

Diversity is the hard currency of ecologists. Various statistics have been developed for summarizing the diversity of an ecological community. A commonly adopted summary statistic is the Shannon-Weiner index: H = −Σpilnpi, where pi is the frequency of the ith species. In addition, species richness (the number of different species) often is reported, and recent work emphasizes the importance of accurate estimates of species richness when ecological communities and processes that affect the composition of communities and the function of ecosystems are described (5). The significance of diversity is often inferred by comparing communities characterized from different environments. Typically, such comparisons rely on standard measures of overlap, including the percentage of species shared by two communities or similarity indices. One of the indices used is Sorensen's index: S = S12/[0.5(S1 + S2)], where S12 is the number of species common to both sites and Si is the number of species found at site i.
A limitation of traditional statistics for describing and comparing diversity is that species (or operational taxonomic units [OTUs]) are defined inconsistently. For instance, Kroes et al. (6) defined an OTU as a 16S ribosomal DNA (rDNA) sequence group in which sequences differed by less than 1%. By contrast, the definition of McCaig et al. (11) included sequences that were less than 3% different, and other studies have used 5% as the magic number. The lack of consensus limits the comparative utility of statistics based solely on identification of species (or OTUs). A second, and perhaps more important, limitation of the standard statistics of diversity is that OTUs are counted equivalently even though some may be highly divergent and phylogenetically unique, whereas others may be part of a closely related group of species and are therefore phylogenetically redundant (4). The contrast can be illustrated by comparing two hypothetical communities in which the numbers of species, the richness profiles of species, and the rarefaction profiles are identical but which differ in the magnitude of phylogenetic diversity (i.e., the degree of divergence among the sampled sequences). Standard ecological statistics of diversity would miss the genetic difference between the two communities, and ecologists would most likely consider the two communities equally diverse when, in fact, one community harbors more genetic diversity (or disparity) than the other. Because genetic variation and phenotypic variance often are positively correlated in populations of animals (12), plants (7), and microbes (15), descriptions of microbial communities based on DNA data should include information about diversity and disparity. This is especially important in light of studies demonstrating an association between ecosystem function and community diversity (14, 28).
In this review I introduce various statistics borrowed from population genetics and systematics for describing and comparing the diversity evident from samples of gene sequences. I briefly introduce the statistics and methodological underpinnings of tests for differences between communities, and I use the methods to analyze well-described microbial communities. I show that information gained from analysis of DNA sequences provides the basis for statistical analysis of communities in ways that advance inferences about the processes that may govern the compositions and functions of microbial communities. Furthermore, the advocated analytical approaches make it possible to accomplish broad comparisons of ecological communities. The methods of analysis explored in this paper are meant to be complementary to other methods, such as the robust estimation of richness advocated by Hughes et al. (5) and approaches for estimating functional properties of bacteria from phylogenetic inference (16).
ESTIMATING GENETIC DIVERSITY
When DNA sequence information is considered, diversity is described by the variation in the nucleotide sequences among individuals. All sampled sequences are related through common ancestry; therefore, the total genetic variation, referred to as theta (θ), can be visualized as a phylogenetic tree with dimensions that depend on the number of replicating lineages, the nucleotide substitution rate, and the time over which the sampled sequences have evolved.
Various statistics have been invented to estimate θ. Average sequence divergence is the number of nucleotide differences between two randomly chosen sequences from a population. It is calculated as follows:
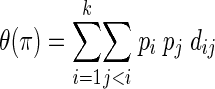 |
where k is the number of distinct sequences, pi is the frequency of the ith sequence, pj is the frequency of the jth species, and dij is the number of differences between two sequences (27). If we divide θ(π) by the length of the sequences being compared, we obtain the average nucleotide diversity, the probability that two randomly chosen sequences differ at a single base position. Values of θ(π) provide an estimate of the total variation in a sample of sequences; moreover, equations for the variance of θ(π) exist (27), providing the possibility of statistical comparison of different samples of sequences. A drawback of this statistic is that, like the Shannon-Weiner index, accurate estimation of average sequence divergence depends on estimates of sequence frequencies. Estimation of gene frequency from surveys of community DNA libraries can be biased because of PCR drift and selection (18, 19, 24). Consequently, the relative abundance of sequences amplified by PCR may not accurately estimate the relative abundance of DNA sequences in nature.
