

Abstract
We suggest that the evolution of the population structure of microbial pathogens is influenced by that of modern humans. Consequently, the timing of hallmark changes in bacterial genomes within the last 100,000 yr may be attempted by comparison with relevant human migrations. Here, we used a lineage within Mycobacterium tuberculosis, a Beijing genotype, as a model and compared its phylogeography with human demography and Y chromosome-based phylogeography. We hypothesize that two key events shaped the early history of the Beijing genotype: (1) its Upper Palaeolithic origin in the Homo sapiens sapiens K-M9 cluster in Central Asia, and (2) primary Neolithic dispersal of the secondary Beijing NTF::IS6110 lineage by Proto-Sino-Tibetan farmers within east Asia (human O-M214/M122 haplogroup). The independent introductions of the Beijing strains from east Asia to northern Eurasia and South Africa were likely historically recent, whereas their differential dissemination within these areas has been influenced by demographic and climatic factors.
Intriguing clues about the history of a biological species can be derived from the study of the geographical distribution of the phylogenetic/genealogical lineages, in the approach known as “phylogeography” (Avise et al. 1987). The underlying assumption of human phylogeography is that there is a correspondence between the overall distribution of haplotypes and haplogroups and past human movements. The uniparentally inherited nonrecombining haploid mtDNA and the Y chromosome loci are particularly sensitive to the influences of drift, especially founder effect. Consequently these loci are suitable for assessing the origins of contemporary population diversity and provide context for paleontological hypothesis testing (Foley 1998). The mutation rate of the maternally transmitted mitochondrial genome is ∼10 times higher than that of nuclear DNA, which provides abundance of polymorphic sites but creates difficulties in reconstructing genealogies owing to repeated and reverse mutations. By contrast, the mutation rate of the paternally inherited nonrecombining portion of the Y chromosome (NRY) is comparable to that of nuclear DNA, which means that polymorphisms are more difficult to find but genealogies are easier to reconstruct. In addition, the greater length of the NRY DNA compared with mtDNA compensates in data analysis for its lower mutation rate (Cavalli-Sforza and Feldman 2003).
The rarity of back and recurrent mutations in NRY contributes to the property of displaying the strongest geographic correlation and greatest diversity among, rather than within, populations. To date, NRY binary polymorphisms have been widely used to trace the origin and migration events of modern humans (Underhill et al. 2001). Here, we propose the hypothesis that NRY-based phylogeography of H. sapiens sapiens, offers a convenient spatiotemporal framework for inferring early primary dispersals of those human pathogens that are essentially (1) devoid of horizontal gene transfer and (2) family/household-transmitted. We believe that Mycobacterium tuberculosis makes an ideal model to test this hypothesis.
A 70% excess of male over female tuberculosis (TB) cases are reported globally each year (Uplekar et al. 2001). Although the reasons for this difference are unclear, there exists a strong opinion that claims that there is a gender bias in TB infection (Thomas 2000). Therefore, assuming (1) a predominantly family/household mode of transmission of TB in a preindustrialized time and (2) a likely bias in TB infectivity toward males, the M. tuberculosis transmission seems to resemble the unidirectional inheritance of the paternally transmitted human NRY DNA.
Whereas the M. tuberculosuis complex includes mycobacteria widely differing in terms of their host specificities, M. tuberculosis sensu stricto (M. tuberculosis) is exclusively a human pathogen. The tubercle bacillus has the remarkable ability to persist in the human host in the form of a long-term asymptomatic infection referred to as latent TB. In latent TB, the host immune response is capable of controlling the infection and yet falls short of eradicating the pathogen. One third of the world population is estimated to have a latent or dormant TB infection, which was perhaps the predominant mode of M. tuberculosis coexistence with human host in a preindustrialized time when transmission of the pathogen was mostly family-linked. M. tuberculosis has a clonal population structure, and the circulating strains show strong geographical affinities. Among several genetic families identified within M. tuberculosis (Sola et al. 2001; Baker et al. 2004), the Beijing genotype is marked by genetic homogeneity and geographical omnipresence (Bifani et al. 2002; Glynn et al. 2002). Taken together, these data likely reflect its rapid global spread during the past century, if not the past few decades. For the first time, it was identified in the M. tuberculosis strains isolated in the Beijing area in China, which coined its name (van Soolingen et al. 1995). Subsequent studies have shown that these strains are endemically prevalent in east Asia, South Africa, and northern Eurasia (Bifani et al. 2002; Glynn et al. 2002).
Three chromosomal regions are sufficiently, although not exceedingly, polymorphic to be relevant to the evolutionary scale studies of this genotype (Figs. 1, 2). The first one is the direct repeat (DR) locus which consists of minisatellite alternating exact DRs and variable spacers (Kamerbeek et al. 1997) and represents a large polyphyletic family of DNA repeats found in many bacterial lineages (Jansen et al. 2002). Van Embden et al. (2000) hypothesized that such a locus in M. tuberculosis might have initially presented a region consisting of hundreds of short (36-bp) tandem repeats. Variable spacers emerged and accumulated further during evolution, and subsequent neutral changes in the DR locus in M. tuberculosis have occurred and are still occurring via consecutive deletions of either single units or contiguous blocks, occasionally including IS6110-mediated disruption and recombination (Beggs et al. 2000; van Embden et al. 2000). In the Beijing genotype, most of the DR units were deleted during evolution, perhaps by a single event mediated by IS6110 recombination (Beggs et al. 2000). Thus the characteristic structure of the DR locus is a marker that defines the Beijing genotype and distinguishes it from other families within M. tuberculosis.
Figure 1.

