HCT-8 (ATCC CCL-244; American Type Culture Collection, Rockville, Md.) were grown in 24-well culture plates (Corning Costar, Corning, N.Y.) at the Metropolitan Water District of Southern California. Cells were cultured in RPMI 1640 medium (Sigma Chemical Co., St. Louis, Mo.) supplemented with 4 mM l-glutamine, 30 mM HEPES (pH 7.3), 100 U of penicillin per ml, 100 μg of streptomycin per ml, 100 μg of kanamycin per ml, and 0.25 μg of amphotericin B per ml. Cell culture growth medium was supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, Utah) for maintenance of uninfected cells and with 2% FBS for maintenance of cells following inoculation with oocysts. Cells were incubated in a humidified incubator at 36°C in an atmosphere containing 5% CO2. Caco-2 cells (ATCC HTB 37) were grown under the same conditions as HCT-8 cells by using Eagle's minimal essential medium (Sigma) supplemented with the additives described above, 15% FBS for maintenance of uninfected cells, and 5% FBS for growth following inoculation with oocysts. Oocysts were resuspended in cell culture media, inoculated onto 80 to 100% confluent monolayers, and incubated for 48 h at 36°C.
MDCK cells (ATCC CCL 34) were grown at the Atlanta Veterans Medical Center in serum-free Ultraculture medium (Biowhitaker, Walkersville, Md.) supplemented with penicillin, streptomycin, and l-glutamine as described above in two-well culture chamber slides (Nunc, Naperville, Ill.) and incubated at 37°C in the presence of 5% CO2. Oocysts were resuspended in cell culture media, inoculated onto confluent monolayers, and incubated at 37°C for 48 h.
Detection of infection in cell cultures.
Following incubation of inoculated monolayers for 48 h, infection of HCT-8 and Caco-2 cells was detected by reverse transcriptase PCR (RT-PCR) amplification of a 361-bp fragment of C. parvum-specific heat shock protein (hsp70) mRNA by using a modification of a previously described procedure (24). All glassware used for RNA extraction reagents was baked at 180°C overnight, and all reagents were treated with 1% diethyl pyrocarbonate (DEPC) or made with DEPC-treated sterile distilled water to destroy contaminating RNA-degrading enzymes. DEPC was removed from water and reagents by autoclaving after incubation at room temperature for 8 h. Total RNA was extracted with a S.N.A.P. total RNA isolation kit (Invitrogen, Carlsbad, Calif.), which includes a DNase digestion step to eliminate DNA carryover. Reverse transcription was performed by using an oligo(dT) primer (Applied Biosystems, Foster City, Calif.) and murine leukemia virus RT (Applied Biosystems), and the PCR was performed by using Platinum Taq (Invitrogen) and C. parvum-specific primers (24) with an annealing temperature of 53°C. The amplification reaction mixtures contained dUTP instead of dTTP and uracil DNA glycosylase (Roche Molecular Biochemicals, Indianapolis, Ind.) to prevent carryover contamination of PCR products from earlier reactions. The negative amplification controls included omitting the RT from the RT reaction mixture to ensure that the amplified product originated from RNA and not from DNA and omitting the template from both RT and PCR mixtures to ensure that the amplified product was not the result of laboratory contamination. Extracted RNA was also amplified by RT-PCR by using primers specific for a 600-bp fragment of the human glyceraldehyde phosphate dehydrogenase housekeeping gene (Stratagene, La Jolla, Calif.). This amplicon should have been present in all samples at the same signal strength regardless of the oocyst dose applied to the cells. Amplicons were visualized by agarose gel electrophoresis and ethidium bromide staining.
Infections in MDCK cells were detected by using an immunofluorescence assay (IFA). Slide chambers were removed at 48 h postinfection, and monolayers were washed with PBS, fixed in Bouin's solution, and decolorized with 70% ethanol. Monolayers were washed with PBS and stained with two C. parvum-specific monoclonal antibodies. The first monoclonal antibody, 3C3C, labeled all intracellular life cycle stages and was conjugated to Cy3 (2). The second antibody, OW50, was conjugated to fluorescein isothiocyanate and labeled oocysts, which allowed detection of adherent oocysts. Antibody-labeled culture chambers were examined by epifluorescence microscopy at a magnification of ×400 by using an Olympus BX50 microscope (Olympus America Inc., Lake Success, N.Y.). The entire chamber area was examined for the presence of intracellular parasite stages.
Oocyst treatment prior to cell culture inoculation.
For some infections in cell cultures, oocysts were incubated in 0.75% sodium taurocholate (prepared in RPMI medium) at 37°C for 10 min to trigger excystation and were washed with PBS prior to inoculation of monolayers.
Mouse infectivity assay.
Dams and neonatal CD-1/ICR mice (5 to 7 days old) were obtained from Charles River Laboratories, Inc. (Wilmington, Mass.), and neonates were inoculated by delivering a 10-ml aliquot of each dose of oocysts, suspended in sterile water, to the back of the throat with a calibrated pipette. Seven days after inoculation, infections were detected by examining formalin-fixed, slide-mounted, hematoxylin- and eosin-stained sections (5 μm by 2 to 3 cm) of the terminal ileum that were removed from each animal at necropsy (14). The ileum was chosen because it is the first region of the gut to be colonized by the parasite and harbors the majority of developmental stages (7). Evidence of infection was defined as the presence of C. parvum parasite developmental stages in the microvilli of any of the prepared histological sections.
Oocyst exposure to disinfectants.