An alternative approach for estimating genetic diversity is to summarize the phylogeny as a cumulative function of the number of lineages relative to time. If a tree is drawn such that the branch lengths from the common ancestor of all sequences (the root) to all terminal taxa (the OTUs sampled) are identical, the distribution of divergence times can be summarized as a lineage-per-time plot (13) (Fig. 1). This approach has several advantages. First, the method is not sensitive to gene frequencies, thereby avoiding some of the biases introduced by molecular techniques that plague estimates of species diversity. Second, the null expectation is that the distribution of divergence times is exponentially distributed: the time between successive divergence events progressively decreases with increasing distance from the root of the tree (27). Comparison of observed lineage-per-time plots with best-fit exponential regression equations provides a means of assessing whether communities harbor an excess of highly divergent or closely related species. An excess of divergent lineages suggests that selection may maintain high diversity in the community (Fig. 1). The existence of divergent bacterial consortia, like those that mediate anaerobic oxidation of methane (1), might result in lineage-per-time plots exhibiting the signature of an excess of divergent lineages. By contrast, an excess of closely related lineages might be a fingerprint for a recent selective sweep of one or a few microbial types, as might be expected following antibiotic treatments.
FIG. 1.

Lineage-per-time plots constructed by counting the numbers of lineages present at different time intervals for trees in which branch lengths were optimized by using maximum likelihood, assuming a molecular clock. Time intervals are equal lengths and were defined arbitrarily. The upper tree is one in which there is an excess of highly divergent lineages, yielding a concave lineage-per-time plot. The tree on the lower right is one in which there is an excess of closely related lineages, yielding a convex lineage-per-time plot. Constant rates of birth and extinction of lineages yield exponential lineage-per-time plots, whose signature is indicated by the solid straight line.
DO MICROBIAL COMMUNITIES DIFFER SIGNIFICANTLY?
Ecologists often adopt a comparative framework for establishing the significance of observed phenomenon. For instance, the effect of nitrogen on microbial communities can be evaluated by comparing microbial communities in soils subjected to different fertilization treatments (11). Robust biological inferences depend on rigorous statistical comparison. In the literature, it is standard practice to calculate the percent overlap in species composition of two communities as a means of assessing difference (or similarity). A problem with this approach is that similarity is based only on the fraction of identical species; all sequences that are different are considered equally even though the two communities may harbor very different samples of sequences. For instance, imagine two communities that do not have any sequences in common; however, for every sequence present in one community, there is a closely related sequence in the other community. In this case, the percent overlap based on OTUs is zero, yet the two communities harbor nearly identical phylogenetic diversity (Fig. 2). Thus, phylogenetic information provides more resolution for testing the degree of differentiation between communities.
FIG. 2.

Comparison of the genetic diversities of two communities, one indicated by open boxes and the other indicated by solid boxes. The trees are drawn such that branch lengths are proportional to the amounts of differentiation. The tree on the left shows the two communities combined; none of the species overlap, but each community harbors identical phylogenetic diversity, as shown by the two trees on the right.
One approach for assessing the degree of differentiation between microbial communities compares the genetic diversity within each community to the total genetic diversity of the communities combined. This is explicit in the equation
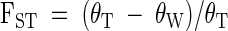 |
where θT is the genetic diversity for all samples (all communities combined) and θW is the genetic diversity within each community averaged over all the communities being compared (3, 22). (Population differentiation by using FST can be accomplished by using a variety of different computer programs; for this paper, I used Arlequin [20].) Consider the two communities described above, namely, two communities that do not have any sequences in common, but for every sequence present in one community, there is a closely related sequence in the other community. In this case, the level of diversity within each community is approximately equal to the level of diversity of the two communities combined (i.e., θW ≈ θT), yielding FST ≈ 0. Thus, even though the species do not overlap, the genetic diversity does. Statistical significance of FST is evaluated by randomly assigning sequences to populations and calculating the FST for 1,000 permutations. Importantly, analysis of variance among communities can be done within particular phylogenetic groups of microbes (e.g., within the α-Proteobacteria).
An alternative approach is to test whether the distribution of unique sequences between different communities exhibits significant covariation with phylogeny. One way to measure covariation is to optimize the presence or absence of particular sequences sampled from multiple communities on the phylogeny by using an objective criterion (e.g., parsimony). For any given set of sequences sampled from multiple communities, parsimony provides an estimate of the minimum number of changes (switch from one community to another) to explain the observed distribution (Fig. 3). The significance of the observed covariation is established by determining the expected number of changes under the null hypothesis that the community from which sequences were sampled does not covary with phylogeny. Maddison and Slaktin (9) showed that null expectation can be estimated by assuming that the community identity of individual sequences remains fixed and that the relationships among sequences are randomized. The number of changes for 1,000 random joining trees represents the expectation for the null hypothesis (the distribution can be determined with the computer program MacClade [8]). If the observed number of transitions from one community to another is less than the null expectation, then the representation of microbial diversity differs significantly between the two communities (Fig. 3). I refer to this evaluation of differentiation as the phylogenetic (P) test.
