

Abstract
Mammalian genomes contain many highly conserved nongenic sequences (CNGs) whose functional significance is poorly understood. Sets of CNGs have previously been identified by selecting the most conserved elements from a chromosome or genome, but in these highly selected samples, conservation may be unrelated to purifying selection. Furthermore, conservation of CNGs may be caused by mutation rate variation rather than selective constraints. To account for the effect of selective sampling, we have examined conservation of CNGs in taxa whose evolution is largely independent of the taxa from which the CNGs were initially identified, and we have controlled for mutation rate variation in the genome. We show that selective constraints in CNGs and their flanks are about one-half as strong in hominids as in murids, implying that hominids have accumulated many slightly deleterious mutations in functionally important nongenic regions. This is likely to be a consequence of the low effective population size of hominids leading to a reduced effectiveness of selection. We estimate that there are one and two times as many conserved nucleotides in CNGs as in known protein-coding genes of hominids and murids, respectively. Polymorphism frequencies in CNGs indicate that purifying selection operates in these sequences. During hominid evolution, we estimate that a total of about three deleterious mutations in CNGs and protein-coding genes have been selectively eliminated per diploid genome each generation, implying that deleterious mutations are eliminated from populations non-independently and that sex is necessary for long-term population persistence.
Comparisons between vertebrate genomes have revealed abundant conserved nongenic sequences (CNGs) showing levels of conservation far above the average for the genome (Frazer et al. 2001; Dermitzakis et al. 2002, 2003, 2004; Margulies et al. 2003; Thomas et al. 2003; Bejerano et al. 2004), and the mean level of conservation frequently exceeds that for vertebrate protein-coding sequences (Dermitzakis et al. 2003; Bejerano et al. 2004). Their status as functional elements has been supported by analysis often revealing strong conservation deep into the vertebrate phylogeny (Dermitzakis et al. 2003, 2004; Thomas et al. 2003; International Chicken Genome Sequencing Consortium 2004). There is evidence that some CNGs are enriched for long-range enhancer sequences (Nobrega et al. 2003), and for them being important in the control of genes involved in vertebrate development (Woolfe et al. 2005). If CNGs are selectively maintained, this would imply that populations suffer a mutation load due to the genetic deaths of individuals carrying deleterious mutations in these regions.
However, quantification of the true extent of CNG functional constraint is difficult for several reasons. First, because data sets of CNGs have been compiled by selecting the most conserved DNA segments between distantly related species from a very large population of possible segments (e.g., all segments exceeding a threshold conservation level from the whole genome or from one chromosome), conservation may be unrelated to selective constraints. Second, variability in the mutation rate (Casane et al. 1997; Matassi et al. 1999; Smith et al. 2002; Keightley et al. 2005) favors the selection of segments having unusually low mutation rates. Third, there is a potentially serious problem with the alignment of noncoding sequences from distantly related taxa: Only noncoding segments that have experienced few insertion-deletion events (indels) can be reliably aligned, so these will tend to be overrepresented. However, there is a positive covariance between indel and nucleotide substitution rates in vertebrates (Hardison et al. 2003), thus alignable segments tend to be sampled from regions having low nucleotide mutation rates. One approach to confirm conservation is to examine orthologous segments of more distantly related species than the species from which the CNGs were identified (Dermitzakis et al. 2003, 2004; Thomas et al. 2003). This has revealed conservation deep into the vertebrate phylogeny, but the number of conserved elements tends to drop as the comparator taxa become more distantly related (Dermitzakis et al. 2003), and the fraction of sequences defined as being conserved is arbitrary. It is also possible to attempt to reconstruct the null distribution of conservation level by measuring the divergence between putatively neutrally evolving sequences, such as transposable elements (Mouse Genome Sequencing Consortium 2002). However, this does not necessarily account for regional variability in the mutation rate.
