

Abstract
The roles of duplicate genes and their contribution to the phenomenon of enzyme dispensability are a central issue in molecular and genome evolution. A comprehensive classification of the mechanisms that may have led to their preservation, however, is currently lacking. In a systems biology approach, we classify here back-up, regulatory, and gene dosage functions for the 105 duplicate gene families of Saccharomyces cerevisiae metabolism. The key tool was the reconciled genome-scale metabolic model iLL672, which was based on the older iFF708. Computational predictions of all metabolic gene knockouts were validated with the experimentally determined phenotypes of the entire singleton yeast library of 4658 mutants under five environmental conditions. iLL672 correctly identified 96%-98% and 73%-80% of the viable and lethal singleton phenotypes, respectively. Functional roles for each duplicate family were identified by integrating the iLL672-predicted in silico duplicate knockout phenotypes, genome-scale carbon-flux distributions, singleton mutant phenotypes, and network topology analysis. The results provide no evidence for a particular dominant function that maintains duplicate genes in the genome. In particular, the back-up function is not favored by evolutionary selection because duplicates do not occur more frequently in essential reactions than singleton genes. Instead of a prevailing role, multigene-encoded enzymes cover different functions. Thus, at least for metabolism, persistence of the paralog fraction in the genome can be better explained with an array of different, often overlapping functional roles.
The genome of the yeast Saccharomyces cerevisiae encodes ∼1500 so-called duplicate genes that exist in multiple copies (Gu et al. 2003), about 496 of which resulted from an ancient genome duplication (Dietrich et al. 2004; Kellis et al. 2004). Their role in compensating knockout mutations—often referred to as genetic network robustness—has been recognized (Pal 2001; Gu 2003; Gu et al. 2003; Blank et al. 2005), although others favor alternative pathways as the main reason that a substantial fraction of gene deletions do not yield a significant phenotype (Wagner 2000). This redundancy-robustness connection or back-up function, however, is not the evolutionary driving force that retains both gene copies. The reigning paradigm on the fate of duplicates predicts that one of the duplicates is either lost or gains a new function. Return to the single-copy state is then prevented by specialization in function, expression, and localization (neo- and subfunctionalization) (Ohno 1970; Kellis et al. 2004; Zhang and Kishino 2004; Presgraves 2005) or increased gene dosage to boost activity of key reactions (Seoighe and Wolfe 1999). More specifically, it has been suggested that many duplicates from the genome duplication played a direct role in the adaptation of S. cerevisiae toward fermentation, and thus were largely selected for in the domestication of yeast (Wolfe 2004).
Since duplicates are highly enriched in S. cerevisiae metabolism (105 duplicate gene families with 295 members) (Conant and Wagner 2002; Kellis et al. 2004), this subgroup has attracted particular attention. Presently, gene dosage function (Papp et al. 2004) or differential regulation of reactions (Ihmels et al. 2004) is advocated as the primary function of metabolic duplicates that prevent their counterselection in yeast. However, a comprehensive classification of duplicates based on the mechanism that may have led to their conservation is missing, because genome-scale experimental analysis would require a presently unavailable multiple knockout library of entire duplicate gene families. Typically, duplicate gene functions are assessed indirectly through genome-wide comparative sequence analysis (Lynch and Katju 2004) or transcriptional profiling (Ihmels et al. 2004; Kafri et al. 2005). In contrast to other cellular processes, however, metabolism-wide functions of duplicate genes are more directly tractable owing to the available single knockout library (Giaever et al. 2002), genome-scale models of metabolism (Förster et al. 2003a; Duarte et al. 2004; Price et al. 2004), and methods for quantitative fluxome analysis (Blank et al. 2005; Fischer and Sauer 2005).
Using yeast metabolism as a model, we attempt a functional classification of duplicate genes to elucidate whether a prevailing role is the basis of their conservation. Systematic categorization of the 295 metabolic duplicates was achieved by a combined approach that includes experimental phenotype data for the entire S. cerevisiae single knockout library, genome-scale in vivo flux data, in silico flux balancing with a genome-scale model, and network topology analysis.
Results
Reconstruction and experimental verification of a genome-scale metabolic model
Elucidation of duplicate gene functions requires knowledge on whether or not the specified reaction is essential or dispensable. Since comprehensive knockout libraries for duplicate gene families are presently not available, we predicted lethality of metabolic mutants with the recently described genome-scale model iFF708 of S. cerevisiae (Förster et al. 2003a) by Flux Balance Analysis (FBA) (Price et al. 2004). The alternative approach of elementary flux mode analysis to predict lethality from stoichiometry does not yet work, unfortunately, at the genome scale (Stelling et al. 2002). For experimental verification, we determined growth phenotypes of the entire single-gene deletion library (Giaever et al. 2002) under five environmental conditions, that is, complex medium (YPD) or minimal medium with glucose, galactose, glycerol, or ethanol as the sole carbon source, with a total of 23,290 experiments in duplicate (Supplemental Table S1). Then, FBA in silico predictions were compared to the 3360 plate growth experiments of all metabolic gene knockouts. The experimentally determined singleton lethality was correctly predicted in 40%-53% of the 79 to 146 cases (Fig. 1). While the more recent in silico strain iND750 includes compartmentalization (Duarte et al. 2004), its accuracy for lethality predictions was lower than that of iFF708 (Förster et al. 2003b). The low accuracy is not overly surprising, since the comparatively small number of lethal mutants is harder to predict than the more frequent viable mutants. Even for a random choice, the odds to forecast true positives are higher than to predict true negatives in this case (Provost and Kohavi 1998; Guda et al. 2004). The apparent bias can be overcome by using the geometric mean as a key number to quantify the trade-off (Provost and Kohavi 1998; Guda et al. 2004) between both accuracies by a single value. This reformulation allows to calculate the overall predictive accuracy (Kubat et al. 1998). For iFF708, the geometric mean of 60%-71% settles the considerable difference in predictive accuracy for viable (90%-96%) and lethal singleton deletions (40%-53%).
Figure 1.

Correct prediction of S. cerevisiae mutant lethality with the in silico strain iFF708 (Förster et al. 2003a) and the in silico strain iLL672 with FBA and MoMA optimization. In the latter case, experimentally determined flux distributions were used as the reference solution (cf. Fig. 4). The sole exception was YPD medium, for which the optimal FBA solution was used. Cases in which a correct prediction is structurally not possible, that is, lethals in single-deletion mutants of duplicate gene families, were not taken into account.