FIG. 3.

Illustration of the P test. The community type (solid or open boxes) is shown for a sample of 15 sequences. Given the observed phylogenetic relationships of the 15 sequences, the distribution of sequences with respect to community type requires two changes to explain how community type and phylogeny covary (changes are indicated by solid circles). The significance of covariation can be assessed by constructing n random trees and, for each replicate tree (r), determining the number of changes required to explain the covariation of community type and phylogeny. Values for random trees can be summarized as a frequency distribution, and the significance of the observed covariation can be established by comparison. The vertical line labeled with an asterisk delimits significance. In this case, hypothetical case A is significant (indicating that two communities harbor distinct groups of microbes), whereas hypothetical case B is not significant.
The FST and P tests yield different information about differentiation between communities (Fig. 4). Significance for both tests signals less genetic diversity within each community than for two communities combined and that the different communities harbor distinct phylogenetic lineages. Insignificance for both tests implies that the sequences sampled from two communities were drawn from the same distribution, resulting in levels of diversity and samples of phylogenetic lineages within each community that are statistically indistinguishable from those for the communities combined. A significant P test coupled with an insignificant FST test implies that two communities harbor high levels of diversity (relative to the combined data) but that the phylogenetic lineages present in each community differ. This can occur when both communities harbor many ancient lineages but the groups of ancient lineages are different in the different communities (Fig. 4). Alternatively, a significant FST test combined with an insignificant P test implies that the average within-community diversity is significantly less than the diversity when two communities are combined, even though the sampling of the phylogenetic diversity is statistically indistinguishable. This can occur if each community harbors unique groups of closely related microbes that are distributed across the tree (Fig. 4).
FIG. 4.

Examples of phylogenetic trees depicting patterns of relationships that would result in the four possible results from the two tests of differentiation discussed in this paper. The open and solid squares represent different communities, and the trees are drawn with branch lengths proportional to the amounts of sequence evolution. Clear differentiation is evident if both FST and P tests are significant. By contrast, insignificance for both tests implies that the samples from two communities are drawn from the same pool of sequences. A significant FST test coupled with an insignificant P test implies that the tree contains several clades of closely related bacteria that are unique to one community or the other but that these clades are interspersed throughout the phylogenetic tree of all samples. Finally, a significant P test coupled with an insignificant FST test might reflect the existence of highly divergent lineages within each community (such that the within-community diversity approaches the total diversity) but indicate that there is significant covariation between community and phylogeny.
The FST and P tests differ from a method recently proposed by Singleton et al. (21) for comparing communities by using DNA sequence data. The method of Singleton et al. compares the sampling coverage of a community (based on OTUs) with the rank order evolutionary distance between sequences. This approach is similar to comparing lineage-per-time plots for two different communities.
CASE STUDIES
Estimators of diversity and differentiation based on microbial sequence data provide promising tools for assessing microbial diversity and the nature of and differences between microbial communities. To investigate the utility of these tools, I analyzed three 16S rDNA data sets used to describe and compare microbial community diversity. The data sets were chosen because each provides a unique view of the phylogenetic diversity of microbial diversity and the three sets provide the opportunity for exploring the full range of possible outcomes. Moreover, two of the three sets of communities were the subject of a recent review that focused on the issue of estimating the diversity of microbial communities (5). For all data sets, the raw sequence data available from GenBank were used for analyses; OTU designations of the original authors were not used when statistics of diversity and differentiation were calculated.
Microbial communities in the human mouth and gut.
Many of the best-sampled microbial communities come from humans. Kroes et al. (6) sampled the bacterial community in the subgingival plaque by using 16S rDNA methods. Suau et al. (23) surveyed the diversity of the human gut using the same techniques. In both cases, a community DNA extract was used as a template for PCR amplification, PCR products were cloned, and a sample of clones was sequenced. Kroes et al. sequenced 264 clones, and Suau et al. sequenced 284 clones; in the two studies, 59 and 63 different sequences from these clones were reported, respectively. The ecological and evolutionary estimates of diversity were remarkably similar for the two environments (Table 1). On average, two randomly sampled sequences were about 30% different, implying that there was a high level of sequence diversity in the samples.
TABLE 1.
