

Abstract
We engage the experimental and computational challenges of de novo regulatory module discovery in a complex and largely unstudied metazoan genome. Our analysis is based on the comprehensive characterization of regulatory elements of 20 muscle genes in the chordate, Ciona savignyi. Three independent types of data we generate contribute to the characterization of a muscle-specific regulatory module: (1) Positive elements (PEs), short sequences sufficient for strong muscle expression that are identified in a high-resolution in vivo analysis; (2) CisModules (CMs), candidate regulatory modules defined by clusters of overrepresented motifs predicted de novo; and (3) Conserved elements (CEs), short noncoding sequences of strong conservation between C. savignyi and C. intestinalis. We estimate the accuracy of the computational predictions by an analysis of the intersection of these data. As final biological validation of the discovered muscle regulatory module, we implement a novel algorithm to search the genome for instances of the module and identify seven novel enhancers.
Characterization of the regulatory logic underlying development and differentiation of multicellular animals remains one of the most formidable challenges in contemporary genomics. High-throughput experiments that use expression arrays and related tools can describe global patterns of gene regulation during development and serve as a basis for discovering regulatory hierarchies (see Furlong et al. 2001; Kim et al. 2001; Montalta-He et al. 2002; Gaudet et al. 2004; Schroeder et al. 2004). A complementary approach, high-resolution structure-function studies of individual regulatory regions, can reveal on a gene-by-gene basis exactly which noncoding portions of a locus are sufficient or necessary for proper regulation. Such studies will ultimately be necessary to obtain a full understanding of the genomic control of development, and there is considerable interest in how computational predictions could enhance their efficiency. We therefore set out to assess the effectiveness of computational predictions and to estimate their sensitivity and specificity, by comparing results from computational analyses against the activities of a large number of regulatory constructs assayed in the ascidian chordate, Ciona (Corbo et al. 1997; Johnson et al. 2004). The culmination of these analyses was a regulatory module characterized in sufficient detail that we were able to obtain genome-wide module predictions and experimentally verify a subset of these as enhancers.
Ciona is uniquely suited for structure-function and computational analyses of gene regulation. Draft genome sequences for C. savignyi (Vinson et al. 2005) and C. intestinalis (Dehal et al. 2002) as well as a sizeable EST sequence and in situ hybridization databases for C. intestinalis (Satou et al. 2002) are available. A small genome (180 Mb) with a number of expressed genes (15,000) similar to that of Drosophila (Dehal et al. 2002) ensures that the search space for noncoding functional elements is smaller than in vertebrates. Considerable functional conservation despite the large evolutionary distance between the Ciona species allows discovery of functional sequence elements by comparative sequence analyses (Johnson et al. 2004). Finally, electroporation of reporter constructs into developing embryos facilitates efficient in vivo expression analyses (see Corbo et al. 1997; Bertrand et al. 2003; Johnson et al. 2004).
We use a combination of experimental and computational approaches to discover and characterize a regulatory module common to 20 Ciona muscle genes. We chose muscle for several reasons. First, the proteins encoded by many muscle-specific genes, most notably those of the muscle fiber strand, are sufficiently conserved that their identification in the Ciona genome by similarity searches is unambiguous. Second, muscle is an easily recognized tissue in the ascidian larva, allowing efficient quantification of expression patterns. Third, the cellular interactions of muscle proteins likely require tight coregulation of their expression, enhancing the likelihood that they are under functionally similar regulatory control that would facilitate identification of a shared regulatory module.
Previous genome-wide computational searches for tissue-specific regulatory elements have been carried out in metazoan model organisms with comprehensive genome annotations and substantial prior knowledge of motifs and regulatory regions from decades of experimentation (see Berman et al. 2002, 2004; Gaudet et al. 2004; Wenick and Hobert, 2004). By contrast, in our study we discovered a tissue-specific regulatory module de novo without prior knowledge of the motifs it contains, and structure-function studies carried out independently allowed us to estimate the predictive power of the module. We then searched for instances of the module on a genome-wide scale and validate the predictions in vivo. The success of this study raises the possibility of genome-wide searches for enhancers with more complex expression patterns, as well as computational searches for tissue-specific elements in the human genome.
Results
Initial characterization of 20 muscle-specific regulatory regions
We selected 20 genes for detailed experimental characterization of regulatory sequences. The genes were chosen based on their strong similarity to proteins known to be involved in muscle physiology or structure (Fig. 1a). We located these genes in the unannotated C. savignyi genome by tBLASTn (Altschul et al. 1997) with vertebrate protein sequences as queries. By conducting in situ hybridizations at representative stages, we show that all of the genes demonstrate strong, specific expression in the embryonic tail muscle (Fig. 1a, grey field; Supplemental Fig. 1). Onset of expression varies among the genes. Occasionally, weak expression occurs at the 64-cell, pre-gastrula stage in two genes, MRLC and CK (cf. Fig. 1a, grey field, for all gene names and abbreviations), and several genes express at the onset of gastrulation at the 110-cell stage.
Figure 1.