Position of the MIRU-VNTR and DR loci on the M. tuberculosis H37Rv chromosome and their structure. In bold are eight polymorphic MIRU loci used to build the Beijing genotype network (Fig. 3). NTF region is specific for Beijing strains and not shown on the map.
Figure 2.

Schematic view and evolutionary scenario for the NTF region in the M. tuberculosis Beijing genotype (based on Plikaytis et al. 1994; Kurepina et al. 1998). Triangles and black arrows depict IS6110 insertions and their orientation; not to scale.
Second, a genetic marker specific to the Beijing genotype is the so-called NTF region that may harbor IS6110 insertion(s) (Plikaytis et al. 1994; Bifani et al. 2002). Three NTF variants of Beijing strains are distinguished based on the presence/absence of IS6110 sequence, thus providing a rough subdivision within this genotype (Fig. 2). A strictly clonal population structure of M. tuberculosis species as a whole further implies a single founder population of one of its lineages (the Beijing strain), thus justifying a straightforward evolutionary scenario and unidirectional evolution of the NTF locus. The W-branch prevalent in the United States harbors two head-to-tail IS6110 insertions separated by a 556-bp noncoding spacer (Kurepina et al. 1998). Most Beijing strains worldwide harbor only one IS6110 insertion (Kurepina et al. 1998; Bifani et al. 2002); we define them as NTF::IS6110 branch. Finally, a low-endemic type in the United States, the N-branch has no IS6110 insertion in NTF region (Kurepina et al. 1998; Milan et al. 2004). Sequencing of the NTF region in N-branch demonstrated that it is intact (Kurepina et al. 1998); that is, it has never harbored IS6110 insertions. In our opinion, this implies that N-branch presents the most ancient or “primordial” group that was isolated from the rest of the Beijing strains at the very beginning of its evolution. In contrast, the W-branch is apparently the youngest and likely originated in situ from the main Beijing NTF::IS6110 lineage imported to the modern United States with immigrants, perhaps from Russia or east Asia.
Third, mycobacterial interspersed repetitive units (MIRU) are polymorphic VNTR (variable number of tandem repeats) loci scattered throughout the bacterial chromosome (Fig. 1) (Supply et al. 2000). The number of repeat copies per locus may vary among strains, and the use of several such loci allows sufficient interstrain differentiation (Supply et al. 2001). The MIRU-VNTR profiles are presented as multidigit numerical codes (complex haplotypes), each digit representing the copy number in a locus (see Supplemental Table). In fact, these MIRU loci present multiple independent genetic markers and therefore ideally suit for phylogeographic analysis.
In the present study, we analyzed the MIRU-VNTR (12 loci) (Supply et al. 2001) variation of the M. tuberculosis Beijing strains from different geographical locations, which correspond to the areas where this genotype is endemically prevalent, namely, east Asia, Russia and South Africa. We compared the resulting M. tuberculosis diversity with known prehistoric and recent human migrations dating from the past 60,000-100,000 yr and human phylogeography based on Y chromosome binary markers.
Results
MIRU-VNTR analysis
In the present study, we included all available MIRU-VNTR profiles of the M. tuberculosis Beijing genotype strains published elsewhere and typed in our laboratories. A Beijing genotype was defined by the DR locus structure revealed as specific spoligoprofile in which signals 1-34 were absent (Fig. 1). A total of 313 strains were thus included in the database. Based on the use of all 12 MIRU loci taken together, 91 types with unique MIRU signatures were identified and designated as MT1 to MT91. A detailed analysis was done for 31 shared types (two or more strains), including at least one strain from at least one of five principal areas of origin (Fig. 3; Supplemental Table).
Figure 3.

The most parsimonious genetic network of the M. tuberculosis Beijing genotype based on MIRU-VNTR data. MIRU alleles (no. of copies per locus) for MIRU types are shown in the Supplemental Table. Each arm in the network represents one event (one change per locus). Circle size is roughly proportional to the number of strains. Numbers inside circle segments are the percentage of strains of a given type of all strains from the same area. Dotted branches depict possible alternatives. Question mark means intermediate node in which no strain was found in our database. In 2004, Sun et al. who reported Beijing strains from Singapore suggested that the majority of the Singaporean population is of Chinese descent and that tuberculosis mainly affects the elderly, many of whom are first- or second-generation immigrants from mainland China. We therefore designated the Beijing genotype strains from Singapore as Chinese.
All 12 loci taken together, the allelic diversity of the local samples varied from high (China, 0.95; Vietnam, 0.88) to moderate (South Africa, 0.75) and low (Russia, 0.65). The estimate for Bangladesh (0.78) is probably biased (underestimated) due to the small sample size (n = 15). At first glance, two principal core types are present at the MIRU-based most parsimonious Beijing network, MT2 and, especially, MT11 to which most other types are directly linked (Fig. 3). These two types are likely to be the initial and most ancient Beijing variants. A closer look at the type distribution in the particular samples reveals that the Chinese strains are located all over the network, at both inner and many terminal nodes. Together with highest diversity, this suggests the Chinese sample to be the most ancient. In contrast, the South African strains are found only in the terminal positions and are probably the youngest sample, that is, comparatively most recently introduced. Finally, Russian strains are located in both core nodes but show the least diversity, a pattern that may reflect their relatively ancient introduction but only a recent dissemination in this area.
To estimate a geographical component of the variation in the Beijing genotype and, ultimately, to partly reconstruct its dispersals, we further compared the four local populations of the Beijing strains (Russia, Vietnam, China, and South Africa). The Bangladesh sample was excluded from this analysis because of too small sample size; in addition, these strains were not confirmed by IS6110-RFLP typing to be truly unlinked strains (Banu et al. 2004).