Comparison of standard ecological and molecular estimates of sequence diversity for the human mouth and gut
| Community | Diversity estimate
|
|||||
|---|---|---|---|---|---|---|
| No. of distinct sequences | No. of OTUsa | Shannon-Weiner index | Gene diversity | Nucleotide diversity | θ(π)b | |
| Mouth | 59 | 123 (93, 180)c | 3.18 | 0.958 ± 0.004 | 0.32 ± 0.15 | 341.7 ± 162.1 |
| Gut | 63 | 135 (110, 170) | 3.50 | 0.960 ± 0.004 | 0.30 ± 0.14 | 154.8 ± 73.6 |
Data from reference 5.
The values are not directly comparable because different numbers of nucleotide positions were examined.
The values in parentheses are the 95% confidence limits.
Perusal of the gene tree for the combined data from the human mouth and gut showed that each community harbored unique monophyletic groups that were highly divergent (tree not shown). Lineage-per-time plots were similar and showed remarkable correspondence with trees resulting from constant birth and death rates of lineages; the only detectable difference between the two communities was an apparent excess of divergent lineages in the mouth, although the difference was not tested for significance. The analysis of differentiation based on genetic diversity when only single representatives of each unique sequence were used was significant (FST = 0.11, P < 0.00001). In addition, the P test statistic was highly significant (P < 0.001). These results indicated that although the two communities harbored nearly identical diversity (Table 1), they were highly disparate, exhibiting little phylogenetic overlap. These results provided unambiguous evidence of significant and substantial differentiation between the microbial communities of the mouth and the gut.
Microbial diversity associated with improved and unimproved grasslands.
McCaig et al. (11) surveyed bacterial diversity in improved and unimproved pasture communities by using 16S rRNA analysis. The unimproved plots sampled represented natural grassland that had never received fertilization but was grazed by sheep during summer months. By contrast, the improved sites were planted with an introduced grass and clover (a nitrogen-fixing plant) and received 40-20-20 fertilizer twice each year. Sheep grazed these plots from spring to fall. Both community types were from the same area in Scotland (the Sourhope Research Station).
Species diversity and species richness were slightly, but not significantly, higher in the unimproved soils than in the improved soils (Table 2) (5). The estimates of nucleotide diversity were identical, and values for θ were not significantly different. Overall, an immense diversity of microbes was sampled, spanning several different divisions of bacteria (11). Lineage-per-time plots also were remarkably similar for the two types of plots and were nearly identical to theoretical predictions if diversification corresponded to a constant birth and death model (Fig. 5). These results imply that the two communities harbor similar high levels of genetic diversity even though they may have different numbers of OTUs (Table 2). Although “the (taxonomic) diversity was slightly greater in the unimproved soil libraries than in the improved soil libraries” (11), the two communities exhibited nearly identical genetic diversity.
TABLE 2.
Comparison of standard ecological and molecular estimates of sequence diversity for unimproved and improved grassland soil communities
| Community | Diversity estimate
|
|||||
|---|---|---|---|---|---|---|
| No. of distinct sequences | No. of OTUsa | Shannon-Weiner index | Gene diversity | Nucleotide diversity | θ(π) | |
| Unimproved | 138 | 590 | 2.04 | 1.0 | 0.37 ± 0.18 | 121.4 ± 52.4 |
| Improved | 137 | 467 | 2.01 | 0.960 | 0.37 ± 0.18 | 114.8 ± 49.6 |
Data from reference 5. The estimated numbers of OTUs were not significantly different.
FIG. 5.

Lineage-per-time plot for the improved (•) and unimproved (□) grassland microbial communities. The ordinate is the log of the number of lineages, and the abscissa is the time (in arbitrary units) measured from the common ancestor. See the legend to Fig. 1 for a description of how the plot was constructed. The phylogenetic diversities of the two communities are virtually identical.