(a) Overview of functional analysis for 20 muscle-specific genes. Transcript expression patterns are all specific to primary and secondary tail muscle and are strongly expressed by mid-tailbud stage (Supplemental Fig. 1). All initial native-promoter fusion constructs express in the muscle by mid-tailbud, and many demonstrate expression in other tissues (TVC indicates trunk ventral cells; HS, trunk hot spot; NT, notochord; AE, anterior ectoderm; and CNS, central nervous system). The average and median percentage of conservation increases between the initial constructs and the smallest functional constructs. Original indicates the initially tested, long construct with which muscle expression was ascertained; smallest, the shortest construct that gives expression comparable to the original construct. All of the original constructs show expression in the mesenchyme (ME, pink arrows), and some show expression in the CNS (b,c,f). Fragments of the initial constructs of five different loci show notochord expression (NT, green arrow, d,e). Eight constructs express as early as gastrulation (g).
For each gene, we built an initial reporter construct in which a putative native promoter, a putative start codon, and a varying amount of endogenous protein-coding sequence are fused in-frame to LacZ (see Methods) (Johnson et al. 2004). Expression is therefore driven by endogenous tissue-specific elements and a native promoter. All constructs demonstrate consistent expression in the larval tail muscle, with a median of 74% of the animals staining (Fig. 1a, green field). As expected, there is reproducible variation among the constructs in expression strength and amount of mosaicism (see Corbo et al. 1997; Johnson et al. 2004). Some constructs, such as that for Acetylcholinesterase (AChE), are consistently weaker with considerable mosaicism, while others, such as Troponin T (TnT), are consistently stronger with rare mosaicism (Fig. 1a, green field). The time at which staining is first evident generally corresponds to the in situ data (cf. Supplemental Fig. 1 and Fig. 1g).
While the in situ hybridizations show that the genes are expressed specifically in the tail muscle, our initial reporter constructs often result in expression in other tissues (Fig. 1a, green field, b,c). Two of these tissues, mesenchyme and trunk ventral cells (heart precursors) (Davidson and Levine 2003), arise in the mesoderm lineage from the same blastomeres that give rise to tail muscle. The reporter constructs of all genes show expression in the mesenchyme, which is consistent with previous observations that ectopic mesenchyme staining occurs extremely often (Harafuji et al. 2002; Johnson et al. 2004). The reporter constructs of five genes show expression in the TVC. Finally, five of the reporter constructs show expression in a central nervous system “hot spot” that was noted in an earlier study (Harafuji et al. 2002). These ectopic expression patterns are reproducible. We speculate that the constructs are missing repressor elements that keep the endogenous genes off in these tissues of the developing embryo; alternatively, they may be due to an artifact of unknown cause. Regardless of these complexities, at each locus we unambiguously located sequences that are sufficient for expression in the embryonic tail muscle. As our goal was not to comprehensively locate all regulatory sequences but rather to find as many sequences as possible that express strongly in embryonic tail muscle, we reasoned that these constructs would be suitable for the subsequent structure-function analyses that would identify subregions containing regulatory elements.
High-resolution in vivo analysis of 20 muscle-specific regulatory regions
By using the same in vivo reporter assay, we then identified regions within the initial constructs that are sufficient for muscle expression. In subsequent analyses, these subregions will be compared to computational predictions. Most of the constructs that we tested were truncations and/or deletions of the initial native promoter constructs (Fig. 1a, blue field; Supplemental Fig. 1). We also used heterologous promoter constructs, which contain a heterologous promoter and a heterologous translation start codon and, therefore, do not require the native promoter or start codon (Harafuji et al. 2002; Bertrand et al. 2003; Johnson et al. 2004). Heterologous constructs are particularly useful for confirming short regulatory sequences distant from the first exon. We tested an average of 11 constructs per gene (Fig. 1a, salmon field). To demonstrate the resolution of our in vivo analysis, we list the “shortest construct” that retains an expression level comparable to the initial native promoter construct at each locus (Fig. 1a, blue field). The average shortest construct is only 289 bp, or 22% of the size of the initial native promoter construct, yet it retains most the activity (63% of embryos staining vs. 71%).
Some of these shortened constructs drive ectopic expression patterns that were not found in the initial native promoter constructs. For example, constructs of four genes (TnT, TPM2, MA-like, and MBP) show moderate (∼5%) reproducible expression in the secondary notochord (see Fig. 1e). A heterologous Brachyury fusion construct for a fifth locus, CK, shows very strong (>90%) primary and secondary notochord expression (Fig. 1d). A heterologous Brachyury fusion construct for MBP shows activity in muscle, anterior ectoderm, central nervous system, and spinal cord (Fig. 1f).
Translation of in vivo results into binary data for subsequent analyses
To facilitate subsequent analyses in which we compare the functional data to computational predictions, we devised a method to translate the functional data into a binary data set. The result of this transformation is that every base in the original regulatory regions is either part of a functional element or not. We define a positive element (PE) as the shortest sufficient and non-overlapping sequence that drives strong expression in muscle (Table 1). Only constructs that give a positive result are considered; negative results are not considered because reporter constructs may be nonfunctional for a variety of reasons unrelated to the function of positive regulatory elements (e.g., disruption of the transcription start site or spurious introduction of negative regulatory elements). This does not exclude the possibility that functional sequences reside outside of PEs, nor does it suggest that every base within the PE is necessary for function. Note that the set of PEs (Table 1) greatly overlaps with, but is not identical to, the “shortest constructs” (cf. Fig. 1a, blue field), for the following reasons: (1) we exclude two genes from PE analysis—TnC, because our deletions did not narrow the region sufficiently, and AChE, because the expression levels of all constructs, including the original one, are too low; and (2) three genes have more than one non-overlapping functional sequence and therefore have more than one PE but only one “shortest construct.”
Table 1.