Here, we have estimated the fraction of conserved nucleotides in a set of CNGs that were originally identified in a comparison of the sequence of the long arm of human Chromosome 21 and the mouse genome (Dermitzakis et al. 2002, 2003). These sequences belong to a class of elements having low transcription potential (Dermitzakis et al. 2002, 2003). In our analysis, we control for mutation rate variation by comparing nucleotide substitution rates in CNGs with substitution rates in sequences flanking CNGs. We use multiple species alignments to attempt to obtain less biased estimates of selective constraints, as advocated by Margulies et al. (2003). Selective constraint is the estimated fraction of mutations that have been removed by natural selection. We analyze sequence data from two pairs of species (human-chimp and rat-mouse) for which evolution under a neutral model can be expected to be largely independent from human-mouse divergence. In the lineages leading to our ingroup species (either chimpanzee or rat), evolution is independent from evolution in the human and mouse lineages under a neutral model. Furthermore, evolution from the human-chimp (mouse-rat) common ancestor to human (mouse) is also nearly independent from human-mouse under a neutral model because this lineage is short relative to that of human-mouse.
Results
We compiled data sets of human-chimp and mouse-rat orthologs along with their flanking sequences, and estimated sequence divergences and levels of selective constraints.
Rates of evolution of CNGs and their flanking regions
We estimated evolutionary rates for human-chimpanzee and mouse-rat CNGs and flanking regions by the method of Tamura and Nei (1993). Rates of evolution are substantially slower in CNGs than their flanking regions in both murids and hominids (Table 1). In the flanking regions, there is also a noticeable reduction in evolutionary rate close to both the 5′- and 3′-ends of CNGs (Fig. 1). This indicates that conserved regions extend somewhat beyond the CNGs originally identified by Dermitzakis et al. (2002). Beyond ∼2 kb on either side of CNGs, evolutionary divergences show essentially no change for distances up to 10 kb from CNGs. For subsequent analysis of constraint, we therefore chose to use as neutrally evolving reference sequences the regions flanking CNGs from 3-10 kb on either side.
Table 1.
Numbers of nucleotide differences and nucleotide divergences in CNGs and their flanking DNA segments of up to 10 kb at non-CpG-prone sites
| Species pair | DNA category | Sites | Differences | Proportion of differences (SE) | Evolutionary rate (SE) |
|---|---|---|---|---|---|
| Human-chimp | CNG | 215,888 | 1859 | 0.00862 (0.00021) | 0.00866 (0.00022) |
| CNG 5′-flank | 5,350,833 | 64,995 | 0.0121 (0.00010) | 0.0123 (0.00009) | |
| CNG 3′-flank | 5,269,495 | 63,740 | 0.0121 (0.00012) | 0.0122 (0.00010) | |
| Mouse-rat | CNG | 188,225 | 13,342 | 0.0709 (0.00098) | 0.0747 (0.00108) |
| CNG 5′-flank | 2,446,742 | 361,742 | 0.148 (0.00057) | 0.167 (0.00074) | |
| CNG 3′-flank | 2,341,277 | 345,408 | 0.148 (0.00047) | 0.167 (0.00076) |
Figure 1.

Evolutionary rate calculated by the Tamura-Nei method in sequences flanking CNGs in (A) hominids and (B) murids. 95% confidence limits are shown in gray.
Selective constraints in CNGs
We estimated constraint as a function of numbers of substitutions observed in a sequence segment and the number expected if the sequence had the same mutation rate as a linked sequence that was assumed to be evolving neutrally. Sequence divergence between human and chimp and between mouse and rat (Table 1) and estimates of evolutionary constraints for CNGs (Table 2) suggest that CNGs are quite strongly selectively constrained, that is, CNGs evolve 29% and 53% slower than their 3-10-kb upstream and downstream flanking sequences in hominids and murids, respectively. Notably, the constraint estimate for murine CNGs is about twice as high as for hominids. In both hominids and murids, levels of constraints in CNGs are therefore lower than levels previously reported for nondegenerate sites of protein-coding genes (typically of the order of 0.8) (see, e.g., Eyre-Walker et al. 2002) and higher than for sequences within 1-2 kb of the start or stop codon, in which regulatory elements are believed to be concentrated (Mouse Genome Sequencing Consortium 2002).
Table 2.