Tests of differentiation based on levels of genetic diversity (FST) were insignificant (Table 3), implying that the within-community variation was nearly identical to the total diversity sampled when the two communities were combined. However, the P test was significant (Table 3), implying that the two communities harbored different groups of bacteria. McCaig et al. (11) noted that “α-proteobacterial clones were more diverse in the unimproved than in the improved soil samples.” To assess the influence of potential differences in the representation of α-proteobacterial lineages between the two environments, FST and P tests were performed without these taxa. Lack of differentiation implied that the difference between the communities was mainly attributable to differential representation of α-proteobacterial lineages. Both the FST and P tests were significant when they were applied to the α-proteobacteria alone (Table 3). This is an important extension of the conclusions of McCaig et al. (11). Not only did the two communities exhibit differences in diversity (based on phylotypes), but they were also phylogenetically disparate. The strong signal of differentiation was lost when clade B was omitted from the analysis (Table 3). This clade was represented by relatively few sequences (10). Most of the sequences were highly divergent from other sampled sequences, and eight of nine sequences were sampled from the unimproved grassland. These results suggest that much of the differentiation between the communities can be attributed to the differential representation of clade B in the two communities. However, when I examined differentiation for one of the distinct α-proteobacterial clades (identified as clade A in Fig. 6), the FST test was significant, but the P test was not significant. This result can be explained by the presence of several groups of relatively closely related bacteria in each community, so that the average within-community diversity was less than the diversity in the two communities combined, coupled with the fact that each unique group was interspersed in the tree. Assessment of the differentiation for sequences in clade C revealed an insignificant FST test and a significant P test, implying that the two communities harbored different phylogenetic lineages, although the degree of phylogenetic differentiation was relatively slight (Table 3). When considered together, the tests for differentiation for the α-proteobacteria supplement the evaluation of differentiation offered by McCaig et al. (11) based on analysis of phylotypes. Although differentiation was evident at several hierarchical scales within the α-proteobacteria, most of the signal was attributable to the differential distribution of clade B. Phylogenetic analysis of close matches from the GenBank database indicated that clade B contains sequences related to Rhodobacter, Paracoccus, and Aquabacter. Attempts to gain an understanding of functional differentiation between microbial communities inhabiting fertilized and unfertilized soils might begin by focusing on the biology of this group of bacteria.
TABLE 3.
Summary of FST and P tests for differentiation between improved and unimproved grassland communities
| Group |
P valuea
|
|
|---|---|---|
| FST test | P test | |
| All sequences | NS | 0.002 |
| Without α-proteobacteria | NS | NS |
| All α-proteobacteria | 0.00879 | 0.018 |
| α-Proteobacteria without clade Bb | NS | NS |
| Clade Ab | <0.00000 | NS |
| Clade Cb | NS | 0.032 |
NS, not significant.
See Fig. 6.
FIG. 6.

16S rDNA gene tree for the α-proteobacteria sampled from improved (solid boxes) and unimproved (open boxes) grasslands. The letters indicate specific clades that were subject to tests of differentiation (Table 3). The tree was produced by neighbor-joining clustering of genetic distances corrected for multiple substitutions by using a HKY + G + I model of evolution with PAUP* (25). (For details about the construction of phylogenetic trees from sequence data, see reference 26.) Optimization of the branch lengths was done by using the maximum-likelihood method (using the HKY + G + I model) subject to the constraint that all sampled sequences were contemporary (i.e., molecular clock was enforced).
Microbial diversity at different depths in an anaerobic water column.
Madrid et al. (10) obtained microbial community samples from depths of 320, 500, and 1,310 m below the surface in the anoxic zone of the Cariaco Basin. This basin has restricted water circulation, and no oxygen exists below a depth of about 240 to 320 m. 16S rDNA analysis was used to survey the diversity of microbes at different depths. Because the environment is stable, only the amount of substrate delivered from the surface water varies with depth (10). Clone libraries were constructed from water samples taken at each depth, and different portions of the 16S rDNA were sequenced. In contrast to the other two studies, the amount of sequence data obtained per clone varied among the clone libraries. The sampling efforts per library were similar, however (51, 56, and 65 clones were surveyed from the 320-, 500-, and 1,310-m samples, respectively).
Table 4 shows the diversity estimate published by Madrid et al. (10) for each environment compared with diversity statistics borrowed from population genetics. Several of the statistics show similar patterns. For instance, the values for gene diversity, the Shannon-Weiner index, and nucleotide diversity show similar patterns; the shallowest sample harbored the least diversity, and the two deeper samples harbored more diversity and were similar. Similarly, the average nucleotide diversity mirrors this pattern. Despite the dramatic difference in diversity between the shallow- and deeper-water samples, estimates of θ(π) were similar at all sites. The difference does not reflect similarity in the levels of diversity but reflects differences in the number of bases determined for different isolates. At the shallow site, 839 bp were comparable across all sequences sampled; by contrast, at the 500- and 1,310-m depths, only 282 and 294 bp were comparable. Thus, the estimates of θ from the shallow site are not directly comparable to the estimates of θ from the deeper sites.
TABLE 4.