To clarify the logic for identification of PEs, we present detailed functional data for three of the 20 genes (Fig. 2). The CK locus (Fig. 2a) contains one PE that contributes to strong embryonic muscle expression. While several native promoter constructs that include this region show tail muscle expression (e.g., clones 412 and 413), the shortest construct that drives reproducible expression, and which therefore defines this PE, is clone 526. This clone is a heterologous promoter construct using the Brachyury minimal promoter. The Mbp locus contains two PEs (Fig. 2b). One PE is defined by the heterologous promoter construct 691, while another corresponds to native promoter construct 123. Either region is sufficient, and they do not overlap each other. The TnT locus (Fig. 2c) also has two PEs, defined by clones 291 (native promoter) and 530 (heterologous promoter), which are non-overlapping and sufficient for muscle expression. The same logic was applied for the remaining 15 loci to give at least one PE for each of them (Fig. 3; Supplemental Fig. 2). In total, these structure-function studies narrow the initial regulatory regions of the 18 genes that entered this analysis from 23,620 nonexonic base pairs to 6050 bp contained within PEs. Thus, we have converted the functional data to a binary data set, where each base pair is either contained within a PE or not.
Figure 2.

Detailed annotations of the loci Creatine Kinase (a), Myosin Binding Protein (b), and Troponin T (c), illustrate the functional and computational analyses carried out in this study. The x-axis represents base pair position in the C. savignyi sequence, with the origin at the first base of the original construct. The black line is sequence identity between C. savignyi and C. intestinalis, the plum line is the CisModule score of predicted module instances (CMs). Blue bars at 1.0 represent conserved windows at 75% for at least 20 bp (CEs). Green shading represents exons, and yellow shading represents PEs. Bars above these annotations represent the positions of DNA constructs electroporated in vivo, with red representing strong activity, green representing weak activity (<10%), and blue representing constructs that never showed activity. The construct name and percentage of staining tail muscle are listed to the right of each construct. Most plasmids are native fusions, although some are heterologous promoter fusions (denoted by bra or fkh, where appropriate).
Figure 3.

Summary of functional and computational annotations at each of the 20 loci. The key is the same as in Figure 2, except that detailed functional annotations are omitted (for those, see Supplemental Fig. 2). Here, light green shading represents exons from neighboring genes, and position 0 is the position of the forward primer for the original construct. Orange bars link PEs that have internal deletions from the original functional constructs and are therefore noncontiguous on the identity plot.
Identification and motif composition of a muscle regulatory module
Embedded within the upstream sequences of our muscle genes must be regulatory sequences that contain binding sites (motifs) for tissue-specific transcriptional activator proteins. Clusters of single motifs are often indicative of regulatory sequences (Markstein et al. 2002), as are clusters of several distinct motifs (see Berman et al, 2002). Accordingly, by reasoning that it is likely that more than one motif contributes to muscle-specific activation of target genes, we chose to use CisModule (Zhou and Wong 2004) to identify likely regulatory sequences (see Methods). Cis-Module implements Bayesian analysis to discover clusters of overrepresented motifs in a suite of coregulated sequences. These motif clusters (modules) consist of up to K distinct motifs that occur in spatial proximity and are predicted de novo from the input sequences (we used K = 4; see Methods). Instances of the predicted cis-module (CM) are then annotated in the input sequences. In all subsequent analyses, we will disregard those CMs that are >50% contained within exons, since CisModule predictions within exons may be spurious. All of our initial functional regulatory regions have at least one statistically significant CM; five genes have two highly significant modules, and two genes have three (Table 2; Fig. 3; Supplemental data).
Table 2.

CisModule outputs position-specific scoring matrices (PSSMs) for four motifs that are not overrepresented in our background sequences (Fig. 4a). Motif 1 (Mf1) is a palindrome that resembles the cyclic AMP response element (CRE) motif, which has been previously described to be able to drive muscle expression in Ciona in combination with a muscle-gene basal promoter (Kusakabe et al. 2004). Motif 2 (Mf2) resembles a GC-core E-box that may be the binding site for Ciona's bHLH myoD-like transcription factor (Meedel et al. 2002; Johnson et al. 2004; Kusakabe et al. 2004). Motif 3 (Mf3) has a GGCG core and resembles no known binding site, though the GC richness is reminiscent of an SP1 site. Motif 4 (Mf4) is an 11-bp CT-rich sequence also without resemblance to known transcription factor binding sites.
Figure 4.

(a) Motifs of the muscle module, their abundance in all CMs, and their abundance in CMs that overlap PEs over >50% of their length. (b) Counts of CMs that overlap PEs over >50% of their length by the motifs they contain. Most CMs contain two or three of the motifs, but all combinations (except Mf2/Mf3) are observed at least once.
To ascertain which of these CM motifs are likely to be functional, as opposed to false positives, we determined their abundance in all predicted CMs versus CMs that overlap PEs at >50% (Fig. 4b). Note that CisModule only reports highly significant instances of the motifs. Mf1 and Mf2 are common, averaging more than one instance per CM. Mf3 is slightly less common but is still present at almost one instance per CM. Mf4 stands out as being least common. More than three fourths of instances of Mf1, Mf2, and Mf3 occur in the CMs that overlap PEs, but there are only three Mf4 instances in the 17 CMs that overlap PEs. We conclude that Mf1, -2, and -3 are likely real motifs, whereas Mf4 is probably spurious. Given that we found what appears to be a spurious motif, it is likely that Mf1, Mf2, and Mf3 are all the motifs that are shared among these muscle-specific regulatory regions. However, to be certain that we had not missed other motifs, we masked all significant motif instances in the 20 functionally annotated sequences and reran CisModule with the same parameters. This run returned no significant motifs. Other motifs may be present in small subsets of the genes that fulfill functions not shared among all the regions, but these would not be detected here.