Oocysts were exposed to medium-pressure UV light by using a continuous-wave 1-kW UV lamp mounted above a collimating tube (inside diameter, 6.4 cm). Oocysts of the Moredun and TAMU isolates were suspended in PBS at a concentration of 1 × 105 oocysts/ml and were exposed to UV doses ranging from 2.2 to 8 mJ/cm2. Details of the UV equipment and the procedures used to measure UV fluence have been described previously (19). Oocysts of the Moredun and Iowa isolates were also exposed to ozone by using a simple 200-ml batch reactor. Ozone was delivered into a solution of 0.05 M phosphate buffer (pH 7.0) with a V5-0 ozone generator (Osmonics, Minnetonka, Minn.) to obtain a dissolved ozone concentration of approximately 10 mg/liter. Aliquots of this stock solution were then added to 0.05 M phosphate buffer containing C. parvum oocysts to obtain ozone concentration × time (CT) values of 1.25 to 16 mg·min/liter. Residual ozone was measured by an indigo colorimetric method (5), and following incubation, residual ozone was quenched with sodium thiosulfate. Following exposure to UV or ozone, oocysts were diluted in PBS and inoculated into CD-1 mice and HCT-8 cell monolayers to measure the remaining infectivity.
Data analysis.
To ensure that the results obtained with the different infectivity models were directly comparable, the same general procedure was used to quantitate the level of infection in all models. For animal infectivity experiments, each dose of oocysts was inoculated into the mice in a maximum of six cages (10 to 12 mice per cage), and infectivity was expressed as the proportion of animals that became infected at each dose. Similarly, for cell cultures, each dose of oocysts was inoculated into a maximum of seven replicate sets of cell monolayers (6 to 10 cell culture wells per replicate). The proportion of cell culture wells or animals that became infected at each dose was transformed by using a response logit to ensure linearity of the dose-response curves (11):
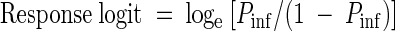 |
where Pinf is the proportion of mice or cell culture wells infected at each dose of oocysts. The logit function does not allow logarithmic transformations of 0 and 100% responses, so any cage of animals or set of monolayers that exhibited 0 or 100% infection (Pinf = 0 or 1) was excluded from the logit response model. The logit dose-response method is an accepted analytical tool for expressing parasite infectivity and has been used previously for interlaboratory comparisons of C. parvum infectivity in mice (14). Solving the least-squares regression of log10 oocyst dose against the response logit for a logit value of 0 generated ID50 values. Solving for the logit following disinfection of oocysts and comparison with a dose-response curve generated by nonexposed oocysts allowed calculation of the levels of inactivation.
The data sets were analyzed for normality by using the Shapiro-Wilk test for sample sizes of less than 2,000. For the 70 data sets analyzed (five isolates in mouse and cell culture infectivity models), the data were normally distributed (P > 0.05) in 87% of the cases. Since the majority of the data were normally distributed, a parametric two-tailed t test was used to compare infectivities in different models. A two-tailed test was used because there was no a priori expectation that one method would generate consistently higher levels of infection than any other method. A parametric chi-square test was used to determine whether there were significant differences in the percentages of infectivity resulting from different doses of each isolate. Dose-response relationships were compared by using one-way analysis of covariance (ANCOVA) with percentages of infectivity to determine whether there were significant differences among the different infectivity models for each isolate. The means and standard deviations of replicate values for percentages of infectivity for each infection detection method, C. parvum isolate, and oocyst dose were used as input for a standard power analysis (α = 0.05, β = 0.80) to determine the number of replicates of mice or cell culture wells needed to allow detection of specific differences in percentages of infectivity.
RESULTS
Comparison of infectivity assays performed with fresh oocysts of genotype 2 isolates.
Overall, the results obtained with fresh oocysts from five genotype 2 strains of C. parvum demonstrated that there was good agreement between the mouse and cell culture infectivity assays in terms of the shape of dose-response curve (Fig. 1), the range of ID50s (Table 2), and the level of variability. Chi-square contingency analyses demonstrated that there were significant differences in the infectivities of different oocyst doses for all isolates and all infectivity models (P < 0.01) except for the Moredun and Glasgow isolates with MDCK cells with infection detected by IFA (P = 0.34). The data indicated that in the latter experiments, increasing the oocyst dose did not lead to a significant increase in the level of infectivity. This is illustrated by the data obtained for the Moredun isolate with MDCK cells (Fig. 1D), which showed that the level of infectivity was 75% with 50 oocysts and increased to just 85% with 500 oocysts. For all other isolates and infectivity models, an expected dose-response relationship was observed (i.e., an increase in the oocyst dose led to an increase in the amount of infection). The lack of a significant dose response in the MDCK IFA for the Moredun and Glasgow isolates was also reflected by the very low ID50s (1.2 and 1.5 oocysts) (Table 2) and the significant differences in the slopes of the dose-response regressions (ANCOVA, P < 0.05) compared to the results obtained with other infectivity models for these two isolates. With these two exceptions, ANCOVA of the data used to generate the dose-response curves for the Iowa, Moredun, Maine, and Glasgow isolates demonstrated that the slopes of the regression lines were not significantly different (range of P values, 0.11 to 0.98). Two-tailed t tests of replicate percentages of infectivity for each dose of oocysts of each isolate (a total of 26 comparisons) demonstrated that HCT-8 cell cultures yielded significantly higher levels of infection than CD-1 mice in four instances (range of P values, <0.01 to 0.04). Conversely, CD-1 mice exhibited higher levels of infection in six instances (range of P values, <0.01 to 0.03). The differences were distributed evenly over all isolates. There was no significant difference between HCT-8 cells and CD-1 mice (range of P values, 0.06 to 0.91) for the remaining 16 comparisons.
FIG. 1.

Dose-response curves for five genotype 2 isolates of C. parvum in CD-1 mice (A), HCT-8 cell monolayers (B), Caco-2 cell monolayers (C), and MDCK cell monolayers (D). The ID50 is the point at which the regression line crosses the logit response 0 axis. Infectivity was measured for the Iowa (○, ——), Moredun (□, - - — - -), TAMU (▵, — — —), Maine (⋄, - - - - -), and Glasgow (∗, — - —) isolates. Each data point is the logit of the mean proportional infectivity (Pinf) for three to six replicate cages of mice (9 to 12 mice per cage) or for two to seven replicate sets of cell cultures (6 to 10 monolayers per set) at each dose of oocysts.
TABLE 2.