The Neighbor-Joining (NJ) tree showed a topology that strikingly resembled geographical distances (Fig. 4A). It may be noted that using the NJ method, mixing between populations shortens the mixed branch and moves it toward the tree's origin (Cavalli-Sforza 2001). Therefore, a short distance between Vietnamese and Chinese populations (Fig. 4A) may result from their geographical proximity. Conversely, a long South African branch (Fig. 4A) may be due to a very small number of founders.
Figure 4.

Relationships of the four geographical populations of the M. tuberculosis Beijing strains estimated as NJ tree (A) and MDS graph (B) based on Fst (Θ) distances calculated from MIRU-VNTR data.
A representation in the form of phylogenetic tree may over-simplify relations between complex populations, and we, therefore, also estimated them by principal component (PC) analysis by means of multidimensional scaling (MDS) (Fig. 4B). The Chinese population is placed in the central position in both the NJ tree and the MDS graph and, thus, may be interpreted as the most ancient and ancestral compared with the other three populations corroborating the above network analysis. The PC method has been shown in many studies to be very useful in resolving superimposed human migrations since PCs are statistically independent from one another and so can isolate independent expansions (Cavalli-Sforza 2001). At the same time, PCs cannot distinguish two expansions having the same area of origin and taking place at different times. In this view, although the first PC of MIRU-VNTR loci explains virtually all observed geographical variation of the Beijing genotype (i.e., four populations from the major high-prevalence endemic areas), this result should be interpreted with caution since it does not determine how many expansions emanated from east Asia, bringing the Beijing genotype to Russia and South Africa.
NTF locus analysis
Multiplex PCR analysis of the NTF region revealed that 42 of 44 Russian Beijing strains harbored one IS6110 insertion. The two remaining Russian Beijing strains had no IS6110 insertions in this region and hence belonged to the ancestral N-branch. These two strains (IS6110-RFLP profiles) were previously shown to represent 7% of the Beijing strains circulating in Russia and were also defined as ancient by use of other genomic markers, such as the IS1547 and Rv3135-PPE sequences (Mokrousov et al. 2002).
It should be noted that the NTF region was not analyzed in the previously published MIRU studies that we used for our analysis, and conversely, published data on the NTF variation were not accompanied by MIRU profiles. In particular, MIRU-VNTR data are not yet available for the W-branch strains and therefore are not included in our MIRU network analysis. As a consequence, we could not directly superpose NTF alleles onto the whole Beijing network based on MIRU s, except for the Russian M. tuberculosis populations (Fig. 3). This comparison placed one of two ancient Beijing strains from Russia into the core MIRU type MT11. Consequently, this type MT11, to which many other types are directly linked, may indeed be considered as the ancestral one to which all the network may be rooted (Fig. 3). However, further studies on both NTF and MIRU loci in more Beijing strains from diverse geographical areas are undoubtedly required to confirm this hypothesis.
Discussion
Thirty years ago, William H. McNeill (1976) reviewed how human history appeared influenced by various local and global epidemics, the Black Death being the most notorious example. Here, we propose a reverse hypothesis that (1) the population structure of microorganisms may be partly shaped by history of their human carriers, and consequently, (2) the timing of hallmark changes in bacterial genomes within the past 100,000 yr may be attempted by comparison with relevant human migrations. Here, we used as a model one globally dispersed genetic lineage within Mycobacterium tuberculosis, the Beijing genotype. Currently, these strains attract great attention worldwide, because they demonstrate some important pathogenic features: increased virulence in the BCG-vaccinated mice (Lopez et al. 2003), the ability to more rapidly multiply in human macrophages (Zhang et al. 1999), and a presumably easier adaptation to changing environments due to mutator alleles of the mutT genes (Rad et al. 2003).
In an attempt to confirm and date the routes of expansion of the Beijing genotype suggested from its phylogeography and to trace back its origin, we compared our results with known prehistoric migrations of modern humans, since primary “out-of-Africa” dispersals supported by NRY binary markers (Underhill et al. 2001). In brief, a single most parsimonious phylogeny was recently constructed for the 153 human NRY haplotypes, and a hierarchical nomenclature system, which superseded and unified past nomenclatures, was suggested (Y Chromosome Consortium 2002). Two complementary nomenclatures of NRY were proposed. The first one is hierarchical, and capital letters (A-R) are used to identify the major clades constituting front symbols of all subsequent subclades. Alternatively, or rather complementarily, haplogroups can be named by the “mutations” that define lineages rather than by the “lineages” themselves. Thus, a second nomenclature retains the major haplogroup information (i.e., 19 capital letters) followed by the name of the terminal mutation that defines a given haplogroup.
We sought to define a human host population in which the most recent common ancestor of the Beijing primordial N-branch (currently, endemic North American) and the ancient MIRU types MT11 and MT2 (radiating through a presumably Chinese primary expansion) appeared. This most recent ancestor could not be in the initial group of humans migrating from Africa, nor it could be in next step Levantine populations since this genotype is not endemic in Africa as a whole or in the Middle East and Europe (Bifani et al. 2002; Glynn et al. 2002). It could not have arisen in China (east Asia), since Chinese isolates already had one IS6110 insertion in the NTF locus (Bifani et al. 2002) and presented a second step in the Beijing evolution. It seems unlikely that the North American N-branch (i.e., initial Beijing variant) emerged in situ, since we can hardly imagine any significant human gene flow from there to east Asia or Eurasia as a whole. Rather, the low-level endemicity of the most ancient Beijing N-branch in North America demonstrates that it was brought to this continent from Eurasia with a small human group, an event that corresponds to the first entry of humans to this continent.