To improve lethality predictions, we reformulated the biomass composition, by considering ergosterol, thiamin, folate, and porphyrin as biomass components. These comparatively minor modifications improved the model predictions in the corresponding biosynthesis pathways. Further analysis of the metabolite balance equations revealed 151 metabolites that were either not produced or not consumed. Such dead-end metabolites were involved in 143 reactions, of which 110 were removed and 33 were connected based on new biological knowledge, thus closing gaps in the biosynthetic pathways (Supplemental Tables S2 and S3; http://www.gmm.gu.se/YSBN/models.htm). Examples for new gene functions are the roles of ALD2 and ALD3 in β-alanine synthesis (White et al. 2003) and elucidation of the sphingolipid biosynthesis pathway (Obeid et al. 2002).
This reconciled stoichiometric model, henceforth referred to as iLL672, includes 672 genes (95 of which participate in 24 enzyme complexes) that catalyze 579 biochemically distinct reactions and an additional 166 reactions that are not (yet) associated with any gene. Of these 745 reactions, 180 were involved in various transport processes and 105 reactions were encoded by 295 duplicate genes. Members of 18 duplicate gene families were present in two different compartments (Table 1; Huh et al. 2003). In total, iLL672 comprises 636 metabolites and 1038 reactions, which include isoenzyme reactions and others in the 745 biochemical reactions. The stoichiometric matrix of the reconciled network illustrates the overall structure of metabolism, where most metabolites occur in only few reactions (the diagonal in Fig. 2) and cofactors can be exchanged between remote parts of the network (Fig. 2; Csete and Doyle 2004). The high connectivity of common metabolic currency metabolites, such as the end products of cofactor (e.g., NADH, NADPH) or nucleotide (e.g. ATP, ADP, GTP) metabolism, is highlighted in this representation by clusters of dots in the horizontal dimension. Generally, duplicate gene-encoded reactions exhibit no particular clustering pattern, but are randomly distributed throughout metabolism. An exception is their overrepresentation in central carbon metabolism and in certain uptake reactions, for example, the 17-member hexose transporter family (Wieczorke et al. 1999). On the basis of the 3360 plate growth experiments of the 672 single-gene deletion mutants that are represented in the genome-scale metabolic model (Supplemental Table S1), the predictive capability of iLL672 with the FBA algorithm improved to 96%-98% for viable and 68%-80% for lethal singletons (Fig. 1), which results in a geometric mean of 81%-89%.
Table 1.
Compartmentalization of duplicate gene families in S. cerevisiae metabolism
| Duplicate families | ORF | Localizationa | Growth phenotypeb | Essential reactionc | Duplication eventd |
|---|---|---|---|---|---|
| AAT1 | YKL106W | mit | +/+/+/+/+ | -/+/+/+/+ | |
| AAT2 | YLR027C | cyt | +/+/+/+/+ | ||
| ACC1 | YNR016C | cyt | -e | +/+/+/+/+ | Genome duplication |
| HFA1 (ACC2) | YMR207C | mit | +/+/+/+/+ | Genome duplication | |
| ADH1 | YOL086C | cyt | +/+/+/+/+ | -/-/-/-/+ | |
| ADH2 | YMR303C | cyt | +/+/+/+/+ | ||
| ADH3 | YMR083W | mit | +/+/+/+/+ | ||
| ADH4 | YGL256W | mit | +/+/+/+/+ | Genome duplication | |
| ADH5 | YBR145W | cyt | +/+/+/+/+ | Genome duplication | |
| ADK1 | YDR226W | cyt | +/+/+/+/- | +/+/+/+/+ | |
| ADK2 | YER170W | mit | +/+/+/+/+ | ||
| ALD4 | YOR374W | mit | +/+/+/+/+ | -/-/-/-/+ | |
| ALD5 | YER073W | mit | +/+/+/+/+ | ||
| ALD6 | YPL061W | cyt | +/+/+/+/+ | ||
| ALT1 | YLR089C | mit | +/+/+/+/+ | -/-/-/-/- | Genome duplication |
| ALT2 | YDR111C | cyt | +/+/+/+/+ | Genome duplication | |
| BAT1 | YHR208W | mit | + | +/+/+/+/+ | Genome duplication |
| BAT2 | YJR148W | cyt | +/+/+/+/+ | Genome duplication | |
| CIT1 | YNR001C | mit | +/+/+/+/+ | -/+/+/+/+ | Genome duplication |
| CIT2 | YCR005C | cyt | +/+/+/+/+ | Genome duplication | |
| CIT3 | YPR001W | mit | +/+/+/+/+ | ||
| DLD1 | YDL174C | mit | +/+/+/+/+ | -/-/-/-/- | |
| DLD2 | YDL178W | mit | +/+/+/+/+ | ||
| DLD3 | YEL071W | cyt | + | ||
| GLO2 | YDR272W | cyt | +/+/+/+/+ | -/-/-/-/- | |
| GLO4 | YOR040W | mit | +/+/+/+/+ | ||
| GPD1 | YDL022W | cyt | +/+/+/+/+ | -/-/-/-/- | Genome duplication |
| GPD2 | YOL059W | cyt/mit | +/+/+/+/+ | Genome duplication | |
| IDP1 | YDL066W | mit | +/+/+/+/+ | -/-/-/-/- | |
| IDP2 | YLR174W | cyt | +/+/+/+/+ | Genome duplication | |
| IDP3 | YNL009W | cyt | +/+/+/+/+ | Genome duplication | |
| LEU4 | YNL104 | cyt | +/+/+/+/+ | -/-/-/-/- | Genome duplication |
| LEU9 | YOR108W | mit | +/+/+/+/+ | Genome duplication | |
| MDH1 | YKL085W | mit | +/+/+/+/+ | -/-/-/-/+ | |
| MDH2 | YDL078C | cyt | +/+/+/+/- | ||
| MDH3 | YOL126C | cyt | +/+/+/+/+ | ||
| IPP1 (PPA1) | YBR011C | cyt | - | +/+/+/+/+ | |
| PPA2 | YMR267W | mit | +/+/-/-/- | ||
| PSD1 | YNL169C | cyt | +/+/+/+/+ | -/-/-/-/- | |
| PSD2 | YGR170W | mit | +/+/+/+/+ | ||
| SHM1 | YBR263W | mit | +/+/+/+/+ | -/-/-/-/- | |
| SHM2 | YLR058C | cyt | +/+/+/+/+ | ||
| TRR1 | YDR353W | cyt | + | -/-/-/-/- | Genome duplication |
| TRR2 | YHR106W | mit | +/+/+/+/+ | Genome duplication |
According to Huh et al. (2003). (cyt) cytosol; (mit) mitochondria
Experimental phenotype on YPD, glucose, galactose, glycerol, or ethanol-containing media
In silico phenotype predictions on YPD, glucose, galactose, glycerol, or ethanol as the carbon source
According to Dietrich et al. (2004)
According to SGD (http://www.yeastgenome.org/)
Figure 2.