Estimates of evolutionary constraint in CNGs and their 3′ and 5′ 1000-base flanking sequences in hominids and murids
| Species comparison | Sequence category | Constraint (SE) |
|---|---|---|
| Human-chimp | CNGS | 0.293 (0.020) |
| 5′- and 3′-flanks | 0.0502 (0.011) | |
| Mouse-rat | CNGs | 0.529 (0.0081) |
| 5′ and 3′-flanks | 0.0970 (0.008) |
Constraint is estimated from 1 - ΣO/ΣE, where O is the observed number of substitutions in a sequence and E is the number expected based on the number of substitutions in flanking sequences 3-10 kb upstream and downstream from each CNG. These flanking sequences exclude other CNGs and their flanks, and coding sequences. The summations are over CNGs. See Methods for details.
Selective constraints in sequences immediately flanking CNGs
To infer levels of constraints in regions flanking CNGs, we computed mean evolutionary constraint in 500-bp blocks upstream and downstream from CNGs using the sequences 3-10 kb upstream and downstream of CNGs as the neutrally evolving standard. To maintain independence among loci, we analyzed data up to the midpoint between adjacent CNGs. Like the CNGs themselves, mean constraint in flanking sequences is substantially higher in murids than in hominids (Fig. 2). In both taxa mean constraint drops to values close to zero by ∼2 kb upstream and downstream from CNGs. In the 1-kb flanks, in which constraint is significant in both taxa, the total numbers of constrained nucleotides in flanking sequences are 162 and 76 in murids and hominids, respectively. These figures are approximately two times higher than the numbers of constrained nucleotides in CNGs themselves.
Figure 2.

Constraint in sequences flanking CNGs in (A) hominids and (B) murids. 95% confidence limits are show in gray.
Pattern of polymorphism in CNGs
These results are unlikely to be due to local fluctuations in the mutation rate. Empirical data suggest that domains of local similarity of mutation rates extend over megabase scales (Lercher et al. 2001, 2004; Webster et al. 2003; Keightley et al. 2005), and, thus, substantial changes in mutation rate over the kilobase scales examined in this study seem unlikely. In addition, currently we know of no mechanism that could produce mutational “cold spots” sufficiently extreme as to fully explain the pattern we observe. Such mechanisms would also have to operate independently of, and have little impact on, sequence composition, since the CNGs that have been analyzed consist of unique sequences.
To further investigate whether conservation of CNGs is due to purifying selection or low regional mutation rate, we analyzed human polymorphisms in CNGs. Purifying selection operating on CNGs should increase the proportion of rare polymorphic variants (variants with low allele frequencies) compared to neutrally evolving regions, whereas local fluctuations in mutation rate would not be expected to affect the allele frequency spectrum. We compared allele frequency distributions for human SNPs in CNGs and SNPs in putatively neutrally evolving genomic regions, and determined the ancestral and derived alleles based on orthologous nucleotides in the chimpanzee genome sequence. We used a random set of SNPs identified by the Perlegen data set in 71 Americans of European, African, and Asian ancestry (Hinds et al. 2005). The fraction of SNPs having derived allele frequencies <20% was higher in CNGs compared to nontranscribed, nonrepetitive sequences of Chromosome 21 in all three populations (Table 3). Furthermore, SNP density in CNGs in non-CpG-prone sites is ∼20% lower than in flanking regions. This reduction is smaller than the corresponding reduction in human-chimpanzee divergence rate, which would not be expected if conservation is solely due to low mutation rate, and is therefore also indicative of selection against deleterious alleles.
Table 3A.
Comparison of density and allele frequency spectra for SNPs within CNGs and within putatively neutrally evolving regions
|
SNPs in CNGs
|
SNPs in neutral genomic regions
|
||||||
|---|---|---|---|---|---|---|---|
| Population | No. with derived allele frequency <20% | No. with derived allele frequency ≥20% | Percentage with derived allele frequency <20% | No. with derived allele frequency <20% | No. with derived allele frequency ≥20% | Percentage with derived allele frequency <20% | p-value |
| European-American | 85 | 152 | 36.00 | 2903 | 6897 | 30.00 | 0.02 |
| African-American | 119 | 150 | 44.00 | 3895 | 6748 | 37.00 | 0.007 |
| Han Chinese | 74 | 145 | 34.00 | 2650 | 6495 | 29.00 | 0.07 |
Table 3B.