Summing up, it appears that most parsimoniously the Beijing genotype (specific deletion in the DR locus) (van Soolingen et al. 1995) might have originated in the human population of NRY K-M9 haplogroup that emerged in central Asia in humans migrating from the Middle East during a second out-of-Africa migration in the Upper Palaeolithic (Fig. 5A; Underhill et al. 2001; Y Chromosome Consortium 2002). This K-M9 human cluster diversified into several lineages that generally moved eastward to Siberia and east Asia (Underhill et al. 2001). The M45 mutation founded haplogroup P in the northeast direction, while the M214 mutation founded haplogroup O in the southeast direction in northern China (Fig. 5A; Underhill et al. 2001; Deng et al. 2004). The first major split within the Beijing genotype likely involved alteration in the NTF genome region and took place in these small founding (human) subpopulations. The ancestral Beijing lineage (intact NTF) must have remained in the human P-M45 population, which then moved further through Siberia, eventually reaching Beringia and entering North America prior to the Last Glacial Maximum (LGM), a relatively brief period at the end of the Upper Palaeolithic, 17,000-20,000 yr ago (Fig. 5B). Native Americans are known to have experienced two episodes of reduced population size: one with the peopling of the Americas and the other with European contact (Mulligan et al. 2004). Either or both might have contributed to the currently low-level endemicity of the initial Beijing N-branch in North America, and further studies are needed to elucidate this issue. It is noteworthy that two N-branch strains with intact NTF region were found in our collection of Russian Beijing strains and that Beijing strains, defined as ancestral by other markers, have previously been described, although as low-endemic, in modern Russia and the United States (Mokrousov et al. 2002; Rad et al. 2003). Thus, they may represent relic strains left on the first passage of the Beijing primordial sublineage (intact NTF) through Siberia to North America 20,000-30,000 yr ago.
Figure 5.

Hypothesized chronology of the early evolution of the M. tuberculosis Beijing genotype superimposed onto human prehistoric migrations and NRY based phylogeography (Underhill et al. 2001). Arrows indicate human migrations. In bold are human haplogroups and mutations according to the NRY hierarchical nomenclature system (Y Chromosome Consortium 2002). Bj indicates Beijing genotype defined by specific signature of the DR locus (absence of spoligosignals 1-34) (Bifani et al. 2002; Glynn et al. 2002); Kya, thousand years ago.
The history of the second Beijing lineage marked by the first IS6110 insertion in the NTF locus (Fig. 2) may be related to the Neolithic agriculture revolution that occurred independently in different continents following rapid global warming and the retreat of glaciers at the end of LGM (Mithen 2004). One of the main derivatives of the K-M9 Asian cluster is haplogroup O, which achieved very high frequency in east Asia (Underhill et al. 2001). The haplogroup O-M214/M175 lineage seems to have originated in Northern China. Its distribution may reflect the impact of the millet and rice agriculture on east Asian demographic history (Cohen 1998; Shelach 2000; Underhill et al. 2001; Bettinger et al. 2005), displacing to a great extent all other NRY variants with a clinal frequency from the expected Chinese area of origin (Underhill et al. 2001). The Beijing NTF::IS6110 subpopulation was likely brought by K-M9/O-M214 humans to northern China ∼20,000-30,000 yr ago. After the LGM, this ancient population (O-M214/M175/M122) who lived in the upper-middle Yellow River basin, expanded mirroring the radiation of the Proto-Sino-Tibetan languages (Su et al. 2000; Deng et al. 2004). This population expansion brought the Beijing NTF::IS6110 lineage to all areas of east Asia (Fig. 5C), whereas the already achieved divergence within the Beijing genotype was differentially transferred to new areas by distinct human subpopulations. The absence of the MIRU types shared by the Beijing strains from Vietnam and Bangladesh (Fig. 3) suggests that these areas may have been contaminated in the early Neolithic when human subpopulations were small enough to exert a founder effect of the particular Beijing variants (MIRU types) differentially carried by the host populations.
As we mentioned above, the M. tuberculosis Beijing genotype is neither frequent nor endemic variant in Africa as a whole. The exception is South Africa, namely, the Cape Town area. The introduction of the Beijing genotype (NTF::IS6110 branch) to South Africa appears to have occurred relatively recently. Strictly speaking, migrations from China itself are not likely to have contributed to the genotype's importation here. Currently, the Chinese human population constitutes only 30,000 individuals in South Africa (http://www.nationmaster.com/encyclopedia/South-Africa). Significant, but extremely transient, Chinese gene flow to South Africa occurred only 100 yr ago, when ∼50,000 Chinese workers were imported for the Rand gold mines in 1903-1907 (http://61.1911encyclopedia.org/S/SO/SOUTH_AFRICA.htm). However, by 1910 they were all repatriated (http://61.1911encyclopedia.org/S/SO/SOUTH_AFRICA.htm), and their impact on the health situation in South Africa was likely to be negligible. Recently, van Helden et al. (2002) suggested that Beijing strains might have been introduced to South Africa following the sea trade route from east Asia to Europe that started 400 yr ago. Indeed, in the 17th and 18th centuries, Dutch colonists at the Cape of Good Hope largely imported slaves from Indonesia, Madagascar, Mozambique, and India. Descendants of these slaves, who often married with Dutch settlers, later became known as “Cape Coloreds” or “Cape Malays,” and presently form the majority of the 4.7 million population of the Western Cape Province (http://www.nationmaster.com/encyclopedia/South-Africa). Therefore, it is likely that the Beijing strains were historically recently brought to South Africa, not directly from its primary focus of origin (China), but from the secondary one (Indonesia). The historical evidence is supported by genetic data: In the Beijing genetic network all main South African (Indonesian?) types are found at distant nodes and hence appear to be the youngest.