Stoichiometric network representation of the reconciled S. cerevisiae metabolic network iLL672. Metabolites (y-axis) and biochemical reactions (x-axis) are grouped into specific functional categories. Duplicate genes and singleton gene-encoded reactions are marked in black and gray, respectively. The number of dots in the horizontal direction highlights the reaction in which the corresponding metabolite is involved.
Phenotype prediction by MoMA from genome-scale flux data
Although the predictive capability was clearly improved, central metabolic deletions such as PGI1 and FBP1 were often falsely predicted to be viable. This in silico viability was caused by biologically irrelevant bypass reactions around the lesion. To reduce such artifacts, we used the Minimization of Metabolic Adjustment (MoMA) algorithm (Segre et al. 2002), which weighs the deviation from the wild-type flux distribution by minimizing the Euclidean distance between both solutions. While MoMA suppresses major flux rerouting, it is particularly sensitive to the accuracy of the reference flux distribution. Originally it was suggested to base MoMA on the reference fluxes of an FBA solution with maximum growth yield (Segre et al. 2002), but the underlying flux solutions are typically not unique (Mahadevan and Schilling 2003). To obtain a reference flux vector with high biological relevance, we estimated intracellular fluxes from 13C experiments (Blank and Sauer 2004; Fischer et al. 2004; Blank et al. 2005) or from quantitative physiological data (Fig. 4 below; Supplemental Table S4; Varma and Palsson 1994). These fluxes were used to constrain the FBA solution space. A particular flux solution was then identified by minimizing the overall intracellular flux to largely exclude alternate optima. This modified MoMA analysis further improved the in silico predictions under several conditions by another 3% when compared with FBA (Fig. 1). The thus verified iLL672 model was used in the following to predict lethality of entire duplicate family deletion mutants by MoMA analysis. The relatively small improvement of MoMA compared to FBA also demonstrates that the choice of search algorithm was not overly critical for a high predictive accuracy, which largely was a function of the model structure.
Figure 4.

Relative distributions of absolute carbon fluxes through S. cerevisiae central carbon metabolism. Flux distributions were obtained with (A) 13C-constrained flux analysis based on the data of Blank and Sauer (2004) (glucose) and stoichiometric flux balancing with (B) galactose; (C) glycerol; and (D) ethanol. Flux values are relative to the specific substrate uptake rate.
In the absence of a duplicate knockout library, we assessed the predictive quality of in silico duplicate knockouts from a subgroup of our phenotype data. Since 22 single-gene knockouts of the 295 duplicates were experimentally lethal, they can be used to verify in silico duplicate lethality predictions because the encoded reactions were obviously essential. With four of these 22 as falsely predicted nonessential reactions, the predictive accuracy was in the range obtained for singleton knockouts. A more qualitative cross-check with 16 published duplicate phenotypes further underlines the predictive accuracy of our calculations (Table 2).
Table 2.

Essential reactions
Back-up of important or essential functions with duplicate genes plays an important role in genetic network robustness (Gu et al. 2003; Blank et al. 2005), where null mutations often do not result in observable phenotypes. If, indeed, duplicate genes were selected for this function during evolution, one would expect them to be enriched in reactions that are essential for growth. To elucidate whether duplicate genes are statistically overrepresented in these essential metabolic reactions, we compared the fraction of lethal mutants in singletons and duplicate genes of S. cerevisiae. For any given condition, 159-171 singletons and 38-42 duplicate gene family mutants were predicted to be lethal.
To obtain a biologically meaningful proportion of essential duplicates and singletons, their quantity should be related to the number of active reactions under a given condition. Therefore, we identified all active reactions in the wild type for each condition, defined by carrying a nonzero flux, from the genome-scale flux solutions (Supplemental Table S4). About 52%-56% of all reactions were inactive under each of the four conditions, which agrees favorably with a recent estimate (Papp et al. 2004). For growth on a single carbon source, there was no significant difference in the fraction of lethal phenotypes among active singleton (63%-71%) and active duplicate (53%-74%) genes (Fig. 3). Thus, essential reactions are not more likely to be encoded by duplicate genes than by singleton genes, which indicates that genetic redundancy is not maintained by evolutionary selection.
Figure 3.

Relative occurrence of singleton (gray bars) and duplicate (black bars) genes in essential reactions during growth on four different single carbon substrates. The numbers are normalized to the active reactions under each growth condition.
Back-up function
While the above results demonstrate that evolution does not generally favor maintenance of duplicate genes to back up essential reactions, the results do not exclude, however, a potential role of individual duplicates in the compensation of genetic dysfunctions (Pal 2001; Gu 2003; Gu et al. 2003; Hurst and Pal 2005). If a gene exhibits such a back-up function, single-gene deletions in duplicate-encoded essential reactions should be viable. Hence, we compared our experimental singleton phenotypes with the in silico phenotype predictions of complete deletions of duplicate gene families. Of the 52 essential duplicate families, 32 were experimentally viable when a single gene member was knocked out, but lethal when the entire duplicate gene family was deleted in silico. This indicates that the remaining enzymes in these families compensate the loss of function of the deleted gene, which is, in turn, a very strong indicator for backup function of these duplicate genes. This back-up function does not necessarily imply that it is the primary reason why both copies were retained and may simply result from a gradual subfunctionalization, recent duplication events, or reprogramming of the duplicate family members (Kafri et al. 2005). Indication for such subfunctionalization was, indeed, obtained for the two duplicate gene families LAG1/LAC1 and ADK1/2, which exhibited back-up function under only two and three conditions, respectively.
In the remaining 18 essential duplicate gene families, a single member was essential for growth. This indicates that the other members have acquired a specialized function, restricted expression, or localization pattern that precludes functional complementation. Such duplicates are henceforth referred to as of specialized function, defined as genes that encode an essential reaction, yet lack the capability to back up the deletion of another family member. Largely in contrast to the above 32, the 18 duplicate gene families of this group exhibit highly imbalanced protein expression levels, where one member accounts for >95% of the entire enzyme population of this family (Ghaemmaghami et al. 2003). The only duplicate families with back-up function and unbalanced protein numbers of >90% are the transketolase (TKL1p accounts for 99.6%) and pyruvate decarboxylase families (PDC5p accounts for 97.5%).
Gene dosage
Another hypothesis on duplicate gene function is gene dosage, meaning occurrence in pathways that catalyze high fluxes to boost activity of critical enzymes (Papp et al. 2004). To test this hypothesis, we related the experimentally determined flux data (Fig. 4) to the localization of duplicate-catalyzed reactions. High flux was defined as 5% or higher of the substrate uptake rate (Table 3). Based on these genome-scale flux solutions (Supplemental Table S4), only 30 of all duplicate families were localized in high flux reactions. In several cases, however, a single major isoform was essential, hence excluding gene dosage function. For the example of growth on glucose, 21 duplicate gene families encode high flux reactions but ACO1, CDC19, ENO2, and GPM1 are essential, lowering the number of gene duplicate families with potential gene dosage function to 17. Overall, 19 duplicate families were categorized to exhibit a potential dosage function under at least one of the four conditions for which flux solutions were available (Fig. 5). It should be noted, that this number is an upper bound that has to be verified experimentally for each case.