Comparison of ID50s for five genotype 2 isolates of C. parvum in mouse and cell culture-based infectivity models
| Isolate | Oocyst doses | CD-1 mice
|
HCT-8 cell RT-PCRa
|
Caco-2 cell RT-PCRa
|
MDCK cell IFAb
|
||||
|---|---|---|---|---|---|---|---|---|---|
| ID50 (oocysts)c | 95% confidence interval (oocysts)d | ID50 (oocysts) | 95% confidence interval (oocysts) | ID50 (oocysts) | 95% confidence interval (oocysts) | ID50 (oocysts) | 95% confidence interval (oocysts) | ||
| Iowa | 50-1,000 | 347 (220) | 128-942 (18) | 106 (77) | 52-154 (13) | 629 (327) | 237-1,669 (12) | 0.5 (24)e | 0.02-13 (14) |
| TAMU | 5-25 | 23 (25) | 17-32 (15) | 27 (34) | 21-34 (11) | 31 (58) | 17-33 (11) | 18 (17) | 6-53 (11) |
| Moredun | 25-500 | 16 (28) | 9-25 (25) | 30 (26) | 18-38 (21) | 45 (40) | 28-70 (22) | 1.2 (5) | 0.6-2 (16) |
| Maine | 25-250 | 21 (22) | 10-40 (21) | 86 (84) | 66-114 (22) | 118 (104) | 89-156 (20) | 13 (20) | 2-78 (11) |
| Glasgow | 25-250 | 21 (27) | 13-34 (20) | 81 (81) | 60-110 (22) | NDf | ND | 1.5 (3) | 0.4-7 (11) |
Infection was detected by RT-PCR.
Infection was detected by immunofluorescence microscopy.
ID50s were calculated by solving the logistic regression of each dose-response curve for a logit response of 0, using individual percentages of infectivity for each replicate dose of oocysts or by first averaging the percentages of infectivity for all replicates and then performing the logit transformation (values in parentheses).
The 95% confidence intervals of the ID50s were calculated from the logistic regression by using individual percentages of infectivity for each replicate dose of oocysts. The numbers in parentheses are the numbers of cages of mice or sets of cell culture wells used to calculate the ID50 and the 95% confidence intervals from the logistic regression by using individual percentages of infectivity for each replicate dose of oocysts. The total numbers of animals or wells of cell cultures used to generate the values can be approximated by multiplying these numbers by 10.
Iowa oocysts were incubated in 0.75% sodium taurocholate prior to inoculation of MDCK cell monolayers.
ND, not done.
The dose-response curves in Fig. 1 were generated by performing the logit transformation on the average percentage of infectivity for all replicates at each oocyst dose, allowing 0 and 100% infectivity to be included in the calculation of averages as long as at least one of the replicates did not yield a value of 0 or 100%. However, dose-response curves could also be generated by using the logit transformation of each individual percentage of infectivity at each oocyst dose without averaging (Fig. 2). This approach was adopted for calculating the primary ID50s in Table 2 and the 95% confidence interval of the ID50 for all isolates. Since all 0 and 100% infectivity data points were excluded when we used information from each individual replicate without averaging, the ID50s varied from the values calculated by using the averaged infectivity data (Table 2). There was considerable variation between replicates in the level of infection detected at each oocyst dose by all infectivity models. An example of this variability is shown in Fig. 2 for the Iowa and Moredun isolates assayed by using HCT-8 cell cultures combined with RT-PCR. Nonaveraged infectivity data are not presented graphically for all isolates and all methods because the graphs would be difficult to interpret due to overlapping symbols. However, ID50s and 95% confidence intervals were calculated for all isolates and assay methods by using the nonaveraged approach (Table 2). Similar levels of variability were displayed for all assays; the average standard deviations for percentages of infectivity for all replicates of all isolates were 13.9, 11.5, 13.2, and 10.7% for CD-1 mice, HCT-8 cells, Caco-2 cells, and MDCK cells, respectively. The ranges of standard deviations for percentages of infectivity between replicates for all isolates were 3.7 to 32.6% for CD-1 mice and 0 to 34.1% for the three cell culture assays combined. With an average standard deviation for all infectivity methods of 11.7%, standard power analysis demonstrated that with six replicates consisting of 10 mice or 10 cell monolayers each (equivalent to a total of 60 mice or wells of cell cultures) at each oocyst dose, the smallest detectable difference in percentages of infectivity was 16%. An example of the variability in infectivity measured in HCT-8 cell cultures between replicates at each oocyst dose is shown for two isolates (Iowa and Moredun) in Fig. 2. This figure also demonstrates that plotting individual logit values, rather than logit transformations of the average percentage of infectivity at each dose, resulted in different ID50s.
FIG. 2.

Infectivities of the Iowa (○) (R2 = 0.57) and Moredun (□) (R2 = 0.62) isolates of C. parvum in HCT-8 cell monolayers based on individual logit calculations for each oocyst challenge dose. The ID50s based on these dose-response curves were 106 oocysts for the Iowa isolate and 30 oocysts for the Moredun isolate.
When just the smaller oocyst challenge doses (5 to 25 oocysts) of the TAMU isolate were used (Fig. 3), the ID50s ranged from 18 oocysts for MDCK cells to 31 oocysts for Caco-2 cells (Table 2), and there was not a significant difference between the dose-response curves for HCT-8 cells and CD-1 mice (P = 0.18). However, using the entire TAMU isolate data set obtained with oocyst challenge doses of 5 to 250 oocysts to generate dose-response curves (Fig. 1) resulted in ID50s of 19 oocysts for CD-1 mice (n = 25; 95% confidence interval, 15 to 23 oocysts), 67 oocysts for HCT-8 cells (n = 27; 95% confidence interval, 18 to 234 oocysts), and 73 oocysts for Caco-2 cells (n = 27; 95% confidence interval, 25 to 216 oocysts), and there were significant differences between the slopes of the dose-response curves for HCT-8 cells and CD-1 mice (P < 0.01).
FIG. 3.