Sensitivity and specificity of module predictions
As the CMs were predicted independently from the functional analyses, we asked to what extent the predictions are correct by analyzing overlaps between CMs and PEs. The purpose of this analysis is twofold: (1) to validate or refute the module on the basis of the functional data; and (2) if the module is validated, to assess sensitivity and specificity of the CisModule predictions. We again turn to the three representative loci (Fig. 2) to illustrate the data. The CK gene (Fig. 2a) has two CMs, one of which is fully contained in the single PE of the region. The MBP gene (Fig. 2b) has a single CM, more than three fourths of which overlaps one of the two PEs. The TnT (Fig. 2c) gene has three CMs, two of which are almost exactly congruent with the two PEs of the gene; a third CM does not overlap a PE. For subsequent statistical analyses, we will call a CM prediction a “true positive” if at least 50% of it is contained in a PE.
Across all 18 PE-containing loci (Fig. 3; Supplemental Fig. 2), the sensitivity of CisModule predictions (defined as the number of CMs within PEs, divided by the total number of PEs) is remarkably high at 17 out of 21, or ∼80% (Fig. 5a). Given the total size of the noncoding regions of >20 kb, and the median size of ∼200 bp of the CM predictions, >100 CMs could have been predicted in the total space of the regions. The high sensitivity of CisModule predictions therefore does not come at the expense of specificity, as only eight of these >100 possible CMs are outside of PEs (Fig. 5a).
Figure 5.

Overlap analyses of PEs versus CMs at the element level (a), PEs versus CMs at the base pair level (b), PEs versus CEs at the element level (c), and PEs versus CEs at the base pair level (d). Sensitivity and specificity calculations are based on these figures.
In order to further define the predictive success of CMs, we converted the data into base pair counts (Fig. 5b). The size of the 18 initial regulatory regions for which PEs were obtained is 23,304 bp, and the number of bases in PEs is 6050, or 26% of the total. Each base from a CM prediction therefore has a 26% random chance of occurring in a PE (Fig. 5b). However, in this data set, 58% of all CM bases are contained within a PE, a substantially greater-than-expected overlap between the two classes. Estimates of sensitivity and specificity of the CM predictions (using standard definitions) (cf. Sokal and Rohlf 1995) at the base pair level also sheds additional light on CisModule's predictive power. Sensitivity, defined as True Positives/(True Positives + False Negatives), is the number of PE bases contained within CMs divided by the total number of bases in PEs (Fig. 5b): 2811/6050 = 46.5%; random expectation for sensitivity is simply the fraction of bases in CMs, or 4860/23304 = 20.9%. Specificity, defined as True Negatives/(True Negatives + False Positives), is the number of bases that are not in PEs or in CMs, divided by the number of bases that are not in PEs (Fig. 5c): 15,205/17,254 = 88%. Therefore, the high sensitivity of the CisModule predictions is not due to low specificity.
One caveat to these calculations is that we do not have perfect experimental ascertainment of PEs at the base pair level. Some bases in PEs are certainly not functional, and other bases outside of PEs are likely functional. Thus, while our experimentally determined PEs are enriched for truly functional bases, the exact numbers are subject to uncertainty. Nonetheless, the main conclusions are supported by the overlap analyses both at the element level (Fig. 5a) and at the base pair level (Fig. 5b): (1) CisModule predictions are highly specific and very sensitive, and (2) the discovered muscle regulatory module is genuine.
Analysis of evolutionarily conserved regions within PEs
Every PE contains at least one highly conserved element (CE), defined here as at least 20 bp of at least 75% identity between the two Cionas (Figs. 2, 3, light blue line). In addition, PEs have a higher median fraction of such conserved regions, and a higher median percentage of identity, than do the original functional constructs (Fig. 1; Table 1). We therefore sought to address how much of the noncoding sequence identity remaining since the last common ancestor of the two Ciona species could be explained by functional conservation on PEs. There are 63 nonexonic CEs that occupy 3200 (13.7%) of the base pairs of the regions. By chance, ∼16 of these CEs would be expected to reside in PEs, and 47 outside of PEs, but we observe 32 within and 31 outside PEs. This indicates that CEs are enriched in areas with transcriptional activation function compared with that of other noncoding sequence (Fig. 5c). If the unusually highly and continuously conserved upstream region of LIM is excluded, then there are 30 conserved elements in PEs versus 22 outside of PEs, while random expectation would be 14 in PEs versus 38 outside. We conclude that a substantial fraction of conserved regions can be explained by conservation on the positive activator functions that were assayed in this study.
Those CEs that do not reside within PEs may have a variety of functions. Some may be exons missed due to incomplete annotation of the two genomes. Others may have regulatory roles other than activator functions, such as repressors or insulators, which would not be reliably detected by our assays. Repressor functions are particularly attractive candidates, as we did occasionally observe reproducible ectopic expression by constructs carrying internal deletions or truncations (Fig. 1).