Using random Perlegen Class A SNPs
| Genomic regions | Total number of non-CpG-prone sites in reliable alignment | Number of SNPs | Dsnp, SNP density per nucleotide | Dsnp/Dsnpo |
|---|---|---|---|---|
| “Neutral” | 7,194,108 | 8081 | 0.00112 | 1 (by definition) |
| CNG | 211,204 | 195 | 0.00092 | 0.82 |
Discussion
We have found that mean evolutionary constraint in CNGs and their flanking sequences is ∼50% lower in hominids than murids. This is consistent with similar observations for protein-coding genes and noncoding sequences close to genes (Eyre-Walker et al. 2002; Keightley et al. 2005). We previously examined several factors that might explain the lower levels of constraints in hominids (Keightley et al. 2005). Rates of sequence error between human and chimp, estimated for data compiled by our methods are ∼10-3, and this would have only a small impact on estimated levels of constraints (Keightley et al. 2005). The mouse-rat divergence is considerably greater than that of human-chimp, but this would not in itself lead to differences in constraints unless the sequences have evolved different functions. A high proportion of selectively driven substitutions in hominids would also lead to reduced constraints, but does not seem to explain the low level of conservation in hominid noncoding DNA close to genes, since the proportion of adaptive substitutions, inferred by comparing human polymorphism levels with human-chimp nucleotide divergence, is small. More plausibly, the lower long-term effective population size (Ne) of hominids could lead to the accumulation of mildly deleterious mutations with selection coefficients in the range 1/Ne (murids) to 1/Ne (hominids), and therefore reduced levels of sequence conservation. If this is the correct explanation for the lower levels of constraint in hominids, many mutations in CNGs have selection coefficients smaller than 10-4, since 1/Ne for hominids is of that order (Rannala and Yang 2003), and we estimate that ∼2 × 107 slightly deleterious mutations in CNGs have become fixed in the human and chimpanzee lineages since they diverged from their common ancestor. Presumably, these have been compensated for by adaptive substitutions, and/or any absolute fitness declines have not been relevant for the evolutionary fates of the species. In both taxa there is likely to be a mixture of constraint and unconstrained nucleotides in CNGs and their flanks.
The conservation of CNGs and their flanking sequences imply that populations bear a mutation load due to selective elimination of deleterious mutations. In addition, there is a mutation load associated with more weakly conserved sequences, not included in the set of CNGs analyzed here. Numbers of constrained nucleotides in CNGs, estimated for the long arm of Chromosome 21 and extrapolated to the whole genome, are shown in Table 4. Locations of CNGs are negatively correlated with genes, that is, CNGs are concentrated on human Chromosome 21 in the AT-rich gene-poor proximal region (Dermitzakis et al. 2002). We therefore regressed numbers of CNGs in 1-Mb blocks on Chromosome 21 on the numbers of coding sequences in each block (Fig. 3), and used the slope and intercept to predict the frequency of CNGs for the average frequency of coding sequences in the genome, under the assumption that there are 24,000 coding sequences and that the genome is 3068 Mb (build 35 of human genome). This yielded a predicted genomic number of CNGs of 202,000 (the uncorrected estimate is only slightly smaller at 198,000). How does the predicted number of constrained nucleotides in CNGs compare to the number in protein-coding genes? By assuming estimates of mean constraint at nondegenerate sites of genes are 0.69 and 0.84 for hominids and murids, respectively (Eyre-Walker et al. 2002), we conclude that numbers of constrained nucleotides associated with CNGs are one and two times higher than in coding sequences in hominids and murids, respectively (Table 4). Some constrained nucleotides in CNGs may be associated with unannotated protein-coding gene sequences, although the fraction of these is likely to be small since the CNGs analyzed here do not have properties of exonic sequences and have low transcriptional potential (Dermitzakis et al. 2002, 2003). Assuming generation intervals and evolutionary divergence times for hominids and murids from Keightley and Eyre-Walker (2000), estimates of the genome-wide deleterious mutation rate per diploid (U) in protein-coding loci are 1.3 in hominids and 0.15 in murids. If deleterious mutations in CNGs are added to these figures, estimates for U become 2.9 in hominids and 0.47 in murids. Under a multiplicative model, these U values imply that 94% and 37% of individuals would undergo genetic death as a consequence of selective elimination of deleterious mutations (Kondrashov 1988). For hominids, this predicted mutation load seems to be too high for a species with such a low reproductive rate, and implies that deleterious mutations are nonindependently removed by selection, and that sex is necessary for long-term population persistence (Kondrashov 1988).