Infectivity of the TAMU isolate as measured by CD-1 mouse (□), HCT-8 cell culture RT-PCR (○), Caco-2 cell culture RT-PCR (⋄), and MDCK cell culture IFA (▵) assays based on logit calculations from average percentages of infectivity with oocyst doses of 5 to 25 oocysts.
When data for all isolates were combined, the cell culture system that generated infectivity results most similar to the CD-1 mouse assay results was the RT-PCR procedure with HCT-8 cells (r = 0.88) (Fig. 4). The IFA method with MDCK cells resulted in generally higher levels of infection than the levels in CD-1 mice, while the RT-PCR assay with Caco-2 cells produced lower levels of infection. Although the relatively large variability in the data resulted in few significant differences between infectivity models, when differences did occur, they consistently demonstrated that the levels of infection of Caco-2 cells were lower than the levels of infection of either of the other two cell lines or CD-1 mice. A direct comparison between HCT-8 and MDCK cells by using the same method to detect infections (RT-PCR) was performed only with the Glasgow isolate. The ID50 for HCT-8 cells was 81 oocysts (n = 22; 95% confidence interval, 60 to 110 oocysts), compared to 49 oocysts (n = 27; 95% confidence interval, 35 to 67 oocysts) for MDCK cells. Although the average ID50 determined by RT-PCR was lower for MDCK cells than for HCT-8 cells, the 95% confidence intervals of the ID50s overlapped, and there was no significant difference between the dose-response curves based on an ANCOVA (P = 0.48).
FIG. 4.

Overall comparison of infectivity measurements obtained with mice and cell cultures for untreated fresh oocysts of all isolates combined. Symbols: ○, CD-1 mice versus HCT-8 cells (R2 = 0.77); □, CD-1 mice versus Caco-2 cells (R2 = 0.67); ▵, CD-1 mice versus MDCK cells (R2 = 0.67). Each data point represents the average for all replicate analyses performed at each dose for the various isolates. All lines are best-fit linear regressions.
Incubation of oocysts in 0.75% sodium taurocholate prior to inoculation of cell monolayers resulted in increased infectivity in cell cultures. Direct comparisons of infectivity with the Iowa isolate were made by using untreated and sodium taurocholate-treated oocysts and both HCT-8 and Caco-2 cell monolayers. In contrast to the ID50s of untreated oocysts (106 and 629 oocysts for HCT-8 and Caco-2 cells, respectively) (Table 2), the ID50s for taurocholate-treated oocysts were both <1 oocyst. A direct comparison was not made for MDCK cells, but the ID50 of sodium taurocholate-treated oocysts of the Iowa isolate was also <1 oocyst as determined by the IFA with MDCK cells (Table 2).
Comparison of infectivity models when oocysts were exposed to disinfectants.
For the purposes of this comparative study, oocyst inactivation was evaluated by using the CD-1 mouse and HCT-8 cell culture RT-PCR assays only. There was a significant correlation (r = 0.89, n = 29) between infectivity in HCT-8 cells and infectivity in CD-1 mice for oocysts that had been exposed to UV doses ranging from 2 to 8 mJ/cm2 and ozone CTs up to 16 mg·min/liter (Fig. 5). The mean level of inactivation obtained with 4 mJ of UV light per cm2 was 97.6% ± 1.1% (equivalent to 1.6 ± 0.2 log10) as measured by the HCT-8 cell culture method, compared to 96% ± 2.1% (1.4 ± 0.3 log10) when the level was measured by the CD-1 mouse infectivity assay. The amount of inactivation obtained with an ozone CT of 16 mg·min/liter in a simple batch reactor was 99.4% (2.2 log10) when it was measured by the HCT-8 RT-PCR assay, compared to 99.7% (2.5 log10) when it was measured by the CD-1 mouse assay. The average standard deviations for replicate measurements of infectivity in HCT-8 cells and CD-1 mice were 11.9 and 13.3%, respectively, for oocysts exposed to disinfectants. This shows good concordance with the average standard deviations for HCT-8 cell culture and mouse data (11.5 and 13.9%) for all of the data shown in Fig. 1.
FIG. 5.

Comparison between infectivity in CD-1 mice and infectivity in the HCT-8 cell culture RT-PCR assay (R2 = 0.80) (solid regression line) for oocysts of the Iowa, TAMU, and Moredun isolates exposed to various doses of UV (•) and ozone (○). The horizontal and vertical error bars indicate the average standard deviations for all replicate analyses with oocysts exposed to disinfectants for CD-1 mice and HCT-8 cells, respectively. The dashed line indicates the theoretical 1:1 correlation. The same oocyst challenge doses were inoculated into CD-1 mice and HCT-8 monolayers for each dose of disinfectant.
Comparison of infectivity models with genotype 1 isolates.
Although the focus of this study was to compare infectivity assays with multiple genotype 2 isolates, a limited amount of work was also done with genotype 1 oocysts. In agreement with previous findings (22), neither isolate of genotype 1 C. parvum used in this investigation was able to infect either the calves used for oocyst propagation or CD-1 mice. Dose-response curves obtained with flow cytometry-sorted genotype 1 oocyst challenge doses of 100 to 1,000 oocysts of two isolates generated an average ID50 of 631 oocysts for HCT-8 cell monolayers (results not shown). However, these data cannot be directly compared with the ID50s for the genotype 2 isolates because of differences in oocyst age and storage conditions. Most of the data for the dose-response curves for the genotype 2 isolates were generated by using oocysts that were stored in saline antibiotic solution following purification and were less than 18 days old at the time of infection. In contrast, the genotype 1 isolates collected from human immunodeficiency virus patients were stored in potassium dichromate, were transported across continents, and were more than 60 days old at the time of infection.