We converted the CE data into base pair counts to estimate sensitivity and specificity, in analogy to, and for comparison with, the CM predictions. Sensitivity is here the number of PE bases contained within CEs divided by the total number of bases in PEs (Fig. 5d): 1378/6050 = 22.8%; random expectation is the overall fraction of CE bases (13.7%). Specificity is here the number of bases that are not in PEs or in CEs, divided by the number of bases that are not in PEs (Fig. 5d): 15,105/16,927 = 89%. Compared with CMs, CEs are therefore less sensitive but are comparably specific for PE prediction.
Search of the genome for conserved muscle modules reveals novel muscle enhancers.
We next wanted to use the new information of the muscle-specific module to find enhancers on a genome-wide scale. Our previous analyses demonstrated that CEs and CMs have a correspondence with PEs. To estimate the potential predictive power of our computational methods, we calculated the positive predictive value (PPV) (Sokal and Rohlf, 1995), defined as True Positives/All Predictions. The PPV is 67.2% for bases that are both CE and CM; 55% for bases that are CM but not CE; and 29.2% for bases that are CE but not CM. Given this strong PPV of combining CisModule predictions and sequence conservation, we set out to search genome-wide for instances of the regulatory module that overlap with conserved elements.
To this end, we devised and implemented a novel algorithm (CisModScan) that identifies candidate clusters of given motifs on a genome-wide scale (see Methods). By using CisModScan, we searched the C. savignyi genome for clusters of Mf1, Mf2, and/or Mf3. We found 1183 predictions that contained at least two of the three motifs. We aligned all of these to their orthologous genomic regions in C. intestinalis. Many of the regulatory module predictions contained more than half of their nucleotides in C. intestinalis predicted exons (664/1183, or 56%). Of the remaining module predictions, 52%, or 269/519, have at least one highly conserved element. Of the predictions with at least one highly conserved element, a median 46% of the bases were contained within conserved elements (http://mendel.stanford.edu/supplementarydata/johnson2005/).
We chose 23 module predictions that contained at least one conserved element and were located <2 kb 5′ or 3′ to a predicted first exon. We assayed the function of these sequences with the same heterologous Brachyury promoter that was used for the initial structure-function studies. We found seven novel enhancers, each with distinct expression patterns (Fig. 6). Six of these enhancers express in tail muscle, while a seventh (AS794, Fig. 6c) expresses in central nervous system, papillae, and anterior ectoderm (with a single transgenic embryo showing weak muscle expression). AS788 expresses in the endodermal strand and in the secondary muscle (Fig. 6a). AS808 (Fig. 6e) also shows a complex expression pattern, with LacZ staining in the central nervous system, notochord, and secondary muscle. AS792 (Fig. 6b) and AS817 (Fig. 6g) express very strongly in the tail muscle and also in the notochord. AS816 (Fig. 6f) shows strong expression in tail muscle. Though weak, tail muscle staining in AS805 is above background (∼10% staining vs. 0% staining with random genomic DNA). The discovery of seven novel enhancers, in conjunction with the functional dissection of the regulatory regions of 20 genes, represents a substantial step forward in the characterization of metazoan tissue-specific regulatory mechanisms.
Figure 6.

Expression patterns for novel enhancers identified from a subset of predictions from a genome-wide computational scan for the muscle module. (a) AS788, expressed in endodermal strand (ES) and muscle (MU). (b) AS792, expressed in MU and notochord (NT). (c) AS794, expressed in apical ectoderm (AE) and rarely in MU. (d) AS805, expressed in MU. (e) AS808, expressed in central nervous system (CNS), NT, and MU. (f) AS816, expressed in MU. (g) AS817, expressed in MU and NT. (h) For each enhancer, percent sequence identity between C. intestinalis and C. savignyi, its strength of expression in each tissue, and the nearest gene.
Discussion
A number of previous studies have correctly predicted regulatory sites using conservation (Ghanem et al. 2003; Kellis et al. 2003; Johnson et al. 2004), de novo computational motif prediction (GuhaThakurta et al. 2004; Kusakabe et al. 2004), or conservation of predicted motifs (Wenick and Hobert 2004). Other studies have predicted regulatory modules on a genome-wide scale (Berman et al. 2004; Gaudet et al. 2004; Schroeder et al. 2004). However, our work is unique in leveraging a combination of these approaches to characterize a tissue-specific regulatory module de novo in a metazoan, and then use the knowledge of this module to conduct a genome-wide enhancer search and validate a subset of the enhancer predictions in vivo. Our success rate was similar to a genome-wide search for clusters of some of the most intensely studied binding sites in all of developmental biology (Berman et al. 2004). Our search for tissue-specific enhancers is also significantly more powerful than are less directed methods, such as a screen of random DNA fragments. For example, a recent functional screen of 138 random C. intestinalis genomic DNA fragments (average size, 1.7kb; ∼240kb total) (Harafuji et al. 2002) yielded only five tissue-specific enhancers. In contrast, we identified seven novel enhancers in ∼8 kb of tested DNA.
Metazoan gene regulation is inherently complex, since proper expression patterns often depend upon activators and repressors, interactions of tissue-specific elements with basal promoters, and other functional sequences that are often specific to each individual locus. We clearly have not described all functional elements contained within the 20 original genes or within the positive enhancers from the genome-wide search. The complexity of gene regulatory structures is underscored by the variation exhibited by the seven new enhancers and the initial high-resolution screen for PEs: Five enhancers drive expression outside of the embryonic tail muscle, although most of the enhancers express strongly in muscle. In addition, certain constructs from the initial high-resolution screen for PEs exhibit ectopic expression not only in the notochord (a tissue that shares pre- and post-gastrulation cell lineages with muscle) (Nishida, 1987) but also in tissues embryologically unrelated to muscle, such as the central nervous system, ectoderm, or endodermal strand. We conclude that many of the endogenous loci from which we obtained regulatory regions in this study also contain important repressor elements that fine-tune the expression pattern to the appropriate locations in the developing embryo.