Comparison of standard ecological and molecular estimates of sequence diversity for Cariaco Basin water samples
| Community | Diversity estimate
|
||||
|---|---|---|---|---|---|
| No. of distinct sequences | Shannon-Weiner index | Gene diversity | Nucleotide diversity | θ(π)a | |
| 320 m | 9 | 0.32 | 0.28 | 0.10 ± 0.05 | 83.9 ± 36.6 |
| 500 m | 44 | 1.33 | 0.95 | 0.30 ± 0.15 | 84.9 ± 37.2 |
| 1,310 m | 49 | 1.38 | 0.94 | 0.30 ± 0.15 | 88.7 ± 38.8 |
The values are not directly comparable because different numbers of nucleotide positions were examined.
The phylogenetic tree of all sequences combined revealed tremendous genetic diversity within each sample (Fig. 7); however, the lineage-per-time plots showed differences in the degree of differentiation among communities (Fig. 8). The deepest sample harbored an excess of highly divergent lineages compared to the two shallower samples. Moreover, all three communities exhibited a pattern that differed from the patterns for the human and soil communities examined previously. The excess of divergent lineages in the deeper water and the step pattern exhibited by the two shallow-water communities may reflect a limited number of disparate ecological niches (lower ecosystem complexity). This may be especially true for the shallowest sample, which was dominated by a single sequence.
FIG. 7.

16S rDNA gene tree for Cariaco Basin samples. The boxes identify eubacteria sampled from different environments, as follows: open boxes, 1,310 m; solid boxes, 500 m; shaded boxes, 320 m. The tree was generated by using the methods described in the legend to Fig. 6. None of the groups exhibited a restricted distribution. The asterisk indicates the one sequence (phylotype) that accounted for significant differentiation between the shallow-water sample (320 m) and the two deeper-water samples based on the FST test when all sequences were analyzed. Scale bar = 0.05 substitution per site.
FIG. 8.

Lineage-per-time plots for the three environments sampled from Cariaco Basin. A comparison across communities suggests that an excess of highly divergent microbial lineages exists at 1,310 m compared to the lineages at the shallower sites.
Genetic differentiation among the sampled microbial communities was assessed by using FST. Significant differentiation was evident between the shallow-water sample (320 m) and the two deeper-water samples (P < 0.0001 for both comparisons). The 500- and 1,310-m community samples did not differ significantly, however (P = 0.093). Most of the differentiation between the shallow-water and deep-water communities was due to the occurrence of a single, very abundant sequence at 320 m. When I examined the differentiation among the communities based on single representatives of each distinct bacterial sequence, the FST test was not significant (data not shown), a result mirrored by the P test (P > 0.50)
Madrid et al. (10) concluded that “The composition of the 320m library was markedly different from the compositions of the other two libraries” and that the 320-m “microbial community…is substantially different from the communities in the 500- and 1310-m samples.” While these statements are true based on assessments of diversity that consider the frequency of different sequences, the phylogenetically based methods of analysis did not detect significant differences among communities; none of the environments sampled harbored phylogenetically unique groups. Moreover, the significant genetic differentiation between the 320-m community and the deeper-water communities was attributable to the high relative abundance of only one sequence (Car153a), whose closest relative has not been cultivated. Madrid et al. (10) noted, though, that Car153a groups with species of ɛ-proteobacteria are capable of sulfate reduction (17). Overall, while the diversity analysis revealed differences between the shallow-water and deeper-water samples, analysis of the genetic diversity suggested that the microbial species present in all three communities were organisms from the same pool of diversity. Most interesting was the inference that all communities, especially the community in the deepest sample, harbored more divergent lineages than closely related lineages, a pattern that differed markedly from the patterns for the communities from humans and grassland soils described above.
DISCUSSION
Most studies of microbial communities in which 16S rRNA sequences are used rely on standard estimates of diversity (i.e., the Shannon-Weiner index). This approach is informative, but it ignores important information about the disparity among the sampled sequences. In this study I used standard quantitative methods of analysis borrowed from population genetics and systematics for describing and comparing microbial communities. Information gained from analysis of DNA sequences provided the basis for statistical analysis of communities in ways that advance inferences about the processes that may govern the compositions and functions of microbial communities. Furthermore, the analytical approaches advocated here make it possible to accomplish broad comparisons of ecological communities. For instance, a comparison of lineage-per-time plots across a diverse set of ecosystems might reveal differences in the phylogenetic compositions of ecological communities that would be invisible with standard ecological statistics that ignore the magnitude of genetic differences among sampled sequences.
These statistical approaches still depend on robust sampling of diversity. It is important, for instance, that comparisons of diversity involve the same (or broadly similar) sequences. Some studies focus on only a small portion of the 16S rDNA gene, while others survey the variation in the entire gene. Appropriate comparative studies of these two unevenly sampled communities would require only comparisons of overlapping sequences because of marked differences in the levels of variability across the 16S rDNA molecule. Therefore, complete or nearly complete 16S rDNA sequences should be determined to facilitate broad comparative analyses. Of course, it is necessary to balance a broad survey of microbial diversity with sequencing effort, suggesting that the length of sequence is sacrificed in favor of surveying more clones.