DISCUSSION
There are a variety of in vitro surrogate methods for determining the viability of C. parvum oocysts, but it has been demonstrated that these methods do not always correlate with infectivity assays. A previous comparison of the mouse infectivity assay with four in vitro surrogate viability assays (excystation and staining with 4′,6′-diamidino-2-phenylindole-propidium iodide, SYTO-9, and SYTO-59) demonstrated that the in vitro surrogate methods consistently overestimated oocyst viability and could not be used to accurately assess oocyst inactivation following disinfection (3). The authors concluded that neonatal mouse infectivity should be considered the gold standard for evaluating oocyst disinfection. In addition, it has been suggested that in vitro viability assays should not be used to determine oocyst inactivation by ozone or UV (4). Consequently, assays that can determine the ability of oocysts to cause infection, rather than simply detect some aspect of metabolism or the presence of particular molecules, are essential. In vitro cell culture has been proposed as a potential alternative to animal assays, but rigorous comparisons have not been made. This is the first large-scale, statistically robust comparison of in vitro cell culture with a mouse assay for measuring infectivity of multiple isolates of C. parvum. The objective of this study was not to maximize the sensitivity of infection in cell culture and then compare the optimized assay with a mouse assay. Instead, the intent was to directly compare the shapes of the dose-response curves and the levels of variability in cell culture assays and mice by using oocysts that had been handled and treated in the same way for both types of assay. Although no single cell culture assay matched the CD-1 mouse assay for all C. parvum isolates, the results obtained with fresh, untreated oocysts demonstrated that the use of in vitro cell cultures was generally equivalent to the standard CD-1 mouse model in terms of sensitivity, dose-response relationships, and variability among replicates. The sensitivity of infection in a cell culture can be increased by incubating oocysts in 0.75% sodium taurocholate prior to inoculation of cell monolayers, and in the present study this reduced the ID50 by a factor of approximately 100. In addition, pretreatment with 10% bleach (0.53% sodium hypochlorite) is a routine procedure for simultaneous oocyst surface sterilization and maximizing the sensitivity of infection in cell cultures (29). IFA combined with a most-probable-number analysis generated an ID50 of approximately 10 oocysts for 10% bleach-pretreated oocysts with HCT-8 cells (28), although oocyst concentrations were determined by serial dilution of an enumerated stock, thereby introducing a potential source of error into the ID50 determination. However, no studies have been performed to determine whether such oocyst treatments have an effect, synergistic or antagonistic, when infectivity studies are used to measure oocyst inactivation. Consequently, the overall comparisons between cell culture and mouse assays in this investigation were performed by using oocysts that were introduced directly into each model (cell cultures or mice) without sodium taurocholate or bleach pretreatment. Infectivity assays with maximized sensitivity will be important for determining whether low numbers of oocysts in environmental water samples are capable of causing infection. However, the most important characteristics for assays that will be used to measure the efficacy of disinfectants are (i) the use of oocysts that have not been exposed to any potential stress or agent other than the disinfectant being evaluated and (ii) a well-established dose-response relationship.
Some reports have indicated that infectivity in mice can be maximized by incubating oocysts in sodium hypochlorite prior to inoculation. In one study, bleach-treated oocysts were inoculated into suckling mice, which resulted in 73% infectivity with one oocyst and 100% infectivity with 25 oocysts when infections were detected by flow cytometry of intestinal homogenates (8). However, the authors reported that oocyst doses were prepared by serial dilution of a hemacytometer-enumerated stock, and the difficulty of reliably obtaining low oocyst doses by serial dilution suggests that this reported sensitivity may not be accurate.
The ID50 of the Iowa isolate for CD-1 mice was 347 oocysts when it was calculated by using individual replicate data and 220 oocysts when replicate percentages of infectivity were averaged prior to logit calculations (Table 2). These values are considerably higher than those typically obtained by us for this isolate and strain of mouse. The ID50 of the Iowa isolate for CD-1 mice has been reported to be 60 to 87 oocysts (11, 14). The average ID50 of this isolate for CD-1 mice, based on 22 separate analyses over a 3-year period with oocysts obtained from 10 calves and a total of approximately 1,700 mice, was 100 oocysts, and the 95% confidence interval was 84 to 119 oocysts (15). However, the values obtained in the present investigation, 347 and 220 oocysts, fall within the 80% prediction limits (26 to 390 oocysts) of the earlier study and should therefore be considered valid values for this lot of oocysts in this experiment. This variation in ID50s reflects the variability inherent in infectivity assays even when the gold standard animal model is used. Various other mouse models have been used to measure the infectivity of C. parvum, with ID50s ranging from 60 to 1,000 oocysts (17).
The ID50s of the five genotype isolates in this study ranged from 13 to 629 oocysts for the various cell and mouse models. However, considering the variability displayed by all infectivity assays, it is not clear whether these figures represent genuine differences in the levels of infectivity between different isolates or if they simply reflect a combination of sources of assay variation. The RT-PCR assay with HCT-8 cells generated the narrowest range of ID50s for all isolates, with a 3.9-fold difference between the highest ID50 (Iowa) and the lowest ID50 (TAMU), compared to 12-, 21-, and 26-fold differences for the MDCK IFA, CD-1 mouse, and Caco-2 RT-PCR assays, respectively. The TAMU isolate appeared to be more infectious than the Iowa isolate in CD-1 mouse, HCT-8 cell culture, and Caco-2 cell culture assays, with an average ID50 for all assays of 25 oocysts, compared to 355 oocysts for the Iowa isolate, a 14.2-fold difference. Although studies have been limited by difficulties in obtaining sufficient suitable volunteers, C. parvum infection has also been studied in humans. The ID50 of the Iowa isolate in human volunteers was 132 oocysts (10), compared to 9 oocysts for the TAMU isolate (21), a 14.6-fold difference. These comparisons in humans, mice, and cell cultures indicate that the TAMU isolate is more infectious than the Iowa isolate.