Table 4.
Estimates of numbers of constrained bases per CNG and extrapolation of the number of constrained bases to the whole genome
|
Constrained bases per chromosome or genome
|
||||||
|---|---|---|---|---|---|---|
|
Constrained bases per CNG
|
||||||
| Taxon | In CNGs | In flanksa | Total | CNGs, Chromosome 21b | CNGs, genomec | Coding, genomed |
| Hominids | 45 | 76 | 121 | 2.7 × 105 | 2.4 × 107 | 1.9 × 107 |
| Murids | 81 | 162 | 243 | 4.9 × 105 | 4.9 × 107 | 2.3 × 107 |
The number of bases is calculated by summing over the contributions from the 1000-base flanking segments 5′ and 3′ of each CNG according to No. of constrained bases = ΣCifi, where Ci is the average constraint and fi is the average fraction of the I bases in the 1000-base segment, averaged over the set of CNG loci
The set of 2262 CNGs on the long arm of human Chromosome 21
Based on predicted numbers of CNGs from regression of CNG density on gene density for Chromosome 21. See text for details
Assumes that there are 24,000 mammalian protein-coding genes of average length 1500 bases, that three-quarters of nucleotide substitutions in a gene lead to an amino acid substitution, and that constraint levels at amino acid sites are 0.69 and 0.84 for hominids and murids, respectively
Figure 3.

Relationship between CNG density and coding sequence (CDS) density on the long arm of Chromosome 21, and the linear regression line.
Methods
Compilation of sequence data
The set of 2262 conserved human-mouse nongenic segments described by Dermitzakis et al. (2002) was kindly provided by E. Dermitzakis. Essentially, the data set comprises the most conserved segments of ≥100 bp from 35 Mb of the long arm of human Chromosome 21 that do not have obvious properties of protein-coding genes. The average length of the human CNGs is 153 bases (standard deviation 57 bases). We identified the human CNGs in Chromosome 21 build 35 of the human genome, and extracted 10 kb of flanking sequence on each side. Chimpanzee sequences orthologous to the human CNGs and their flanking sequences were identified by reciprocal best-hits BLAST against the draft chimpanzee genome, and alignments were filtered for obviously nonhomologous segments as described previously (Keightley et al. 2005). We used BLAST to identify the contigs containing each mouse CNG in build 33 of the mouse genome assembly, and extracted 20 kb of 5′- and 3′-flanking sequences. We used reciprocal best-hits BLAST to identify the rat contigs containing the orthologs of the mouse CNGs in build 2 of the rat genome assembly, and also extracted 10 kb of rat sequence flanking each CNG. The mouse-rat flanking DNA sequences were aligned initially by MAVID (Bray and Pachter 2004); then alignments were refined using MCALIGN under a model of indel evolution appropriate to murine noncoding DNA (Keightley and Johnson 2004) in segments of ∼500 bp. The resulting alignments were filtered for obviously nonhomologous segments in two ways. Regions in which each of 30 or more consecutive windows showed a mean divergence >30% were masked. In addition, regions that contained short aligned blocks (<20 bp) surrounded by multiple large gaps (>40 bp) were considered unlikely to be truly orthologous and were also masked off. Annotated hominid and murid coding sequences were excluded from any analysis. Microsatellite loci were masked from all alignments prior to further analysis.
Data on human polymorphism positions and frequencies were extracted from NCBI dbSNP build 123. Only random SNPs identified by Perlegen (class A SNPs according to their terminology) were used in the analysis. Ancestral alleles of human polymorphisms were determined from human/chimpanzee pairwise genomic alignments obtained from the UCSC Genome Browser. The overall spectrum does not match theoretical expectation because it has a slightly lower proportion of low-frequency SNPs than expected. However, comparison of SNP frequencies in different functional categories should be robust with respect to the overall shape of the distribution.