Table 3.
Potential gene dosage function of duplicate genes in highly active reactions
| Medium | High average carbon flux (>5% of substrate uptake rate) |
|---|---|
| Reactions encoded by duplicate genes | |
| YPDa | 22 (35)b |
| Glucose | 21 (34) |
| Galactose | 23 (45) |
| Glycerol | 25 (48) |
| Ethanol | 14 (30) |
Substrate uptake rate defined as the uptake rate of glucose
“Total high-flux reactions”
Figure 5.

Duplicate gene families with potential gene dosage function. Gene duplicate family occurrence was mapped onto four genome-scale flux distributions (glucose, galactose, glycerol, and ethanol minimal medium) (Supplemental Table S4) to elucidate gene dosage function in metabolism. Reactions carrying >5% of the substrate uptake flux were considered for gene dosage function. Gene duplicate families in which a single member is lethal (on any of the four media) were excluded, since every gene duplicate family member involved in gene dosage must suffice for growth.
Regulatory role of duplicate genes
Generally, the positioning of duplicate genes at key points of the network topology provides circumstantial evidence for differential regulation of the encoded isoenzymes. Genome-scale analysis of the reconciled model with the flux coupling finder (Burgard et al. 2004) revealed between 65 and 67 coupled reaction sets that consisted of at least three consecutive reactions. Two-thirds of these putatively coregulated pathway subsets were part of anabolic pathways that catalyze biosynthesis of biomass components. In 18 cases, duplicate family-encoded reactions were located at the beginning or end of such linearly coupled reaction sets, indicating differential regulation of the isoenzymes. Further support comes from promoter motif analysis since the motif-content overlap (Kafri et al. 2005) showed very little agreement between members of duplicate families located at the beginning or end of biosynthetic pathways (Supplemental Table S5). This strongly indicates that these duplicate families are, indeed, regulated differentially. A prominent example of proven biological relevance is the superpathway of aromatic amino acids biosynthesis (Hartmann et al. 2003) that links the upstream duplicate genes ARO3 and ARO4 with two linear pathways downstream of the prephenate branch point (Fig. 6). Thus, we have evidence that at least 18 of the 105 duplicate gene families in metabolism have a potential role in differential regulation of pathways.
Figure 6.

Localization of duplicate genes in aromatic amino acid synthesis of S. cerevisiae metabolism. Linear pathways (dashed boxes) were identified by evaluation of the full genome size metabolic model iLL672 with the Flux Coupling Finder. The correlations between duplicate genes and identified regulation knots were carried out manually.
Discussion
Despite extensive research on the functional role of duplicate genes in yeast, no general consensus has been reached to date and typically the prevalence of a particular function as the selective pressure for duplicate retention was favored (Ihmels et al. 2004; Papp et al. 2004). By integrating experimental and computational analysis, we show that the 105 yeast duplicate families in metabolism do not have a single major but rather an array of different, often overlapping functions (Fig. 7A). While the capability for back-up can hardly be seen as the selective pressure that retains both copies, 32 of the 105 metabolic duplicates were capable of functionally replacing one another under all conditions tested. Another two could substitute the deleted family member under at least two tested conditions. Although catalyzing essential reactions, another 18 duplicate families do not exhibit any back-up activity under the five conditions tested. One copy of these paralogs has probably evolved new (specialized) functions that are sufficiently different to preclude functional complementation or restrict it to rare cases. Such specialized functions (Fig. 7A) may be nonmetabolic functions, an extremely different expression pattern that first needed to be reprogrammed (Hurst and Pal 2005; Kafri et al. 2005), or simply activity in a different compartment. Indeed, six of the 18 duplicate families with specialized function are located in different compartments (Supplemental Table S6). The exposed location of duplicates at the beginning or end of linear pathways indicates a putative regulatory function for at least 18 duplicate families. Little or no overlap in the promoter motifs of these genes (Kafri et al. 2005) further supports such a regulatory differentiation. Based on genome-scale in vivo flux data, we show that maximally 19 duplicate families exhibit a putative gene dosage function to boost activity. Since this is an upper bound, we did not find evidence for gene dosage as the general selection mechanism of duplicate gene retention, as was previously suggested (Seoighe and Wolfe 1999; Papp et al. 2004).
Figure 7.

Functional categorization of the 105 duplicate gene families in S. cerevisiae metabolism. (A) Duplicate distribution of the major functions among all 105 families. The functional roles are gene dosage to boost enzyme activity, regulation by specific gene expression, back-up capability for knockout compensation, and specialized functions where knockout of one family member could not be compensated by the other(s). (B) Functional categorization of individual gene families. Each family is represented by a 3.5° angle, as indicated for PFK1/PFK2. The central circle specifies the computationally predicted essentiality of family knockouts. The first ring represents the experimentally determined single knockout phenotypes in each family. A single lethal knockout indicates a specialized function (gray) of the other(s), while viable single knockouts in a family are separated into those that clearly exhibit back-up activity (black) and those where no clear distinction can be made (white) either because they are inactive or alternative pathways could potentially compensate. The second ring specifies the experimentally determined activity of a reaction under any of the five investigated growth conditions. The third ring represents the enzyme activity modulation level. Specifically, gene dosage (black) and potentially different genetic regulation pattern (gray) can be estimated by the present data.
Thus, only few of the classified duplicate genes appear to be characterized by a single metabolic function, but not all combinations may occur (Fig. 7B; Supplemental Table S6). On a given functional level, different duplicate functions are mutually exclusive (the concentric rings in Fig. 7B), for example, back-up and specialized function that are directly determined from single knockout phenotype data. At the activity modulation level, enzyme activity may either be boosted through parallel expression (gene dosage) or differentially expressed through alternative regulation mechanisms. Across the functional level, however, duplicate genes may exhibit multiple functions with gene dosage and back-up as a particularly frequent combination among essential reactions. This is in contrast to ribosomal duplicates, which have gene dosage function, but can only maintain growth if both copies of the encoding gene are present (Seoighe and Wolfe 1999). While identification of gene dosage and regulatory function are not mutually exclusive, we found no overlap between both groups (Fig. 7A). This is not overly surprising since fine-tuning via regulation and boosted activity are clearly antagonistic. Duplicate gene families with a regulatory role, however, frequently also exhibit other functions (Fig. 7A), which agrees with experimental data demonstrating back-up (Kafri et al. 2005) and specialized function (van den Berg et al. 1996) for paralogs of this group.