Unlike for South Africa, the exact timing of the first entry and primary dispersals of the Beijing strains in Northern Eurasia (modern Russia) is elusive. The Russian Beijing genotype population is the least diverse of all local Beijing populations. Assuming that more diversity results from longer clonal expansion, this may reflect a most recent dissemination of the currently circulating and locally predominant Beijing strains in Russia compared with the other areas studied. This is readily explained by the cold climate and, until very recently, the extremely low population density in a vast area of Russia and Siberia (Christian 1998) compared with the warmer conditions and the fast growing and denser population in east Asia and South Africa. Both network (Fig. 3) and PC (Fig. 4B) analyses suggest that Russia and South Africa were infected with distinct subsets of Beijing strains. However, a strong founder effect may have played a crucial role in the evolution of the Beijing genotype's population structure in South Africa and would consequently generate the unordinately long branch in the NJ tree (Fig. 4A). Since the Beijing genotype is not a European endemic variant, the published PC analysis of European human populations allows us to rule out those migrations that equally concerned both Russia and Europe as sources of the Beijing strains. These are defined by Finno-Ugric (PC2) (Cavalli-Sforza 2001), Scythe (PC3) (Cavalli-Sforza 2001), and Hun (Christian 1998) expansions. We may further speculate that trade contacts as such, even long-lasting ones, are not sufficient for an effective dissemination of the M. tuberculosis strains if they are not supported by a kind of demic diffusion of the strains' carriers, manifested as population growth and migration. The Silk Road connected China with Europe for almost two millennia, 2 BC-1600 AD (Christian 1998), and this route may have been opened much earlier, based on the transfer of the first ceramics technology from Japan to the Middle East and Europe at the beginning of agricultural practice (Cavalli-Sforza 2001). However, it is appropriate to reiterate that Beijing strains are not identified as a European endemic variant.
Finally, we suggest the TB spread related to the Genghis (or Chinggiz) Khan invasion to be more plausible. The Mongol empire of the 13th century brought the different parts of Eurasia closer than they had ever been before and created an economic and cultural system embracing much of the Eurasian land mass (Christian 1998). It was also a period of remarkable ethnic mixing since the Mongol army grew by incorporating the armies of many different nations that it had defeated, including Han Chinese (Christian 1998). McNeill (1976) suggested that Mongol invasions also unified Eurasia epidemiologically, allowing the exchange of the disease vectors throughout Eurasia. Genghis Khan did eventually come in the center of Europe, but for a short time. This was sufficient for the dissemination of Yersinia pestis to occur, but not for that of the far less contagious M. tuberculosis. Even if some M. tuberculosis Beijing genotype strains had been brought to Europe in this way, this may not have manifested rapidly. Subsequently, the Black Death that decimated European human populations could have efficiently eliminated rare carriers of the M. tuberculosis Beijing genotype. By contrast, further close interaction between Rus' and Orda was prolonged for three centuries, and it may be possible that the Mongol invasion and the subsequent yoke/cohabitation were indeed the vehicle that brought M. tuberculosis Beijing genotype strains to Russia.
The identity of the immediate ancestor of the Beijing genotype remains open. The peculiar DR locus structure, actually, the Beijing genotype identification, is “abridged” and thereby prevents an unambiguous reconstruction of the DR locus of a “pre-Beijing” strain. A comparison of major extant genetic families within M. tuberculosis (such as Latin-American-Mediterranean, Haarlem, East African-Indian, Delhi, and Beijing) produced an SNP-based star-like tree (Baker et al. 2004), where the Beijing genotype is not derived from any of these types but is instead directly linked to the hypothetical root. If our hypothesis about the early history of the M. tuberculosis Beijing genotype is correct, then we may expect to find a pre-Beijing strain as well as ancient Beijing isolates in the central Asian (e.g., Caspian-Aral) area of origin. New studies targeting MIRU-VNTR and NTF loci in Beijing strains from central Asia, Siberia, and North America (Amerindians) and, eventually, the analysis of fossil (including mummy) DNA samples will better test our hypothesis about the evolutionary history of this bacterial lineage.
To conclude, the implications from such comparative studies appear to be, at least, twofold: (1) timing specific events in the evolution of bacterial genomes as attempted here with one lineage of M. tuberculosis, and (2) tracing hidden patterns of human migrations of which recent studies on Helicobacter pylori (Falush et al. 2003) and polyomavirus JC (Pavesi 2004) are exciting examples. In the case of M. tuberculosis, a more detailed information on many genotypes within this species from many locations is still needed to address this latter issue. Perhaps, a comparison of phylogenies between M. tuberculosis strains and the Y chromosome haplotypes directly sampled from TB patients from non-urban isolated areas (less influenced by recent human migrations) could also give clues to better understanding of our coevolution.
Methods
Bacterial strains
M. tuberculosis strains from Russia (mainly northwest) and Vietnam were recovered from unlinked adult patients with pulmonary TB, 1997-2003. A total of 520 Russian and 125 Vietnamese strains were studied.
DNA fingerprinting
The DNA was isolated following the recommended method and subjected to IS6110-RFLP typing as described previously (van Embden et al. 1993). The variation in the DR locus (absence/presence of 43 different spacers) was studied by the spoligotyping macroarray-based method as described previously (Kamerbeek et al. 1997). In short, the PCR-amplified biotin-labeled DR locus is hybridized against an array of immobilized 43 different DR spacers. Resulting hybridization signals are revealed by chemiluminescence and are visualized as a profile of discrete dots (Fig. 1).