Estimation of selective constraint
We estimated selective constraints in CNGs and their immediate flanking sequence segments. In order to maintain independence between loci, the flanking sequences associated with a CNG were defined as those nucleotides up to the midpoint between the proximal or distal CNG on the chromosome. To estimate selective constraint (C), we used the method of Halligan et al. (2004), which compares the number of substitutions observed in the sequence segment (O) with the number expected (E) if the segment evolved at the same rate as a closely linked putatively neutrally evolving sequence (the neutral standard). We chose to use the concatenated 5′- and 3′-flanking sequences 3-10 kb on either side of each CNG as the neutral standard; if both such 5′- and 3′-flanks were unusable because they were beyond the midpoint of the adjacent CNGs, we used flanking data from an adjacent CNG. In calculating E, we used a model of sequence evolution that assumes an equilibrium GC content of 0.4 (for details, see Halligan et al. 2004). The level of constraint for a CNG or its immediate flanking sequence is C = 1 - O/E, and an estimate of mean constraint for the complete data set of n CNGs is obtained from
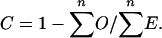 |
It should be noted that our estimates of constraint are conservative if our neutral standard is under a low level of selective constraint. We estimated selective constraint for complete CNGs and for non-overlapping flanking sequence blocks, typically of 500 bp upstream and downstream from CNGs. Standard errors of C were obtained by bootstrapping over CNG loci.
CpG dinucleotide sites are hypermutable in vertebrates, and ascertainment bias leading to apparently higher levels of conservation in CNGs could be partly caused by variation in the frequency of these sites. Furthermore, CpG dinucleotide sites are saturated between mouse and rat, and estimates of sequence divergence are unreliable. In the analysis, we therefore excluded sites that are likely to be part of a CpG dinucleotide by excluding all sites preceded by C or followed by G in CNGs and flanks.
Estimation of the genome-wide number of conserved nucleotides in CNGs
We extrapolated the number of CNGs on Chromosome 21 to the whole genome according to their relative numbers of base pairs. We corrected this number for the relative excess of CNGs on Chromosome 21 due to its AT richness, although this made little difference. The overall number of constrained nucleotides is (the number of CNGs) × (the average length of a CNG) × (the average constraint in a CNG).
Acknowledgments
We are grateful to the genome sequencing centers for the genome sequences used in our analysis and to Manolis Dermitzakis for providing a database of human-mouse CNGs. We thank Adam Eyre-Walker for helpful advice and Alexey Kondrashov and two anonymous reviewers for insightful comments.
[The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: E. Dermitzakis.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3942005.
References
- Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W.J., Mattick, J.S. and Haussler, D. 2004. Ultraconserved elements in the human genome. Science 304: 1321-1325. [DOI] [PubMed] [Google Scholar]
- Bray, N. and Pachter, L. 2004. MAVID: Constrained ancestral alignment of multiple sequences. Genome Res. 14: 693-699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casane, D., Boissinot, S., Chang, B.H.J., Shimmin, L.C., and Li, W.-H. 1997. Mutation pattern variation among regions of the primate genome. J. Mol. Evol. 45: 216-226. [DOI] [PubMed] [Google Scholar]
- Dermitzakis, E.T., Reymond, A., Lyle, R., Scamuffa, N., Ucla, C., Deutsch, S., Stevenson, B.J., Flegel, V., Bucher, P., Jongeneel, C.V., et al. 2002. Numerous potentially functional but non-genic conserved sequences on human chromosome 21. Nature 420: 578-582. [DOI] [PubMed] [Google Scholar]
- Dermitzakis, E.T., Reymond, A., Scamuffa, N., Ucla, C., Kirkness, E., Rossier, C., and Antonarakis, S.E. 2003. Evolutionary discrimination of mammalian conserved non-genic sequences (CNGs). Science 302: 1033-1035. [DOI] [PubMed] [Google Scholar]
- Dermitzakis, E.T., Kirkness, E., Schwarz, S., Birney, E., Reymond, A., and Antonarakis, S.E. 2004. Comparison of human chromosome 21 conserved nongenic sequences (CNGs) with the mouse and dog genomes shows that their selective constraint is independent of their genic environment. Genome Res. 14: 852-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker, A., Keightley, P.D., Smith, N.G.C., and Gaffney, D. 2002. Quantifying the slightly deleterious model of molecular evolution. Mol. Biol. Evol. 19: 2142-2149. [DOI] [PubMed] [Google Scholar]