The back-up function of duplicate genes is tightly connected to the robustness of cellular functions to genetic perturbations, a long-recognized key property of biological systems that is becoming a focal research theme (Kitano 2004; Stelling et al. 2004). While we show here that back-up is not the dominant function of metabolic duplicate genes in S. cerevisiae (Fig. 7), this does not exclude an important contribution of duplicates to genetic robustness. Indeed, it was recently shown that metabolic duplicate genes are the major and alternative pathways the minor mechanistic cause for the robustness of S. cerevisiae metabolism to knockout mutations (Blank et al. 2005), as was also suggested for all duplicate genes in yeast (Gu et al. 2003).
While experimental fluxome analysis (Blank et al. 2005; Fischer and Sauer 2005) and appropriate genome-scale modeling approaches (Förster et al. 2003b; Papin et al. 2004; Price et al. 2004) enable a mechanistic assessment of duplicate functions in metabolism, other cellular processes are less directly accessible. How representative then is the distribution of metabolic duplicate functions for other cellular processes? Generally, there is no reason to believe that they are not representative, because the discussed functions are ubiquitous and all cellular processes are subject to the same evolutionary forces. Not restricted to metabolism, statistical correlation between gene expression similarity and back-up function revealed back-up capability in 53 yeast duplicates (Kafri et al. 2005). Despite the fundamental difference in approaches, 12 of the 18 metabolic duplicate families represented in both studies were consistently assigned a back-up function (Supplemental Table S6). Deviating assignments of the six duplicate families are probably due to different conditions and strain backgrounds. Since neither study provides any evidence for a prevailing function of duplicate genes that might serve as a basis for their conservation, future classifications into functional groups must rely on quantitative data.
Methods
Large-scale experimental lethality testing
For large-scale phenotyping of plate growth under different conditions, we used the entire haploid yeast knockout library of strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (Winzeler et al. 1999) with 4658 mutants. The composition of the yeast minimal medium was, per liter (Verduyn et al. 1992), 5 g of (NH4)2SO4, 3 g of KH2PO4, 0.5 g of MgSO4 · 7H2O, 4.5 mg of ZnSO4 · 7H2O, 0.3 mg of CoCl2 · 6H2O, 1.0 mg of MnCl2 · 4H2O, 0.3 mg of CuSO4 · 5H2O, 4.5 mg of CaCl2 · 2H2O, 3.0 mg of FeSO4 · 7H2O, 0.4 mg of NaMoO4 · 2H2O, 1.0 mg of H3BO3, 0.1 mg of KI, 15 mg of EDTA, 0.05 mg of biotin, 1.0 mg of Capantothenate, 1.0 mg of nicotinic acid, 25 mg of inositol, 1.0 mg of pyridoxine, 0.2 mg of p-amino-benzoic acid, and 1.0 mg of thiamine. The carbon sources (ethanol, galactose, glucose, and glycerol) were added to a final concentration of 20 g/L. Strain auxotrophies were complemented with 20 mg/L histidine, uracil, methionine, and 60 mg/L leucine. About 50 strains of the yeast collection are lysine auxotroph and were independently tested for growth on plates supplemented with 20 mg/L lysine. The YPD medium consisted, per liter, of 10 g of yeast extract, 20 g of peptone, and 20 g of glucose.
The single-gene deletion library was organized in a 384 format that was also used for plate growth testing. Duplicate replica plating was carried out with a Biomek Laboratory Automation Workstation (Beckman Coulter Inc.). The plates were incubated at 30°C for 3 d before scoring growth phenotypes and further incubated for 1 wk to score slow growing mutants. Mutants of uncertain growth phenotypes were re-evaluated by manual streaking on fresh plates. In phenotype experiments of six mutants (YAL012W, YDR300C, YFL018C, YHR018C, YOR184W, and YOR221C), spontaneous suppressor mutations occurred that were characterized by single colonies. These mutants were scored as lethal.
Identification of duplicate genes
To identify all metabolic duplicate genes in the S. cerevisiae genome, we used genes included in the stoichiometric model as bait for translated BLAST analysis (WU-BLAST2, on http://www.yeastgenome.org/). We chose an arbitrary cut-off of P 1e-30 over 80% of the sequence. The results were identical to a recent publication (Kellis et al. 2004), in which the authors used protein, nucleotide, and translation-aware nucleotide alignments to identify all duplicate genes in the genome of S. cerevisiae, of which the 105 duplicate families presented here are a subset.
Stoichiometric network analysis
Flux balance analysis (FBA) (Price et al. 2004) and minimization of metabolic adjustment (MoMA) (Segre et al. 2002) were used to predict mutant lethality. Both methods assume intracellular quasi-steady state, such that the production and consumption of each intracellular metabolite Mi is balanced. This yields the equation
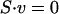 |
1 |
where S corresponds to the stoichiometric matrix and v to the array of metabolic fluxes. Assuming maximization of the growth rate μ as the objective function of cellular behavior in FBA (Edwards and Palsson 2000; Price et al. 2004), a flux distribution v can be obtained by linear programming (LP):
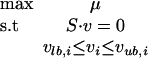 |
2 |
where i = 1,..., M and vlb,i and vub,i correspond to the upper and lower bounds of a specific reaction i. Setting both reaction bounds equal to zero mimics a gene deletion; thus FBA provides a straightforward tool for qualitative mutant phenotype prediction.
Instead of growth rate maximization, MoMA uses the minimization of the Euclidean distance between wild-type reference (vWT) and mutant flux distribution (vmut) as the objective function. This results in a quadratic programming (QP) problem, with
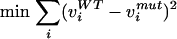 |
3 |
as the objective function. Instead of the l2 norm of the Euclidean distance, we used here the l1 norm, which allows the LP reformulation of MoMA:
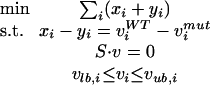 |
4 |
where i = 1,..., M, and xi ≥ 0, yi ≥ 0. Note that minimizing the sum of the strictly positive substitution variables x and y automatically forces the difference between vWT and vmut to zero. Note also that this reformulation is equivalent to minimizing the Euclidean distance as shown in equation 3 or using absolute values as shown in equation 5, but allows them to be optimized with LP solvers. All optimization problems were solved with the open source GNU linear programming kit (Makhorin 2001; www.gnu.org/software/glpk/glpk.html).