The 44 Russian and 43 Vietnamese strains with different IS6110-RFLP profiles and identified as Beijing genotype by spoligotyping (absence of signals 1-34) were selected for MIRU analysis performed as described by Supply et al. (2001) and Mokrousov et al. (2004). Search of the MIRU profiles of the Beijing strains from other studies was done by using the Entrez-PubMed and Google engines, followed by inspection of the retrieved articles for the presence of information on Beijing isolates. It yielded 226 more strains (Supply et al. 2003; Banu et al. 2004; Sun et al. 2004), the largest samples from South Africa (38 strains), Singapore (160 strains), and Bangladesh (15 strains). Information on strain sources and their MIRU profiles is provided in the Supplemental Table. We designated the Beijing genotype strains from Singapore as Chinese because the majority of the Singaporean population is of Chinese descent and TB mainly affects the elderly, many of whom are first- or second-generation immigrants from mainland China (Sun et al. 2004).
NTF locus analysis
A multiplex PCR approach was used to determine possible IS6110 insertion(s) in the NTF region of the M. tuberculosis strains. Primers located within the NTF region and IS6110 sequence as well as PCR parameters were previously described by Plikaytis et al. (1994).
Statistical analysis
The MIRU data were entered into an Excel spreadsheet, and the strains were subdivided into MIRU types with unique 12-loci profiles (see the Supplemental Table). A comparison was done for five areas: Russia, Singapore (i.e., China), South Africa, Vietnam, and Bangladesh. The Hunter-Gaston (Hunter and Gaston 1988) index was used as an estimate of the allelic diversity for specific areas, for the 12 loci taken together. HGDI is a probability that two strains consecutively taken from a given population would be placed into different types by the typing method; in other words, the lower the index value is, the less discriminative is the typing method (and less diverse is the population in a given locus/loci). The HGDI was calculated by using the following formula:
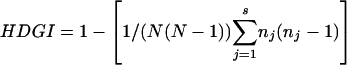 |
where N is the total number of strains in the typing scheme, s is the total number of distinct patterns discriminated by the typing method, and nj is the number of strains belonging to the jth pattern.
The most parsimonious network of the Beijing MIRU types was built by using the PARS routine of PHYLIP 3.6 package (Felsenstein 2004). PARS is a general parsimony program that carries out the Wagner parsimony method with multiple states. It assumes that different characters and different lineages evolve independently and changes to all other states are equally probable. This is applicable to evolution of MIRU-VNTR loci treated as categorical variables. The following conditions were applied to our analysis: (1) only types with two and more strains from the target areas were included, (2) only loci polymorphic in these shared types were included (eight loci), and (3) MIRU alleles were treated as categorical variables (i.e., any changes in copy number were assumed as equivalent). The bootstrapping procedure was not applied to test the robustness of the tree since there were too few number of variable characters (only eight MIRU loci).
Genetic distances between the geographical populations of the Beijing strains (Russia, Singapore [China], Vietnam, and South Africa) were calculated based on the MIRU shared types and eight polymorphic MIRU-VNTR loci as coancestry coefficients (F statistics or Θ values) (Weir 1990) using formula implemented in GDA software (Lewis and Zaykin 2001). The input Nexus file is available upon request. The resulting distance matrix was used to construct a NJ tree with GDA software and for PC analysis by means of multidimensional scaling with Permap software (Heady and Lucas 1997).
Note added in proof
While this manuscript was in review, Kremer (2005) reported the atypical Beijing strains from Southeast Asia. This finding appears to disagree with the previous estimation of Bifani et al. (2002) and, in our opinion, supports the Chinese and more recent origin of the M. tuberculosis Beijing genotype.
Supplementary Material
Acknowledgments
We thank Elena Limeschenko and Anna Vyazovaya for technical assistance and Lidia Steklova for providing us with some of the clinical isolates. We are grateful to the three anonymous reviewers whose invaluable comments and critiques helped us to significantly improve the manuscript.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3840605. Article published online before print in September 2005.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Avise, J.C., Arnold, J., Ball Jr., R.M., Bermingham, E., Lamb, T., Neigel, J.E., Reeb, C.A., and Saunders, J. 1987. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Ann. Rev. Ecol. System 18: 489-522. [Google Scholar]
- Baker, L., Brown, T., Maiden, M.C., and Drobniewski, F. 2004. Silent nucleotide polymorphisms and a phylogeny for Mycobacterium tuberculosis. Emerg. Infect. Dis. 10: 1568-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu, S., Gordon, S.V., Palmer, S., Islam, M.R., Ahmed, S., Alam, K.M., Cole, S.T., and Brosch, R. 2004. Genotypic analysis of Mycobacterium tuberculosis in Bangladesh and prevalence of the Beijing strain. J. Clin. Microbiol. 42: 674-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs, M.L., Eisenach, K.D., and Cave, M.D. 2000. Mapping of IS6110 insertion sites in two epidemic strains of Mycobacterium tuberculosis. J. Clin. Microbiol. 38: 2923-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettinger, R.L., Barton, L., Elston, R.G., Madsen, D.B., Brantingham, P.J., Oviatt, C.G., Wang, H., and Choi, W. 2005. The transition to agriculture in northwestern China. In Food systems and power structures among intermediate scale societies (eds. I. Kuijt and W.C. Prentiss), University of Arizona Press, Tucson, AZ (in press).
- Bifani, P.J., Mathema, B., Kurepina, N.E., and Kreiswirth, B.N. 2002. Global dissemination of the Mycobacterium tuberculosis W-Beijing family strains. Trends Microbiol. 10: 45-52. [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza, L.L. 2001. Genes, peoples and languages. Penguin Books. London.