Hughes et al. (5) advocated adopting methods that enhance the accuracy of estimating the species richness of microbial communities because such information should enhance our understanding of the processes that underlie ecosystem function. These authors noted, however, that inferences about the structures and functions of ecological communities depend, in part, on the criterion employed to define OTUs and that no single objective criterion exists for defining OTUs. The lack of consensus limits the comparative utility of statistics based solely on identification of species (or OTUs). A focus on species richness ignores important information, though. Because genetic differentiation may be more predictive of functional properties than of the number of species, a more informative assessment of diversity is one that incorporates information about the phylogeny of the species sampled. The relevant question is not whether two communities have different numbers of species, but whether the communities harbor different phylogenetic groups and different levels of genetic diversity. For each of these estimates, there is no need to impose an arbitrary objective criterion for defining what constitutes a species. The phylogenetic tree (namely, the relationships and degrees of divergence among sequences) provides the necessary information.
An additional advantage of some phylogenetic methods is that they do not rely on estimation of the frequencies of different sequences. It is well known that enumeration of species by using sequence analysis of cloned community DNA PCR libraries is subject to bias. Depending on the amplification and cloning conditions, some sequences may be overrepresented while others are underrepresented relative to their abundance in nature. This bias compromises the utility of frequency-based estimates of diversity. Standard statistics of diversity (e.g., the Shannon-Weiner index) are highly sensitive to estimates of the frequencies of different species. By contrast, lineage-per-time plots are not sensitive to the accuracy of phylotype frequency estimation. A second source of bias is PCR mutagenesis, namely, the introduction of sequence variation due to PCR replication. Because PCR mutagenesis introduces little sequence variation (typically about 1 change per 1,000 bases [2]), it does not significantly influence assessments of diversity based on OTUs or phylogenetic diversity. PCR mutagenesis should significantly influence θ and the FST and P tests only if certain templates are particularly prone to mutagenesis, resulting in large clusters of closely related phylotypes.
The statistical approaches for characterizing and comparing the diversity of microbial communities advocated in this paper are not new. Population geneticists and systematists have been using these methods for years for the same purpose: to characterize the diversity of populations or groups for inferring processes governing diversification. Because a rich and varied literature exists, incorporation of phylogenetically based assessments of diversity with the more traditional taxon-based estimates should prove to be easy. A key feature of the analyses which I have described is that the information contained in the inferred phylogeny of the sampled sequences is mined for more than gaining putative identities of sequences and defining OTUs for enumerating species richness and comparing communities. In particular, the phylogenetic methods can be used to identify particular groups that account for differences that may exist between communities, and they permit inferences about processes that may regulate the composition of microbial communities.
Acknowledgments
I thank members of the Schmidt lab for their emphasis on microbes in world affairs and Al Meyer for comments on a previous version of the manuscript.
This paper is based on work supported by the National Science Foundation under grant 0084223.
REFERENCES
- 1.Boetius, A., K. Ravenschlag, C. J. Schubert, D. Rickert, F. Widdel, A. Gieseke, R. Amann, B. B. Jorgensen, U. Witte, and O. Pfannkuche. 2000. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407:623-626. [DOI] [PubMed] [Google Scholar]
- 2.Cline, J., J. C. Braman, and H. H. Hogrefe. 1996. PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 24:3546-3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edwards, S. V. 1993. Mitochondrial gene genealogy and gene flow among island and mainland populations of the sedentary songbird, the grey-crowned babbler (Pomatostomus temporalis). Evolution 47:1118-1137. [DOI] [PubMed] [Google Scholar]
- 4.Faith, D. 1994. Phylogenetic pattern and the quantification of organismal biodiversity. Phil. Trans. R. Soc. Lond. B Biol. Sci. 345:45-58. [DOI] [PubMed] [Google Scholar]
- 5.Hughes, J. B., J. J. Hellmann, T. H. Ricketts, and B. J. M. Bohannan. 2001. Counting the uncountable: statistical approaches for estimating microbial diversity. Appl. Environ. Microbiol. 67:4399-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroes, I., P. W. Lepp, and D. A. Relman. 1999. Bacterial diversity within the human subgingival crevice. Proc. Natl. Acad. Sci. USA 96:14547-14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuittinen, H., A. Mattila, and O. Savolainen. 1997. Genetic variation at marker loci and in quantitative traits in natural populations of Arabidopsis thaliana. Heredity 79:144-152. [DOI] [PubMed] [Google Scholar]
- 8.Maddison, W. P., and D. R. Maddison. 1993. MacClade, v. 3. Sinauer Press, Sunderland, Mass.