The success rate for the genome-wide search, at seven positives out of 23 tested, is lower than the predictive value of CMs that contain at least one CE in the original 20 native promoter constructs. One reason for the discrepancy may be that false-positive rates are simply higher for computational searches on a genome-scale, which are inherently more complex than are searches within a moderate number of 5′ regions. Another limitation of the genome-wide scan is the necessity to use a heterologous promoter, which requires the candidate regulatory region to have true enhancer activity in order to give expression: In dissecting the 20 regulatory regions, we often observed elements that were sufficient for expression under the native promoter but were not sufficient under a heterologous promoter (cf. Supplemental Fig. 2b, clones 157, 527, and 528). Given these limitations and complexities, the success rate of the genome-wide search is quite satisfactory, and the discovery of seven novel enhancers with a variety of complex expression patterns underscores that the type of approach we chose will be generally viable.
In the future, painstaking in vivo structure-function studies will be crucial to unraveling the complexity of metazoan gene regulation. Within the initial 20 regulatory regions, there remains significant opportunity for discovery of novel repressors and other functional sequences, as well as for more detailed analysis of regulatory element evolution. On a genome-wide scale, we have only scratched the surface of a complex regulatory network, so future work might validate a larger set of predictions from our genome-wide search. Finally, with future refinements and increases in predictive power, our approach to de novo discovery of modules and to the combined computational and experimental validation will be applicable to systems other than muscle and to organisms other than Ciona.
Methods
Ascidian electroporation, in situ hybridization, and handling
Electroporations were conducted as reported previously (Corbo et al. 1997) with a BioRad GenePulser II or a custom electroporator (R. Zeller, pers. comm.) set at 2000 μF and 20Ω. In situ hybridization was carried out according to standard protocols (Satou et al. 2002) with probes derived from the Ghost EST collection (http://ghost.zool.kyoto-u.ac.jp/indexr1.html). Photographs of all in situ hybridizations and electroporations are available at http://mendel.stanford.edu/supplementarydata/johnson2005/.
Initial construct generation and mutagenesis
We used tailed-end PCR to amplify C. savignyi genomic DNA (http://www.broad.mit.edu/annotation/ciona/) for our initial functional regulatory constructs. We used the djmcs.lacZ plasmid for all native fusion constructs (Johnson et al. 2004), and either pCES (Harafuji et al. 2002) or a Brachyury (Bra) basal promoter plasmid (Bertrand et al. 2003) for the heterologous promoter constructs (the Brachyury plasmid was made available during the course of data collection and, in our hands, has fewer false negatives). Truncations and deletions were carried out as reported previously (Johnson et al. 2004). All clones were verified by sequencing and restriction digest, as reported previously (Johnson et al. 2004). Sequences for all constructs and primers are available at http://mendel.stanford.edu/supplementarydata/johnson2005/.
MLAGAN alignments, conserved element detection, and sequence analysis
Alignments were constructed as reported previously (Johnson et al. 2004). Orthology of three gene families was not clear due to recent gene duplications and/or large sequence gaps in the C. intestinalis assembly (http://genome.jgi-psf.org/ciona4/ciona4.home.html). In these instances, we constructed all pairwise interspecies alignments and chose the alignment with the highest percentage of bases in highly conserved elements. To find conserved elements, we used custom PERL scripts to scan alignments for 20-bp windows that contain at least 75% identity, and then expanded these windows until identity dropped <75%. Detailed functional and computational annotation of each regulatory region is available at http://mendel.stanford.edu/supplementarydata/johnson2005/.
Regulatory module identification
The CisModule algorithm (Zhou and Wong 2004) was used to identify candidate modules within the functionally annotated sequences. CisModule identifies a specified number (K) of motifs that are overrepresented in a set of sequences and that occur in clusters of a specified length (l), and outputs module predictions and a PSSM for each motif. Our input sequences included all of the original construct sequences of the 20 genes, plus 5′ regions from 42 genes whose transcripts show muscle-specific expression (http://ghost.zool.kyoto-u.ac.jp/indexr1.html and http://mendel.stanford.edu/supplementarydata/johnson2005/; we also ran CisModule on the 20 genes dissected here, with identical results). For the latter genes, we included 1 kb upstream of the predicted promoter. In some instances we added conserved sequences between 1 kb and 2 kb, since distal conserved sequences may contain regulatory elements. For our analyses, we used the output from a run with 62 genes as the greater number of genes offers greater sequence depth, and therefore higher quality, for the PSSMs. Our experimental results suggested that muscle regulatory elements are ∼200 bp, so we ran CisModule with l = 200. We ran CisModule with K = 3, 4, and 5. At K = 5, CisModule returned only four significant motifs, so we used a run with K = 4 for our analyses. As a negative control, we ran CisModule with 59 random intergenic regions from the C. intestinalis assembly (http://mendel.stanford.edu/supplementarydata/johnson2005/). None of the four motifs identified in the muscle-specific genes occurred in the background set. We used Weblogo (http://weblogo.berkeley.edu) for visualization of motifs. We did not include exonic CisModule predictions in any of our analyses.