- Frazer, K.A., Sheehan, J.B., Stokowski, R.P., Chen, X.Y., Hosseini, R., Cheng, J.F., Fodor, S.P.A., Cox, D.R., and Patil, N. 2001. Evolutionarily conserved sequences on human Chromosome 21. Genome Res. 11: 1651-1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney, D.J. and Keightley, P.D. 2005. The scale of mutational variability in the murid genome. Genome Res. 5: 1086-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan, D.L., Eyre-Walker, A., Andolfatto, P., and Keightley, P.D. 2004. Patterns of evolutionary constraints in intronic and intergenic DNA of Drosophila. Genome Res. 14: 273-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison, R.C., Roskin, K.M., Yang, S., Diekhans, M., Kent, W.J., Weber, R., Elnitski, L., Li, J., O'Connor, M., Kolbe, D., et al. 2003. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 13: 13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds, D.A., Stuve, L.L., Nilsen, G.B., Halperin, E., Eskin, E., Ballinger, D.G., Frazer, K.A., and Cox, D.R. 2005. Whole-genome patterns of common DNA variation in three human populations. Science 307: 1072-1079. [DOI] [PubMed] [Google Scholar]
- International Chicken Genome Sequencing Consortium. 2004. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432: 695-716. [DOI] [PubMed] [Google Scholar]
- Keightley, P.D. and Eyre-Walker, A. 2000. Deleterious mutations and the evolution of sex. Science 290: 331-333. [DOI] [PubMed] [Google Scholar]
- Keightley, P.D. and Johnson, T. 2004. MCALIGN: Stochastic alignment of noncoding DNA sequences based on an evolutionary model of sequence evolution. Genome Res. 14: 442-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P.D., Lercher, M.J., and Eyre-Walker, A. 2005. Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biol. 3: 282-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov, A.S. 1988. Deleterious mutation and the evolution of sexual reproduction. Nature 336: 435-440. [DOI] [PubMed] [Google Scholar]
- Lercher, M.J., Williams, E.J.B., and Hurst, L.D. 2001. Local similarity in evolutionary rates extends over whole chromosomes in human-rodent and mouse-rat comparisons: Implications for understanding the mechanistic basis of the male mutation bias. Mol. Biol. Evol. 18: 2032-2039. [DOI] [PubMed] [Google Scholar]
- Lercher, M.J., Chamary, J.V., and Hurst, L.D. 2004. Genomic regionality in rates of evolution is not explained by clustering of genes of comparable expression profile. Genome Res. 14: 1002-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies, E.H., Blanchette, M., Haussler, D., and Green, E.D. 2003. Identification and characterization of multi-species conserved sequences. Genome Res. 13: 2507-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matassi, G., Sharp, P.M., and Gautier, C. 1999. Chromosomal location effects on gene sequence evolution in mammals. Curr. Biol. 9: 786-791. [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
- Nobrega, M.A., Ovcharenko, I., Afzal, V., and Rubin, E.M. 2003. Scanning human gene deserts for long range enhancers. Science 302: 413. [DOI] [PubMed] [Google Scholar]
- Rannala, B. and Yang, Z. 2003. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164: 1645-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, N.G.C., Webster, M.T., and Ellegren, H. 2002. Deterministic mutation rate variation in the human genome. Genome Res. 12: 1350-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. and Nei, M. 1993. Estimation of the number of substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10: 512-526. [DOI] [PubMed] [Google Scholar]
- Thomas, J.W., Touchman, J.W., Blakesley, R.W., Bouffard, G.G., Beckstrom-Sternberg, S.M., Margulies, E.H., Blanchette, M., Siepel, A.C., Thomas, P.J., McDowell, J.C., et al. 2003. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424: 788-793. [DOI] [PubMed] [Google Scholar]
- Webster, M.T., Smith, N.G.C., and Ellegren, H. 2003. Compositional evolution of noncoding DNA in the human and chimpanzee genomes. Mol. Biol. Evol. 20: 278-286. [DOI] [PubMed] [Google Scholar]
- Woolfe, A., Goodson, M., Goode, D.K., Snell, P., McEwen, G.K., Vavouri, T., Smith, S.F., North, P., Callaway, H., Kelly, K., et al. 2005. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 3: 116-130. [DOI] [PMC free article] [PubMed] [Google Scholar]