- 9.Maddison, W. P., and M. Slatkin. 1991. Null models for the number of evolutionary steps in a character on a phylogenetic tree. Evolution 45:1184-1197. [DOI] [PubMed] [Google Scholar]
- 10.Madrid, V. M., G. T. Taylor, M. I. Scranton, and, A. Y. Chistoserdov. 2001. Phylogenetic diversity of bacterial and archaeal communities in the anoxic zone of the Cariaco Basin. Appl. Environ. Microbiol. 67:1663-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCaig, A. E., L. A. Glover, and J. I. Prosser. 1999. Molecular analysis of bacterial community structure and diversity in unimproved and improved upland grass pastures. Appl. Environ. Microbiol. 65:1721-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan, K. K., J. Hicks, K. Spitze, L. Latta, M. E. Pfrender, C. S. Weaver, M. Ottone, and M. Lynch. 2001. Patterns of genetic architecture for life-history traits and molecular markers in a subdivided species. Evolution 55:1753-1761. [DOI] [PubMed] [Google Scholar]
- 13.Nee, S., R. M. May, and P. H. Harvey. 1995. The reconstructed evolutionary process. Phil. Trans. R. Soc. Lond. B Biol. Sci. 344:305-311. [DOI] [PubMed] [Google Scholar]
- 14.Norberg, J., D. P. Swaney, J. Dushoff, J. Lin, R. Casagrandi, and S. Levin. 2001. Phenotypic diversity and ecosystem functioning in changing environments: a theoretical framework. Proc. Natl. Acad. Sci. USA 98:11376-11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nubel, U., F. Garcia-Pichel, M. Kuhl, and G. Muyzer. 1999. Quantifying microbial diversity: morphotypes, 16S rRNA genes, and carotenoids of oxygenic phototrophs in microbial mats. Appl. Environ. Microbiol. 65:422-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pace, N. R. 1997. A molecular view of microbial diversity and the biosphere. Science 276:734-740. [DOI] [PubMed] [Google Scholar]
- 17.Phelps, C. D., K. J. Kerkhof, and L. Y. Young. 1998. Molecular characterization of a sulfate-reducing consortium which mineralizes benzene. FEMS Microbiol. Ecol. 27:269-279. [Google Scholar]
- 18.Polz, M. F., and C. M. Cavanaugh. 1998. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 64:3724-3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reysenbach, A.-L., L. J. Giver, G. S. Wickham, and N. R. Pace. 1992. Differential amplification of rDNA genes by polymerase chain reaction. Appl. Environ. Microbiol. 58:3417-3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider, S., J. Kueffer, D. Roessli, and L. Excoffier. 1997. Arlequin ver. 1.l: a software for population genetic data analysis. Genetics and Biometry Lab, University of Geneva, Geneva, Switzerland.
- 21.Singleton, D. R., M. A. Furlong, S. L. Rathbun, and W. B. Whitman. 2001. Quantitative comparisons of 16S rRNA gene sequence libraries from environmental samples. Appl. Environ. Microbiol. 67:4374-4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slatkin, M. 1991. Inbreeding coefficients and coalescent times. Genet. Res. 58:167-175. [DOI] [PubMed] [Google Scholar]
- 23.Suau, A., R. Bonnet, M. Sutren, J.-J. Godon, G. Gibson, M. D. Collins, and J. Dore. 1999. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl. Environ. Microbiol. 65:4799-4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki, M. T., and S. J. Giovannoni. 1996. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62:625-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swofford, D. L. 2000. PAUP*. Phylogenetic analysis using parsimony (*and other methods), v. 4.0. Sinauer Associates, Sunderland, Mass.
- 26.Swofford, D. L., G. J. Olsen, P. J. Waddell, and D. M. Hillis. 1996. Phylogenetic inference, p. 407-514. In D. M. Hillis, C. Moritz, and B. K. Mable (ed.), Molecular systematics, 2nd ed. Sinauer Press, Sunderland, Mass.
- 27.Tajima, T. 1983. Evolutionary relationships of DNA sequences in finite populations. Genetics 105:437-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tilman, D., J. Knops, D. Wedin, P. Reich, M. Ritchie, and E. Siemann. 1997. The influence of functional diversity and composition on ecosystem processes. Science 277:5330-5336. [Google Scholar]