Originally, the wild-type reference flux solution vWT for MoMA was obtained by FBA (Segre et al. 2002). This flux distribution, however, represents the theoretical capabilities of the cell (Edwards et al. 2001) and not a biological meaningful flux estimate, since FBA solutions are typically not unique (Mahadevan and Schilling 2003) and no experimental data are used. Here we used experimentally determined fluxes (vexp) to obtain an experimentally validated reference flux solution vWT for MoMA at the genome scale. For glucose minimal medium, we constrained the model iLL672 with 30 fluxes that were derived from 13C-labeling experiments (Wiechert 2001; Sauer 2004). In particular, we used 13C-constrained flux analysis (Sauer et al. 1997; Fischer et al. 2004) for GC-MS-detected mass isotope distributions in proteinogenic amino acids from a 20% [U-13C] glucose experiment and a compartmentalized yeast model (Blank and Sauer 2004; Blank et al. 2005). For the genome-scale flux solution, we used 20, 24, and 28 flux constraints, for ethanol, galactose, and glycerol growth, respectively. These were calculated from physiological data with a 34-reaction stoichiometric model as was described elsewhere (Nissen et al. 1997; Gombert et al. 2001; Sonderegger et al. 2004). These experimental data were to be kept within an accuracy δ of ±10% when mapping the determined central metabolic fluxes to the genome-scale reference flux solution. To overcome mathematical artifacts such as cycling, that is, a closed loop of fluxes that bring no net change, the original LP problem (equation 2) was modified. A minimization of the l1 norm, that is, the overall intracellular flux, was chosen as the objective function:
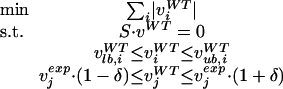 |
5 |
with j as the set of experimentally determined fluxes. This reformulation widely excludes the existence of alternate optima and can be solved according to equation 4.
Metabolic pathway analysis
Approaches for topological network analysis at the genome scale consider either a hierarchical (Gagneur et al. 2003) or modular decomposition (Burgard et al. 2004). We here used the Flux Coupling Finder (Burgard et al. 2004) that elucidates connections between different reactions by solving a sequence of fractional programming problems. By keeping the flux through one reaction constant while maximizing or minimizing another, it is thus possible to detect dependencies between both reactions. The Flux Coupling Finder thus reveals subsets of blocked or coupled enzymes.
Statistical data treatment
The predictive power of the computational model was evaluated by means of a confusion matrix (Provost and Kohavi 1998) that, for a two class identifier, groups the results into correct (true positive [TP], true negative [TN]) and wrong predictions (false negative [FN], false positive [FP]), respectively (Guda et al. 2004). If the number of either total positive or total negative experimental results outperforms the other, the corresponding case will be easier to predict (Kubat et al. 1998), since then the chances for a correct prediction are higher even on a pure random choice. One thus has to consider both accuracies equally. This can be done by the geometric mean (Kubat et al. 1998), which weighs both the positive (viable) and negative (lethal) case identically by multiplying sensitivity and specificity:
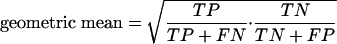 |
6 |
Supplementary Material
Acknowledgments
We are grateful to Marc Sohrmann and Matthias Peter for access and help with the yeast array experiments and to Arend Sidow for critical comments on the manuscript. Lars M. Blank gratefully acknowledges financial support by the Deutsche Akademie der Naturforscher Leopoldina (BMBF-LPD/8-78).
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3992505.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Albers, E., Laize, V., Blomberg, A., Hohmann, S., and Gustafsson, L. 2003. Ser3p (Yer081wp) and Ser33p (Yil074cp) are phosphoglycerate dehydrogenases in Saccharomyces cerevisiae. J. Biol. Chem. 278: 10264-10272. [DOI] [PubMed] [Google Scholar]
- Avendano, A., Deluna, A., Olivera, H., Valenzuela, L., and Gonzalez, A. 1997. GDH3 encodes a glutamate dehydrogenase isozyme, a previously unrecognized route for glutamate biosynthesis in Saccharomyces cerevisiae. J. Bacteriol. 179: 5594-5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basson, M.E., Thorsness, M., and Rine, J. 1986. Saccharomyces cerevisiae contains two functional genes encoding 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Proc. Natl. Acad. Sci. 83: 5563-5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank, L.M. and Sauer, U. 2004. TCA cycle activity in Saccharomyces cerevisiae is a function of the environmentally determined specific growth and glucose uptake rates. Microbiology 150: 1085-1093. [DOI] [PubMed] [Google Scholar]
- Blank, L.M., Kuepfer, L., and Sauer, U. 2005. Large-scale 13C-flux analysis reveals mechanistic principles of metabolic network robustness to null mutations in yeast. Genome Biol. 6: R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgard, A.P., Nikolaev, E.V., Schilling, C.H., and Maranas, C.D. 2004. Flux coupling analysis of genome-scale metabolic network reconstructions. Genome Res. 14: 301-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherest, H. and Surdin-Kerjan, Y. 1978. S-Adenosyl methionine requiring mutants in Saccharomyces cerevisiae: Evidences for the existence of two methionine adenosyl transferases. Mol. Gen. Genet. 163: 153-167. [DOI] [PubMed] [Google Scholar]
- Conant, G.C. and Wagner, A. 2002. GenomeHistory: A software tool and its application to fully sequenced genomes. Nucleic Acids Res. 30: 3378-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csete, M. and Doyle, J. 2004. Bow ties, metabolism and disease. Trends Biotechnol. 22: 446-450. [DOI] [PubMed] [Google Scholar]
- Dietrich, F.S., Voegeli, S., Brachat, S., Lerch, A., Gates, K., Steiner, S., Mohr, C., Pohlmann, R., Luedi, P., Choi, S., et al. 2004. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 304: 304-307. [DOI] [PubMed] [Google Scholar]