Infectivity assays are inherently variable due to the many factors involved in working with multiple living systems (oocysts, propagation animal, cell line or animal infection host). The possible sources of variation for C. parvum infectivity assays include: (i) interisolate variation due to genuine biological differences; (ii) intraisolate variation due to differences in oocyst production and handling procedures; (iii) differences in the health and general condition of oocyst propagation animals; and (iv) interexperimental variation in the susceptibility of the host (either cell monolayers or mice) to infection. Natural lot-to-lot variability of the Iowa isolate was demonstrated by a review of 22 dose-response studies with CD-1 mice over a 3-year period, in which the ID50s ranged from 33 to 476 oocysts (15). Due to the large variability between replicates in the present investigation, standard power analysis demonstrated that 16% was the minimum difference in infectivity of fresh, untreated oocysts that could be detected (with an average standard deviation of 11.7%) when we used six replicate cages containing 10 mice each or six replicate sets consisting of 10 cell monolayers each. Based on the slopes of the regression lines in Fig. 1 for the Iowa isolate as an example, an infectivity difference of 16% equals an average difference in oocyst challenge dose of 0.27 log10 for all methods. This means that a significant difference could be expected when the level of infection produced by 100 oocysts is compared to the level of infection produced by 50 oocysts (difference, 0.3 log10) but not when the levels of infection for 100 and 60 oocysts are compared (difference, 0.22 log10). Therefore, this analysis indicated that C. parvum disinfection experiments performed with either mice or cell culture infectivity models should be limited to discriminating relatively large differences between treatments and should not try to resolve fine differences in the levels of inactivation between small treatment increments.
At least 20 cell lines have been shown to support infection by C. parvum, and a variety of techniques have been used to detect infection in cell culture. Caco-2 cells spontaneously differentiate once they reach confluency, forming microvilli and producing high levels of brush border enzymes (23). Such differentiated cells may represent a more realistic model of the human intestine than nondifferentiating cell lines and may therefore be more appropriate for in vitro infectivity assays. Nevertheless, this study demonstrated that the levels of infection in Caco-2 cell monolayers (average ID50 for all isolates, 216 oocysts) were consistently lower than the levels of infection in HCT-8 cell monolayers (average ID50, 70 oocysts), confirming the results of earlier comparisons (29). Although this study demonstrated that cell culture was equivalent to a standard mouse assay for measuring C. parvum infectivity, it may be necessary to further optimize in vitro cell culture methods to better mimic the human intestine. Almost all of the cell culture assays for C. parvum developed to date have utilized monolayers of host cells grown on microscope slides, coverslips, or permeable membranes or in multiwell plates. However, it has been reported that three-dimensional aggregates of human cells grown in suspension in a low-shear, low-turbulence rotating-wall vessel more accurately model human differentiated tissue than monolayers for studying infection with Salmonella enterica (20). Such a system may be useful for studying C. parvum infection in vitro.
This study was not designed to compare methods for detecting infection in cell culture since the RT-PCR approach with HCT-8 and Caco-2 cells was used in one laboratory (Metropolitan Water District of Southern California) while the IFA technique with MDCK cells was used at a second location (Atlanta Veterans Medical Center). Our study did confirm that the RT-PCR approach targeting C. parvum-specific hsp70 mRNA is a robust and accurate method for detecting infection in cell culture, as demonstrated by its recent use to measure UV inactivation of C. parvum (19). A similar approach using PCR targeting hsp70 DNA was used to demonstrate the occurrence of infectious C. parvum oocysts in 4.9% of untreated water samples (9). However, differences in the present study between the RT-PCR and IFA approaches for detecting infection indicate the necessity of a thorough comparison of detection methods. In addition to the RT-PCR approach described in this paper and adapted from an earlier method (24), previously reported techniques for detecting C. parvum infection in cell culture include a microtiter enzyme-linked immunosorbent assay (31), a chemiluminescence immunoassay (32), IFA (10a, 28), a PCR targeting C. parvum-specific DNA (9), and colorimetric in situ hybridization (26).
Measuring inactivation of C. parvum oocysts by either UV or ozone for treatment plant design purposes was not an objective of this study. Rather, one of the objectives was to compare cell culture and mouse infectivity assays performed with oocysts that had been exposed to disinfectants. Nevertheless, the average levels of inactivation demonstrated by the infectivity assays in this study, 97.6% (1.6 log10) and 96% (1.4 log10) for HCT-8 cells and CD-1 mice, respectively, for oocysts exposed to 4 mJ of medium-pressure UV light per cm2, are in good agreement with the results of previous studies of UV disinfection. An earlier and more extensive evaluation of the efficacy of UV light as a disinfectant, in which the workers used HCT-8 cell cultures and an RT-PCR detection assay similar to the assay used in the present investigation, demonstrated an average level of inactivation of 94% (1.2 log10) with a polychromatic UV dose of 4 mJ/cm2 (19).
Although this study demonstrated that cell culture was equivalent to mouse assays for measuring infectivity and inactivation of C. parvum oocysts, a disadvantage of cell culture is the inability to propagate oocysts in vitro. Consequently, in vivo infection of animals is still necessary for production of the large numbers of oocysts required for experimental infections. Complete life cycle development of C. parvum in cell culture and long-term (25-day) maintenance in HCT-8 cells have been reported (6, 12), but the number of de novo thick-walled oocysts recovered from infected cell cultures never exceeded the size of the inoculum. Consequently, each serial passage of oocysts in cell culture results in lower numbers of fresh parasites. The failure of C. parvum to propagate in cell culture is not yet understood but may be related to nutritional deficiencies in the culture media, inappropriate redox conditions, or death of the host cells following infection. Culture systems that allow three-dimensional cell aggregates to develop, such as the rotating-wall vessel described for S. enterica (20), may be useful for achieving in vitro oocyst propagation. It has been reported that a nonadherent cell line grown in suspension was fully susceptible to C. parvum infection with a continuous asexual life cycle lasting for at least 15 days (16). A disadvantage of standard animal models is their inability to support infection with genotype 1 C. parvum. In contrast, both primary genotypes infect cell cultures, as demonstrated in this and previous studies (26). Transmission and serial propagation of a genotype 1 isolate have recently been demonstrated in gnotobiotic piglets (30), but infection in standard mouse models, immune knockout mice, and propagation animals, such as calves, has not been reported.