Genome-wide module scan
To perform a genome-wide scan for motif clusters, we implemented a novel optimization algorithm (CisModScan, http://www.stanford.edu/group/wonglab/software.html) based on the hierarchical mixture model (HMx model) used in CisModule (Zhou and Wong 2004). A module model is defined by the module length l, the prior probability r of starting a new module, and K distinct motifs with frequencies qk (k = 1,2,...,K). We denote the input sequence by X = x1x2...xL = x[l,L] and the corresponding module locations by Y = y1y2...yL = y[l,L], where L is the full sequence length. Using dynamic programming, we find the optimal Y* that maximize P (X, Y | Ψ, where Ψ denotes all the model parameters. The joint probability of the optimal module locations up to position n(n = 1,2,...,L) is given by
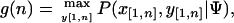 |
which is calculated by the recursion
 |
where P(xn|θ0) is the background probability of observing xn, and P(x[n-l+1,n], y[n-l+1,n] = m|Θ,q) is the probability of observing x[n-l+1,n] in a module, of which the calculation was discussed previously (Zhou and Wong 2004). As we recursively calculate all g(n) to n = L, we reach the global maximum and then generate the optimal module locations for the whole sequence. We score a predicted module by calculating the log-odds over background, i.e.,
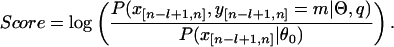 |
We used CisModScan (l = 150; K = 3; r = 0.0001) to predict muscle-specific regulatory modules in the C. savignyi genome (nonredundant supercontigs totaling 220 Mb) using PSSMs for the three most common motifs identified by CisModule. We used a prior expected motif occurrence rate within a module of 0.05 (i.e., one binding site per 200 bp). We then used MLAGAN (Brudno et al. 2003) to align the region surrounding each module prediction with its orthologous C. intestinalis sequence. Since similarity searches against large genomic regions often result in spurious matches, we parsed the C. intestinalis genome into 30-kb fragments. Then we queried the 30-kb C. intestinalis database with C. savignyi genomic sequence 5 kb 5′ and 3′ to each module by using BLASTn (Altschul et al. 1997). We aligned the C. savignyi sequence with the best C. intestinalis match and annotated the output with C. intestinalis predicted genes (http://genome.jgi-psf.org/ciona4/ciona4.home.html). We then used PERL scripts to determine noncoding conservation within each module. We chose 23 predicted modules that contained regions of high conservation and that were located 2 kb 5′ or 3′ to a predicted first exon. To validate these predictions in vivo, we amplified the corresponding sequences plus ∼100 bp of 5′ and 3′ flanking sequence (for an average insert size of ∼350 bp), subcloned them into the Brachyury heterologous basal promoter construct (Bertrand et al. 2003), and assayed activity of the constructs in Ciona as described above.
Supplementary Material
Acknowledgments
Kerrin Small of the Sidow Laboratory provided a nonredundant list of Ciona savignyi supercontigs. Midori Hosobuchi offered critical comments on the manuscript. Thanks to Patrick Lemaire for kindly providing the Brachyury heterologous promoter construct. The laboratories of Julie Baker, Stuart Kim, and Rick Myers provided helpful insights that guided the progress of this work. Thanks to Vy Le, Bob Zeller, David Kroodsma, and Polly Fordyce for assistance with building our custom electroporator.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.4062605. Article published online before print in September 2005.
Footnotes
[Supplemental material is available online at www.genome.org. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: K. Small and P. Lemaire.]
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, B.P., Nibu, Y., Pfeiffer, B.D., Tomancak, P., Celniker, S.E., Levine, M., Rubin, G.M., and Eisen, M.B. 2002. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc. Natl. Acad. Sci. 99: 757-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, B.P., Pfeiffer, B.D., Laverty, T.R., Salzberg, S.L., Rubin, G.M., Eisen, M.B., and Celniker, S.E. 2004. Computational identification of developmental enhancers: Conservation and function of transcription factor binding-site clusters in Drosophila melanogaster and Drosophila pseudoobscura. Genome Biol. 5: R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand, V., Hudson, C., Caillol, D., Popovici, C., and Lemaire, P. 2003. Neural tissue in ascidian embryos is induced by FGF9/16/20, acting via a combination of maternal GATA and Ets transcription factors. Cell 115: 615-627. [DOI] [PubMed] [Google Scholar]
- Brudno, M., Do, C.B., Cooper, G.M., Kim, M.F., Davydov, E., Green, E.D., Sidow, A., Batzoglou, S., and NISC Comparative Sequencing Program. 2003. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13: 721-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo, J.C., Levine, M., and Zeller, R.W. 1997. Characterization of a notochord-specific enhancer from the Brachyury promoter region of the ascidian, Ciona intestinalis. Development 124: 589-602. [DOI] [PubMed] [Google Scholar]