- Cavalli-Sforza, L.L. and Feldman, M.W. 2003. The application of molecular genetic approaches to the study of human evolution. Nat. Genet. Suppl. 33: 266-275. [DOI] [PubMed] [Google Scholar]
- Christian, D. 1998. A history of Russia, Central Asia and Mongolia. Vol. 1: Inner Eurasia from prehistory to the Mongol empire. Blackwell Publishers, Oxford, UK.
- Cohen, D.J. 1998. The origin of domesticated cereals and the Pleistocene-Holocene transition in east Asia. Rev. Archaeol. 19: 22-29. [Google Scholar]
- Deng, W., Shi, B., He, X., Zhang, Z., Xu, J. Li, B., Yang, J., Ling, L., Dai, C., Qiang, B., et al. 2004. Evolution and migration history of the Chinese population inferred from Chinese Y-chromosome evidence. J. Hum. Genet. 49: 339-348. [DOI] [PubMed] [Google Scholar]
- Falush, D., Wirth, T., Linz, B., Pritchard, J.K., Stephens, M., Kidd, M., Blaser, M.J., Graham, D.Y., Vacher, S., Perez-Perez, G.I., et al. 2003. Traces of human migrations in Helicobacter pylori populations. Science 299: 1582-1585. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J. 2004. PHYLIP (Phylogeny Inference Package) version 3.6b. Department of Genome Sciences, University of Washington, Seattle.
- Foley, R. 1998. The context of human genetic evolution. Genome Res. 8: 339-347. [DOI] [PubMed] [Google Scholar]
- Glynn, J.R., Whiteley, J., Bifani, P.J., Kremer, K., and van Soolingen, D. 2002. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: A systematic review. Emerg. Infect. Dis. 8: 843-849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heady, R.B. and Lucas, J.L. 1997. PERMAP: An interactive program for making perceptual maps. Behavior Res. Meth. Instruments Computers 29: 450-455. [Google Scholar]
- Hunter, P.R. and Gaston, M.A. 1988. Numerical index of the discriminatory ability of typing systems: An application of Simpson's index of diversity. J. Clin. Microbiol. 26: 2465-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, R., van Embden, J.D.A., Gaastra, W., and Schouls, L.M. 2002. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 43: 1565-1575. [DOI] [PubMed] [Google Scholar]
- Kamerbeek, J., Schouls, L., Kolk, A., van Agterveld, M., van Soolingen, D., Kuijper, S., Bunschoten, A., Molhuizen, H., Shaw, R., Goyal, M., et al. 1997. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 35: 907-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer, K. 2005. “Genetic markers for Mycobacterium tuberculosis: Characterization and spread of the Beijing genotype.” Ph.D. thesis. University of Amsterdam, Blaricum, The Netherlands.
- Kurepina, N.E., Sreevatsan, S., Plikaytis, B.B., Bifani, P.J., Connell, N.D., Donnelly, R.J., van Soolingen, D., Musser, J.M., and Kreiswirth, B.N. 1998. Characterization of the phylogenetic distribution and chromosomal insertion sites of five IS6110 elements in Mycobacterium tuberculosis: Non-random integration in the dnaA-dnaN region. Tuber. Lung Dis. 79: 31-42. [DOI] [PubMed] [Google Scholar]
- Lewis, P.O. and Zaykin, D. 2001. Genetic Data Analysis: Computer program for the analysis of allelic data. Version 1.0 (d16c). http://lewis.eeb.uconn.edu/lewishome/software.html.
- Lopez, B., Aguilar, D., Orozco, H., Burger, M., Espitia, C., Ritacco, V., Barrera, L., Kremer, K., Hernandez-Pando, R., Huygen, K., et al. 2003. A marked difference in pathogenesis and immune response induced by different Mycobacterium tuberculosis genotypes. Clin. Exp. Immunol. 133: 30-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill, W.H. 1976. Plagues and peoples. Anchor Press. New York.
- Milan, S.J., Hauge, K.A., Kurepina, N.E., Lofy, K.H., Goldberg, S.V., Narita, M., Nolan, C.M., McElroy, P.D., Kreiswirth, B.N., and Cangelosi, G.A. 2004. Expanded geographical distribution of the N family of Mycobacterium tuberculosis strains within the United States. J. Clin. Microbiol. 42: 1064-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithen, S. 2004. After the ice: A global human history 20,000-5000 BC. Phoenix, London.