In summary, this study demonstrated that cell culture-based infectivity was generally equivalent to the infectivity in a standard mouse assay for measuring the infectivity of C. parvum oocysts whether the shape of the dose-response curves, the sensitivity to infection, the range of ID50s, or the level of variability was considered. Therefore, in vitro cell culture-based assays should be regarded as practical alternatives to the use of animals for measuring the infectivity of C. parvum and for assessing the efficacy of some disinfectants. The large variability displayed by both the mouse and cell culture data indicated that infectivity and disinfection experiments should be limited to discerning relatively large differences in the levels of infection or inactivation. Future work should include a similarly rigorous comparison of cell culture and mouse models for measuring oocyst inactivation by a variety of disinfectants, an evaluation of infectivity in cell culture compared to infectivity in human volunteers, and a thorough comparison of the different methods available for detecting and quantitating infections in cell culture.
Acknowledgments
The authors thank the American Water Works Association Research Foundation and the U.S. Government, through the Environmental Protection Agency, for their financial, technical, and administrative assistance in funding and managing the project through which this information was discovered.
The comments and views detailed here may not necessarily reflect the views of the American Water Works Association Research Foundation, its officers, directors, affiliates, or agents or the views of the U.S. Government.
We thank Michael Arrowood (Centers for Disease Control, Atlanta, Ga.), Cynthia Chappell (University of Texas Health Science Center, Houston), Robert Gilman (Johns Hopkins University, Baltimore, Md.), Huw Smith (Scottish Parasite Diagnostic Laboratory, Glasgow, Scotland), and Steve Wright (Moredun Research Institute, Peniculk, Scotland) for providing isolates of C. parvum for this study. We also thank David Polchert and Rebecca Hoffman (Wisconsin State Laboratory of Hygiene) for flow cytometry services, Alex Mofidi and Mark Williams (Metropolitan Water District of Southern California) for assistance with disinfection experiments, and Jose Sobrinho for assistance with statistical analyses. The technical assistance of Katrin Hanley, Leigh Jacobs, Walter Naro, Julie Stewart, Emily Sullivan, and Jennifer Letts is appreciated.
REFERENCES
- 1.Arrowood, M. J., and K. Donaldson. 1996. Improved purification methods for calf-derived Cryptosporidium parvum oocysts using discontinuous sucrose and cesium gradients. J. Eukaryot. Microbiol. 43:89. [DOI] [PubMed] [Google Scholar]
- 2.Arrowood, M. J. 1988. Ph.D. thesis. University of Arizona, Tucson.
- 3.Bukhari, Z., M. M. Marshall, D. G. Korich, C. R. Fricker, H. V. Smith, J. Rosen, and J. L. Clancy. 2000. Comparison of Cryptosporidium parvum viability and infectivity assays following ozone treatment of oocysts. Appl. Environ. Microbiol. 66:2972-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clancy, J. L., Z. Bukhari, R. M. McCuin, T. P. Clancy, M. M. Marshall, D. G. Korich, C. R. Fricker, N. Sykes, H. V. Smith, J. O'Grady, J. P. Rosen, J. Sobrinho, and F. W. Schaefer III. 2000. Cryptosporidium viability and infectivity methods. American Water Works Association Research Foundation, Denver, Colo.
- 5.Clesceri, L. S., A. E. Greenberg, and A. D. Eaton. 1998. Method 4500-O3, p. 4-137-4-139. In Standard methods for the examination of water and wastewater. American Public Health Association, Washington, D.C.
- 6.Current, W. L., and T. B. Haynes. 1984. Complete development of Cryptosporidium in cell culture. Science 224:603-605. [DOI] [PubMed] [Google Scholar]
- 7.Current, W. L., and N. C. Reese. 1986. A comparison of endogenous development of three isolates of Cryptosporidium in suckling mice. J. Protozool. 33:98-108. [DOI] [PubMed] [Google Scholar]
- 8.DeLaunay, A., G. Gargala, X. Li, L. Favennec, and J. J. Ballet. 2000. Quantitative flow cytometric detection of maximal Cryptosporidium parvum oocyst infectivity in a neonatal mouse model. Appl. Environ. Microbiol. 66:4315-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiGiovanni, G. D., F. H. Hashemi, N. J. Shaw, F. A. Abrams, M. W. LeChevallier, and M. Abbaszadegan. 1999. Detection of infectious Cryptosporidium parvum oocysts in surface and filter backwash water samples by immunomagnetic separation and integrated cell culture PCR. Appl. Environ. Microbiol. 65:3427-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DuPont, H. L., C. L. Chappell, C. R. Sterling, P. C. Okhuysen, J. B. Rose, and W. Jakubowski. 1995. The infectivity of Cryptosporidium parvum in healthy volunteers. N. Engl. J. Med. 332:855-859. [DOI] [PubMed] [Google Scholar]
- 10a.Favennec, L., M. Ergaz-Bernard, E. Comby, D. Lemeteil, J. J. Ballet, and P. Brasseur. 1994. Immunofluorescence detection of Cryptosporidium parvum in Caco-2 cells: a new screening method for anticryptosporidial agents. J. Eukaryot. Microbiol. 41:39S. [PubMed] [Google Scholar]
- 11.Finch, G. R., C. W. Daniels, E. K. Black, F. W. Schaefer III, and M. Belosevic. 1993. Dose response of Cryptosporidium parvum in outbred, neonatal CD-1 mice. Appl. Environ. Microbiol. 59:3661-3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hijjawi, N. S., B. P. Meloni, U. M. Morgan, and R. C. A. Thompson. 2001. Complete development and long-term maintenance of Cryptosporidium parvum human and cattle genotypes in cell culture. Int. J. Parasitol. 31:1048-1055. [DOI] [PubMed] [Google Scholar]
- 13.Hunter, P. R., and Q. Syed. 2001. Community surveys of self-reported diarrhoea can dramatically overestimate the size of outbreaks of waterborne cryptosporidiosis. Water Sci. Technol. 43:27-30. [PubMed] [Google Scholar]
- 14.Korich, D. G., M. M. Marshall, H. V. Smith, J. O'Grady, Z. Bukhari, C. R. Fricker, J. P. Rosen, and J. L. Clancy. 2000. Inter-laboratory comparison of the CD-1 neonatal mouse logistic dose-response model for Cryptosporidium parvum oocysts. J. Eukaryot. Microbiol. 47:294-298. [DOI] [PubMed] [Google Scholar]
- 15.Korich, D. G., M. M. Marshall, and C. R. Sterling. 2000. Lot-to-lot variation of Cryptosporidium parvum oocyst viability as assessed by the neonatal mouse infectivity assay. In Proceedings of the American Water Works Association Water Quality Technology Conference. American Water Works Association, Denver, Colo.