- Duarte, N.C., Herrgard, M.J., and Palsson, B.O. 2004. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res. 14: 1298-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, J.S. and Palsson, B.O. 2000. Metabolic flux balance analysis and the in silico analysis of Escherichia coli K-12 gene deletions. BMC Bioinformatics 1: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, J.S., Ibarra, R.U., and Palsson, B.O. 2001. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol. 19: 125-130. [DOI] [PubMed] [Google Scholar]
- Farkas, I., Hardy, T.A., Goebl, M.G., and Roach, P.J. 1991. Two glycogen synthase isoforms in Saccharomyces cerevisiae are coded by distinct genes that are differentially controlled. J. Biol. Chem. 266: 15602-15607. [PubMed] [Google Scholar]
- Fischer, E. and Sauer, U. 2005. Large-scale in vivo flux analysis shows rigidity and suboptimal performance of Bacillus subtilis metabolism. Nat. Genet. 37: 636-640. [DOI] [PubMed] [Google Scholar]
- Fischer, E., Zamboni, N., and Sauer, U. 2004. High-throughput metabolic flux analysis based on gas chromatography-mass spectrometry derived 13C constraints. Anal. Biochem. 325: 308-316. [DOI] [PubMed] [Google Scholar]
- Flikweert, M.T., de Swaaf, M., van Dijken, J.P., and Pronk, J.T. 1999. Growth requirements of pyruvate-decarboxylase-negative Saccharomyces cerevisiae. FEMS Microbiol. Lett. 174: 73-79. [DOI] [PubMed] [Google Scholar]
- Förster, J., Famili, I., Fu, P., Palsson, B.O., and Nielsen, J. 2003a. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 13: 244-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster, J., Famili, I., Palsson, B.O., and Nielsen, J. 2003b. Large-scale evaluation of in silico gene deletions in Saccharomyces cerevisiae. Omics 7: 193-202. [DOI] [PubMed] [Google Scholar]
- Gagneur, J., Jackson, D.B., and Casari, G. 2003. Hierarchical analysis of dependency in metabolic networks. Bioinformatics 19: 1027-1034. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami, S., Huh, W.K., Bower, K., Howson, R.W., Belle, A., Dephoure, N., O'Shea, E.K., and Weissman, J.S. 2003. Global analysis of protein expression in yeast. Nature 425: 737-741. [DOI] [PubMed] [Google Scholar]
- Giaever, G., Chu, A.M., Ni, L., Connelly, C., Riles, L., Veronneau, S., Dow, S., Lucau-Danila, A., Anderson, K., Andre, B., et al. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387-391. [DOI] [PubMed] [Google Scholar]
- Gombert, A.K., Moreira dos Santos, M., Christensen, B., and Nielsen, J. 2001. Network identification and flux quantification in the central metabolism of Saccharomyces cerevisiae under different conditions of glucose repression. J. Bacteriol. 183: 1441-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, X. 2003. Evolution of duplicate genes versus genetic robustness against null mutations. Trends Genet. 19: 354-356. [DOI] [PubMed] [Google Scholar]
- Gu, Z., Steinmetz, L.M., Gu, X., Scharfe, C., Davis, R.W., and Li, W.H. 2003. Role of duplicate genes in genetic robustness against null mutations. Nature 421: 63-66. [DOI] [PubMed] [Google Scholar]
- Guda, C., Fahy, E., and Subramaniam, S. 2004. MITOPRED: A genome-scale method for prediction of nucleus-encoded mitochondrial proteins. Bioinformatics 20: 1785-1794. [DOI] [PubMed] [Google Scholar]
- Hartmann, M., Schneider, T.R., Pfeil, A., Heinrich, G., Lipscomb, W.N., and Braus, G.H. 2003. Evolution of feedback-inhibited β/α barrel isoenzymes by gene duplication and a single mutation. Proc. Natl. Acad. Sci. 100: 862-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh, W.K., Falvo, J.V., Gerke, L.C., Carroll, A.S., Howson, R.W., Weissman, J.S., and O'Shea, E.K. 2003. Global analysis of protein localization in budding yeast. Nature 425: 686-691. [DOI] [PubMed] [Google Scholar]
- Hurst, L.D. and Pal, C. 2005. Dissecting dispensability. Nat. Genet. 37: 214-215. [DOI] [PubMed] [Google Scholar]
- Ihmels, J., Levy, R., and Barkai, N. 2004. Principles of transcriptional control in the metabolic network of Saccharomyces cerevisiae. Nat. Biotechnol. 22: 86-92. [DOI] [PubMed] [Google Scholar]
- Kafri, R., Bar-Even, A., and Pilpel, Y. 2005. Transcription control reprogramming in genetic backup circuits. Nat. Genet. 37: 295-299. [DOI] [PubMed] [Google Scholar]
- Kellis, M., Birren, B.W., and Lander, E.S. 2004. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428: 617-624. [DOI] [PubMed] [Google Scholar]
- Kitano, H. 2004. Biological robustness. Nat. Rev. Genet. 5: 826-837. [DOI] [PubMed] [Google Scholar]
- Kubat, M., Holte, R.C., and Matwin, S. 1998. Machine learning for the detection of oil spills in satellite radar images. Machine Learn. 30: 195-215. [Google Scholar]
- Lynch, M. and Katju, V. 2004. The altered evolutionary trajectories of gene duplicates. Trends Genet. 20: 544-549. [DOI] [PubMed] [Google Scholar]
- Mahadevan, R. and Schilling, C.H. 2003. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 5: 264-276. [DOI] [PubMed] [Google Scholar]
- Makhorin, A. 2001. GNU linear programming kit. Free Software Foundation, Boston.
- Marini, A.M., Soussi-Boudekou, S., Vissers, S., and Andre, B. 1997. A family of ammonium transporters in Saccharomyces cerevisiae. Mol. Cell. Biol. 17: 4282-4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, S., Zimmermann, F.K., and Boles, E. 1997. Mutant studies of phosphofructo-2-kinases do not reveal an essential role of fructose-2,6-bisphosphate in the regulation of carbon fluxes in yeast cells. Microbiology 143: 3055-3061. [DOI] [PubMed] [Google Scholar]
- Nagiec, M.M., Skrzypek, M., Nagiec, E.E., Lester, R.L., and Dickson, R.C. 1998. The LCB4 (YOR171c) and LCB5 (YLR260w) genes of Saccharomyces encode sphingoid long chain base kinases. J. Biol. Chem. 273: 19437-19442. [DOI] [PubMed] [Google Scholar]
- Nissen, T.L., Schulze, U., Nielsen, J., and Villadsen, J. 1997. Flux distributions in anaerobic, glucose-limited continuous cultures of Saccharomyces cerevisiae. Microbiology 143: 203-218. [DOI] [PubMed] [Google Scholar]
- Obeid, L.M., Okamoto, Y., and Mao, C. 2002. Yeast sphingolipids: Metabolism and biology. Biochim. Biophys. Acta 1585: 163-171. [DOI] [PubMed] [Google Scholar]
- Ohno, S. 1970. Evolution by gene duplication. Springer, New York.