- Mokrousov, I., Narvskaya, O., Otten, T., Vyazovaya, A., Limeschenko, E., Steklova, L., and Vyshnevskyi, B. 2002. Phylogenetic reconstruction within Mycobacterium tuberculosis Beijing genotype in northwestern Russia. Res. Microbiol. 153: 629-637. [DOI] [PubMed] [Google Scholar]
- Mokrousov, I., Narvskaya, O., Limeschenko, E., Vyazovaya, A., Otten, T., and Vyshnevskyi, B. 2004. Analysis of the allelic diversity of the mycobacterial interspersed repetitive units in Mycobacterium tuberculosis strains of the Beijing family: Practical implications and evolutionary considerations. J. Clin. Microbiol. 42: 2438-2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan, C.J., Hunley, K., Cole, S., and Long, J.C. Population genetics, history, and health patterns in Native Americans. Annu. Rev. Genomics Hum. Genet. 5: 295-315. [DOI] [PubMed]
- Pavesi, A. 2004. Detecting traces of prehistoric human migrations by geographic synthetic maps of polyomavirus JC. J. Mol. Evol. 58: 304-313. [DOI] [PubMed] [Google Scholar]
- Plikaytis, B.B., Marden, J.L., Crawford, J.T., Woodley, C.L., Butler, W.R., and Shinnick, T.M. 1994. Multiplex PCR assay specific for the multi-drug-resistant strain W of Mycobacterium tuberculosis. J. Clin. Microbiol. 32: 1542-1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rad, M.E., Bifani, P., Martin, C., Kremer, K., Samper, S., Rauzier, J., Kreiswirth, B., Blazquez, J., Jouan, M., van Soolingen, D., et al. 2003. Mutations in putative mutator genes of Mycobacterium tuberculosis strains of the W-Beijing family. Emerg. Infect. Dis. 9: 838-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelach, G. 2000. The earliest Neolithic cultures of northeast Asia: Recent discoveries and new perspectives on the beginning of agriculture. J. World Prehistory 14: 363-413. [Google Scholar]
- Sola, C., Filliol, I., Legrand, E., Mokrousov, I., and Rastogi, N. 2001. Mycobacterium tuberculosis phylogeny reconstruction based on combined numerical analysis with IS1081, IS6110, VNTR, and DR-based spoligotyping suggests the existence of two new phylogeographical clades. J. Mol. Evol. 53: 680-689. [DOI] [PubMed] [Google Scholar]
- Su, B., Xiao, C., Deka, R., Seielstad, M.T., Kangwanpong, D., Xiao, J., Lu, D., Underhill, P., Cavalli-Sforza, L., Chakraborty, R., et al. 2000. Y chromosome haplotypes reveal prehistorical migrations to the Himalayas. Hum. Genet. 107: 582-590. [DOI] [PubMed] [Google Scholar]
- Sun, Y.J., Bellamy, R., Lee, A.S., Ng, S.T., Ravindran, S., Wong, S.Y., Locht, C., Supply, P., and Paton, N.I. 2004. Use of mycobacterial interspersed repetitive unit-variable-number tandem repeat typing to examine genetic diversity of Mycobacterium tuberculosis in Singapore. J. Clin. Microbiol. 42: 1986-1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply, P., Mazars, E., Lesjean, S., Vincent, V., Gicquel, B., and Locht, C. 2000. Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol. Microbiol. 36: 762-771. [DOI] [PubMed] [Google Scholar]
- Supply, P., Lesjean, S., Savine, E., Kremer, K., van Soolingen, D., and Locht, C. 2001. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J. Clin. Microbiol. 39: 3563-3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply, P., Warren, R.M., Banuls, A.-L., Lesjean, S., van der Spuy, G.D., Lewis, L.A., Tibayrenc, M., Van Helden, P.D., and Locht, C. 2003. Linkage disequilibrium between minisatellite loci supports clonal evolution of Mycobacterium tuberculosis in a high tuberculosis incidence area. Mol. Microbiol. 47: 529-538. [DOI] [PubMed] [Google Scholar]
- Thomas, B.E. 2000. Tuberculosis in women. In Status of tuberculosis in India (eds. R. Nayak et al.), pp. 25-32. Society for Innovation and Development, Indian Institute of Science, Bangalore, India.
- Underhill, P.A., Passarino, G., Lin, A.A., Shen, P., Mirazon Lahr, M., Foley, R.A., Oefner, P.J., and Cavalli-Sforza, L.L. 2001. The phylogeography of Y chromosome binary haplotypes and the origins of modern human populations. Ann. Hum. Genet. 65: 43-62. [DOI] [PubMed] [Google Scholar]
- Uplekar, M.W., Rangan, S., Weiss, M.G., Ogden, J., Borgdorff, M.W., and Hudelson, P. 2001. Attention to gender issues in tuberculosis control. Int. J. Tuberc. Lung Dis. 5: 220-224. [PubMed] [Google Scholar]
- van Embden, J.D.A., Cave, M.D., Crawford, J.T., Dale, J.W., Eisenach, K.D., Gicquel, B., Hermans, P., Martin, C., McAdam, R., Shinnik, T.M., et al. 1993. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: Recommendations for a standardized methodology. J. Clin. Microbiol. 31: 406-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Embden, J.D.A., van Gorkom, T., Kremer, K., Jansen, T., van der Zeijst, B.A.M., and Schouls, L.M. 2000. Genetic variation and evolutionary origin of the direct repeat locus of Mycobacterium tuberculosis complex bacteria. J. Bacteriol. 182: 2393-2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden, P.D., Warren, R.M., Victor, T.C., van der Spuy, G., Richardson, M., and Hoal-van Helden, E. 2002. Strain families of Mycobacterium tuberculosis. Trends Microbiol. 10: 167-168. [DOI] [PubMed] [Google Scholar]
- van Soolingen, D., Qian, L., de Haas, P.E.W., Douglas, J.T., Traore, H., Portaels, F., Quing, H.Z., Enkhasaikan, D., Nymadawa, P., and van Embden, J.D. 1995. Predominance of a single genotype of Mycobacterium tuberculosis in countries of east Asia. J. Clin. Microbiol. 33: 3234-3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. 1990. Genetic data analysis: Methods for discrete population data. Sinauer Associates, Sunderland, MA. [DOI] [PubMed]
- Y Chromosome Consortium. 2002. A nomenclature system for the tree of human Y-chromosomal binary haplogroups. Genome Res. 12: 339-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M., Cong, J., Yang, Z., Samten, B., and Barnes, P.F. 1999. Enhanced capacity of a widespread strain of Mycobacterium tuberculosis to grow in human macrophages. J. Infect. Dis. 179: 1213-1217. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.nationmaster.com/encyclopedia/South-Africa; Encyclopedia: South Africa
- http://61.1911encyclopedia.org/S/SO/SOUTH_AFRICA.htm; South Africa
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.