- 16.Lawton, P., M. Naciri, R. Mancassola, and A.-F. Petavy. 1996. Cultivation of Cryptosporidium in a non-adherent human monocytic cell line. J. Microbiol. Methods 27:165-173. [DOI] [PubMed] [Google Scholar]
- 17.Lindsay, D. S. 1997. Laboratory models of cryptosporidiosis, p. 209-223. In R. Fayer (ed.), Cryptosporidium and cryptosporidiosis. CRC Press, Boca Raton, Fla.
- 18.MacKenzie, W. R., N. J. Hoxie, M. E. Proctor, M. S. Gradus, K. A. Blair, D. E. Peterson, J. J. Kazmierczak, D. G. Addiss, K. R. Fox, J. B. Rose, and J. P. Davis. 1994. A massive outbreak in Milwaukee of Cryptosporidium infection transmitted through the public water supply. N. Engl. J. Med. 331:161-167. [DOI] [PubMed] [Google Scholar]
- 19.Mofidi, A. A., H. Baribeau, P. A. Rochelle, R. De Leon, B. M. Coffey, and J. F. Green. 2001. Disinfection of Cryptosporidium parvum with polychromatic UV light. J. Am. Water Works Assoc. 93(6):95-109. [Google Scholar]
- 20.Nickerson, C. A., T. J. Goodwin, J. Terlonge, C. M. Ott, K. L. Buchanan, W. C. Uicker, K. Emami, C. L. LeBlanc, R. Ramamurthy, M. S. Clarke, C. R. Vanderburg, T. Hammond, and D. L. Pierson. 2001. Three-dimensional tissue assemblies: novel models for the study of Salmonella enterica serovar Typhimurium pathogenesis. Infect. Immun. 69:7106-7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okhuysen, P. C., C. L. Chappell, J. H. Crabb, C. R. Sterling, and H. L. DuPont. 1999. Virulence of three distinct Cryptosporidium parvum isolates for healthy adults. J. Infect. Dis. 180:1275-1281. [DOI] [PubMed] [Google Scholar]
- 22.Peng, M. M., L. Xiao, A. R. Freeman, M. J. Arrowood, A. A. Escalante, A. C. Weltman, C. S. L. Ong, W. R. Mackenzie, A. A. Lal, and C. B. Beard. 1997. Genetic polymorphism among Cryptosporidium parvum isolates: evidence of two distinct human transmission cycles. Emerg. Infect. Dis. 3:567-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinto, M., L. S. Robine, M. D. Appay, M. Kedinger, N. Traidou, E. Dussault, B. Lacroix, A. P. Simon, K. Haffen, J. Fogh, and A. Zweibaum. 1983. Enterocyte like differentiation and polarization of the human colon carcinoma cell line Caco-2 in cell culture. Biol. Cell 47:323-330. [Google Scholar]
- 24.Rochelle, P. A., D. M. Ferguson, T. J. Handojo, R. De Leon, M. H. Stewart, and R. L. Wolfe. 1997. An assay combining cell culture with reverse transcriptase PCR to detect and determine infectivity of waterborne Cryptosporidium parvum. Appl. Environ. Microbiol. 63:2029-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rochelle, P. A., E. M. Jutras, E. R. Atwill, R. De Leon, and M. H. Stewart. 1999. Polymorphisms in the β-tubulin gene of Cryptosporidium parvum differentiate between isolates based on animal host but not geographic origin. J. Parasitol. 85:986-989. [PubMed] [Google Scholar]
- 26.Rochelle, P. A., D. M. Ferguson, A. M. Johnson, and R. De Leon. 2001. Quantitation of Cryptosporidium parvum infection in cell culture using a colorimetric in situ hybridization assay. J. Eukaryot. Microbiol. 48:565-574. [DOI] [PubMed] [Google Scholar]
- 27.Rose, J. B., J. T. Lisle, and M. LeChevallier. 1997. Waterborne cryptosporidiosis: incidence, outbreaks, and treatment strategies, p. 93-109. In R. Fayer (ed.), Cryptosporidium and cryptosporidiosis. CRC Press, Boca Raton, Fla.
- 28.Slifko, T. R., D. E. Huffman, and J. B. Rose. 1999. A most-probable-number assay for enumeration of infectious Cryptosporidium parvum oocysts. Appl. Environ. Microbiol. 65:3936-3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Upton, S. J. 1997. In vitro cultivation, p. 181-207. In R. Fayer (ed.), Cryptosporidium and cryptosporidiosis. CRC Press, Boca Raton, Fla.
- 30.Widmer, G., D. Akiyoshi, M. A. Buckholt, X. Feng, S. M. Rich, K. M. Deary, C. A. Bowman, P. Xu, Y. Wang, X. Wang, G. A. Buck, and S. Tzipori. 2000. Animal propagation and genomic survey of a genotype 1 isolate of Cryptosporidium parvum. Mol. Biochem. Parasitol. 108:187-197. [DOI] [PubMed] [Google Scholar]
- 31.Woods, K. M., M. V. Nesterenko, and S. J. Upton. 1995. Development of a microtitre ELISA to quantify development of Cryptosporidium parvum in vitro. FEMS Microbiol. Lett. 128:89-94. [DOI] [PubMed] [Google Scholar]
- 32.You, X., M. J. Arrowood, M. Lejkowski, L. Xie, R. F. Schinazi, and J. R. Mead. 1996. A chemiluminescence immunoassay for evaluation of Cryptosporidium parvum growth in vitro. FEMS Microbiol. Lett. 136:251-256. [DOI] [PubMed] [Google Scholar]