- Davidson, B. and Levine, M. 2003. Evolutionary origins of the vertebrate heart: Specification of the cardiac lineage in Ciona intestinalis. Proc. Natl. Acad. Sci. 100: 11469-11473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehal, P., Satou, Y., Campbell, R.K., Chapman, J., Degnan, B., De Tomaso, A., Davidson, B., Di Gregorio, A., Gelpke, M., Goodstein, D.M., et al. 2002. The draft genome of Ciona intestinalis: Insights into chordate and vertebrate origins. Science 298: 2157-2167. [DOI] [PubMed] [Google Scholar]
- Furlong, E.E., Andersen, E.C., Null, B., White, K.P., and Scott, M.P. 2001. Patterns of gene expression during Drosophila mesoderm development. Science 293: 1629-1633. [DOI] [PubMed] [Google Scholar]
- Gaudet, J., Muttumu, S., Horner, M., and Mango, S.E. 2004. Whole-genome analysis of temporal gene expression during foregut development. PLoS Biol. 2: e352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem, N., Jarinova, O., Amores, A., Long, Q., Hatch, G., Park, B.K., Rubenstein, J.L., and Ekker, M. 2003. Regulatory roles of conserved intergenic domains in vertebrate Dlx bigene clusters. Genome Res. 13: 533-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GuhaThakurta, D., Schriefer, L.A., Waterston, R.H., and Stormo, G.D. 2004. Novel transcription regulatory elements in Caenorhabditis elegans muscle genes. Genome Res. 14: 2457-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harafuji, N., Keys, D.N., and Levine, M. 2002. Genome-wide identification of tissue-specific enhancers in the Ciona tadpole. Proc. Natl. Acad. Sci. 99: 6802-6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, D.S., Davidson, B., Brown, C.D., Smith, W.C., and Sidow, A. 2004. Noncoding regulatory sequences of Ciona exhibit strong correspondence between evolutionary constraint and functional importance. Genome Res. 14: 2448-2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis, M., Patterson, N., Endrizzi, M., Birren, B., and Lander, E.S. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423: 241-254. [DOI] [PubMed] [Google Scholar]
- Kim, S.K., Lund, J., Kiraly, M., Duke, K., Jiang, M., Stuart, J.M., Eizinger, A., Wylie, B.N., and Davidson, G.S. 2001. A gene expression map for Caenorhabditis elegans. Science 293: 2087-2092. [DOI] [PubMed] [Google Scholar]
- Kusakabe, T., Yoshida, R., Ikeda, Y., and Tsuda, M. 2004. Computational discovery of DNA motifs associated with cell type-specific gene expression in Ciona. Dev. Biol. 276: 563-580. [DOI] [PubMed] [Google Scholar]
- Markstein, M., Markstein, P., Markstein, V., and Levine, M.S. 2002. Genome-wide analysis of clustered dorsal binding sites identifies putative target genes in the Drosophila embryo. Proc. Natl. Acad. Sci. 99: 763-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meedel, T.H., Lee, J.J., and Whittaker, J.R. 2002. Muscle development and lineage-specific expression of CiMDF, the MyoD-family gene of Ciona intestinalis. Dev. Biol. 241: 238-246. [DOI] [PubMed] [Google Scholar]
- Montalta-He, H., Leemans, R., Loop, T., Strahm, M., Certa, U., Primig, M., Acampora, D., Simeone, A., and Reichert, H. 2002. Evolutionary conservation of otd/Otx2 transcription factor action: A genome-wide microarray analysis in Drosophila. Genome Biol. 3: research0015. [DOI] [PMC free article] [PubMed]
- Nishida, H. 1987. Cell lineage analysis in ascidian embryos by intracellular injection of a tracer enzyme, III: Up to the tissue-restricted stage. Dev. Biol. 121: 526-541. [DOI] [PubMed] [Google Scholar]
- Satou, Y., Yamada, L., Mochizuki, Y., Takatori, N., Kawashima, T., Sasaki, A., Hamaguchi, M., Awazu, S., Yagi, K., Sasakura, Y., et al. 2002. A cDNA resource from the basal chordate Ciona intestinalis. Genesis 33: 153-154. [DOI] [PubMed] [Google Scholar]
- Schroeder, M.D., Pearce, M., Fak, J., Fan, H., Unnerstall, U., Emberly, E., Rajewsky, N., Siggia, E.D., and Gaul, U. 2004. Transcriptional control in the segmentation gene network of Drosophila. PLoS Biol. 2: E271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal, R.R. and Rohlf, F.J. 1995. Biometry, 3rd ed. W.H. Freeman and Company, New York.
- Vinson, J.P., Jaffe, D.B., O'Neill, K., Karlsson, E.K., Stange-Thomann, N., Anderson, S., Mesirov, J.P., Satoh, N., Satou, Y., Nusbaum, C., et al. 2005. Assembly of polymorphic genomes: Algorithms and application to Ciona savignyi. Genome Res. 15: 1127-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenick, A.S. and Hobert, O. 2004. Genomic cis-regulatory architecture and trans-acting regulators of a single interneuron-specific gene battery in C. elegans. Dev. Cell. 6: 757-770. [DOI] [PubMed] [Google Scholar]
- Zhou, Q. and Wong, W. 2004. CisModule: De novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc. Natl. Acad. Sci. 101: 12114-12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://mendel.stanford.edu/supplementarydata/johnson2005/; Sidow lab Web site with original data generated in this study.
- http://www.stanford.edu/group/wonglab/software.html; Wong lab Web site where CisModule software is available.
- http://ghost.zool.kyoto-u.ac.jp/indexr1.html; C. intestinalis in situ expression database.
- http://www.broad.mit.edu/annotation/ciona/; C. savignyi genome sequence.
- http://genome.jgi-psf.org/ciona4/ciona4.home.html; C. intestinalis genome and annotation.
- http://weblogo.berkeley.edu; Weblogo interface.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.