- Ozier-Kalogeropoulos, O., Adeline, M.T., Yang, W.L., Carman, G.M., and Lacroute, F. 1994. Use of synthetic lethal mutants to clone and characterize a novel CTP synthetase gene in Saccharomyces cerevisiae. Mol. Gen. Genet. 242: 431-439. [DOI] [PubMed] [Google Scholar]
- Pahlman, A.K., Granath, K., Ansell, R., Hohmann, S., and Adler, L. 2001. The yeast glycerol 3-phosphatases Gpp1p and Gpp2p are required for glycerol biosynthesis and differentially involved in the cellular responses to osmotic, anaerobic, and oxidative stress. J. Biol. Chem. 276: 3555-3563. [DOI] [PubMed] [Google Scholar]
- Pal, C. 2001. Yeast prions and evolvability. Trends Genet. 17: 167-169. [DOI] [PubMed] [Google Scholar]
- Papin, J.A., Stelling, J., Price, N.D., Klamt, S., Schuster, S., and Palsson, B.O. 2004. Comparison of network-based pathway analysis methods. Trends Biotechnol. 22: 400-405. [DOI] [PubMed] [Google Scholar]
- Papp, B., Pal, C., and Hurst, L.D. 2004. Metabolic network analysis of the causes and evolution of enzyme dispensability in yeast. Nature 429: 661-664. [DOI] [PubMed] [Google Scholar]
- Presgraves, D.C. 2005. Evolutionary genomics: New genes for new jobs. Curr. Biol. 15: R52-R53. [DOI] [PubMed] [Google Scholar]
- Price, N.D., Reed, J.L., and Palsson, B.O. 2004. Genome-scale models of microbial cells: Evaluating the consequences of constraints. Nat. Rev. Microbiol. 2: 886-897. [DOI] [PubMed] [Google Scholar]
- Provost, F. and Kohavi, R. 1998. Glossary of terms. Machine Learn. 30: 271-274. [Google Scholar]
- Ramos, F. and Wiame, J.M. 1980. Two asparagine synthetases in Saccharomyces cerevisiae. Eur. J. Biochem. 108: 373-377. [DOI] [PubMed] [Google Scholar]
- Sauer, U. 2004. High-throughput phenomics: Experimental methods for mapping fluxomes. Curr. Opin. Biotechnol. 15: 58-63. [DOI] [PubMed] [Google Scholar]
- Sauer, U., Hatzimanikatis, V., Bailey, J.E., Hochuli, M., Szyperski, T., and Wuthrich, K. 1997. Metabolic fluxes in riboflavin-producing Bacillus subtilis. Nat. Biotechnol. 15: 448-452. [DOI] [PubMed] [Google Scholar]
- Schaaff-Gerstenschlager, I., Mannhaupt, G., Vetter, I., Zimmermann, F.K., and Feldmann, H. 1993. TKL2, a second transketolase gene of Saccharomyces cerevisiae. Cloning, sequence and deletion analysis of the gene. Eur. J. Biochem. 217: 487-492. [DOI] [PubMed] [Google Scholar]
- Segre, D., Vitkup, D., and Church, G.M. 2002. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. 99: 15112-15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoighe, C. and Wolfe, K.H. 1999. Yeast genome evolution in the post-genome era. Curr. Opin. Microbiol. 2: 548-554. [DOI] [PubMed] [Google Scholar]
- Sonderegger, M., Jeppsson, M., Hahn-Hagerdal, B., and Sauer, U. 2004. Molecular basis for anaerobic growth of Saccharomyces cerevisiae on xylose, investigated by global gene expression and metabolic flux analysis. Appl. Environ. Microbiol. 70: 2307-2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelling, J., Klamt, S., Bettenbrock, K., Schuster, S., and Gilles, E.D. 2002. Metabolic network structure determines key aspects of functionality and regulation. Nature 420: 190-193. [DOI] [PubMed] [Google Scholar]
- Stelling, J., Sauer, U., Szallasi, Z., Doyle III, F.J., and Doyle, J. 2004. Robustness of cellular functions. Cell 118: 675-685. [DOI] [PubMed] [Google Scholar]
- Stucka, R., Dequin, S., Salmon, J.M., and Gancedo, C. 1991. DNA sequences in chromosomes II and VII code for pyruvate carboxylase isoenzymes in Saccharomyces cerevisiae: Analysis of pyruvate carboxylase-deficient strains. Mol. Gen. Genet. 229: 307-315. [DOI] [PubMed] [Google Scholar]
- Tibbetts, A.S. and Appling, D.R. 2000. Characterization of two 5-aminoimidazole-4-carboxamide ribonucleotide transformylase/inosine monophosphate cyclohydrolase isozymes from Saccharomyces cerevisiae. J. Biol. Chem. 275: 20920-20927. [DOI] [PubMed] [Google Scholar]
- Urrestarazu, A., Vissers, S., Iraqui, I., and Grenson, M. 1998. Phenylalanine- and tyrosine-auxotrophic mutants of Saccharomyces cerevisiae impaired in transamination. Mol. Gen. Genet. 257: 230-237. [DOI] [PubMed] [Google Scholar]
- van den Berg, M.A., de Jong-Gubbels, P., Kortland, C.J., van Dijken, J.P., Pronk, J.T., and Steensma, H.Y. 1996. The two acetyl-coenzyme A synthetases of Saccharomyces cerevisiae differ with respect to kinetic properties and transcriptional regulation. J. Biol. Chem. 271: 28953-28959. [DOI] [PubMed] [Google Scholar]
- Varma, A. and Palsson, B.O. 1994. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl. Environ. Microbiol. 60: 3724-3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verduyn, C., Postma, E., Scheffers, W.A., and Van Dijken, J.P. 1992. Effect of benzoic acid on metabolic fluxes in yeasts: A continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 8: 501-517. [DOI] [PubMed] [Google Scholar]
- Wagner, A. 2000. Robustness against mutations in genetic networks of yeast. Nat. Genet. 24: 355-361. [DOI] [PubMed] [Google Scholar]
- White, W.H., Skatrud, P.L., Xue, Z., and Toyn, J.H. 2003. Specialization of function among aldehyde dehydrogenases: The ALD2 and ALD3 genes are required for β-alanine biosynthesis in Saccharomyces cerevisiae. Genetics 163: 69-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechert, W. 2001. 13C metabolic flux analysis. Metab. Eng. 3: 195-206. [DOI] [PubMed] [Google Scholar]
- Wieczorke, R., Krampe, S., Weierstall, T., Freidel, K., Hollenberg, C.P., and Boles, E. 1999. Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in Saccharomyces cerevisiae. FEBS Lett. 464: 123-128. [DOI] [PubMed] [Google Scholar]
- Winzeler, E.A., Shoemaker, D.D., Astromoff, A., Liang, H., Anderson, K., Andre, B., Bangham, R., Benito, R., Boeke, J.D., Bussey, H., et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901-906. [DOI] [PubMed] [Google Scholar]
- Wolfe, K. 2004. Evolutionary genomics: Yeasts accelerate beyond BLAST. Curr. Biol. 14: R392-R394. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. and Kishino, H. 2004. Genomic background predicts the fate of duplicated genes: Evidence from the yeast genome. Genetics 166: 1995-1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.gmm.gu.se/YSBN/models.htm; The Yeast System Biology Network is a consortium of researchers promoting Systems Biology with the yeast Saccharomyces cerevisiae as a model system. The homepage will present cell models of Saccharomyces cerevisiae besides other information.
- http://www.gnu.org/software/glpk/glpk.html; The GNU Linear Programming Kit (GLPK) is a software package for solving large-scale linear programming (LP) and mixed integer programming (MIP) problems. The GLPK library is written in ANSI C and is part of the GNU project.
- http://www.yeastgenome.org/; SGDTM is a scientific database of molecular biology and genetics of the yeast Saccharomyces cerevisiae, which is commonly known as baker's or budding yeast.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.