

Abstract
Oligonucleotide probes were used to study the structure of anaerobic granular biofilm originating from a pentachlorophenol-fed upflow anaerobic sludge bed reactor augmented with Desulfitobacterium frappieri PCP-1. Fluorescence in situ hybridization demonstrated successful colonization of anaerobic granules by strain PCP-1. Scattered microcolonies of strain PCP-1 were detected on the biofilm surface after 3 weeks of reactor operation, and a dense outer layer of strain PCP-1 was observed after 9 weeks. Hybridization with probes specific for Eubacteria and Archaea probes showed that Eubacteria predominantly colonized the outer layer, while Archaea were observed in the granule interior. Mathematical simulations showed a distribution similar to that observed experimentally when using a specific growth rate of 2.2 day−1 and a low bacterial diffusion of 10−7 dm2 day−1. Also, the simulations showed that strain PCP-1 proliferation in the outer biofilm layer provided excellent protection of the biofilm from pentachlorophenol toxicity.
Bioaugmentation of a natural bacterial community is a potentially efficient approach for maximizing the degradation performance of a biological system. However, the fate of a newly introduced strain strongly depends on its interactions with the members of the indigenous bacterial consortium. As a result, the disappearance of introduced strains was observed as often as their retention (4, 5, 7). Bioaugmentation attempts are most successful when a newly introduced strain establishes commensal or mutualistic relationships within a natural consortium, e.g., by degrading substances otherwise toxic to the consortium.
The anaerobic degradation of chlorinated compounds by indigenous microorganisms often results in incomplete dechlorination and thus can benefit from bioaugmentation. In particular, bioaugmentation of an upflow anaerobic sludge bed (UASB) reactor with Dehalospirillum multivorans led to a transformation of perchloroethylene (PCE) to dichloroethylene, an improvement compared to a noninoculated control, which only transformed PCE to trichloroethene (17). Ahring et al. (1) introduced a 3-chlorobenzoate dechlorination activity in a UASB reactor by bioaugmentation with Desulfomonile tiedjei. Also, Christiansen and Ahring (7) demonstrated inoculation of a UASB reactor containing sterilized anaerobic granules with a pentachlorophenol (PCP)-degrading strain of Desulfitobacterium hafniense.
In our previous work, a UASB reactor degrading PCP was successfully inoculated with the PCP-degrading strain Desulfitobacterium frappieri PCP-1 (28). This strain is the only isolated anaerobic microorganism capable of PCP dehalogenation to 3-chlorophenol (3). Proliferation of strain PCP-1 allowed a substantial increase of the volumetric PCP load from 5 to 80 mg/liter of reaction volume/day with a PCP removal efficiency of 99% and a dechlorination efficiency of not less than 91%. Following inoculation, the density of strain PCP-1 was found to increase from 106 to 1010 cells/g of volatile suspended solids (VSS) as observed by competitive PCR. This enumeration method, however, provided no information on the spatial distribution of strain PCP-1 within the anaerobic granules and the colonization process.
The objectives of the present study were twofold: (i) to experimentally determine the distribution of strain PCP-1 within the anaerobic biofilm with regard to that of Archaea and Eubacteria using fluorescence in situ hybridization (FISH) and (ii) to compare the visualized distribution of strain PCP-1 with computer simulations of strain PCP-1 proliferation in the biofilm based on a bacterial transport and growth biofilm model.
MATERIALS AND METHODS
Microorganisms and enrichment conditions of the UASB bioreactor.
D. frappieri PCP-1 (ATCC 700397) was grown under strict anaerobic conditions at 37°C in a mineral salts medium supplemented with pyruvate (55 mM) and yeast extract (0.1%), as described by Bouchard et al. (3). Anaerobic sludge (62 g of VSS liter−1) was obtained from an industrial UASB reactor treating baby food industry wastewater (Champlain Industries Cornwall, Ontario, Canada). A 5-liter UASB reactor was inoculated with 1.5 liter of anaerobic sludge (93 g of VSS) and 10 ml of the pure strain PCP-1 culture. The initial density of strain PCP-1 in the reactor was 106 cells/g of VSS. The reactor was operated at a temperature of 35°C, which is considered optimal for operation of mesophilic anaerobic reactors (26). Other operational parameters were a pH of 7.3 and a retention time of 28 h. It was fed with a 2-g/liter solution of PCP in NaOH and a nutrient solution which contained (in grams liter−1) sucrose, 304; butyric acid, 96; yeast extract, 7; ethanol (95%), 70; KH2PO4, 6; K2HPO4, 7; NH4CO3, 68, and a chloride-free trace metal solution. A dilution stream contained (in grams liter−1) NaHCO3, 1.36, and KHCO3, 1.74. The PCP loading rate was related to methane production in the reactor, i.e., the toxicity of PCP towards the anaerobic consortium expressed as a normalized methane yield determined the PCP load. More details can be found in the work of Tartakovsky et al. (28).
Oligonucleotide probes.
The Cy3-labeled ARCH915 (22) and Cy3-labeled EUB338 (2) probes hybridized, respectively, to members of Archaea and most Eubacteria. Three probes (Table 1) were designed for the FISH detection of D. frappieri PCP-1 by comparing its 16S rRNA gene (rDNA) sequences with sequences from GenBank (National Center for Biotechnology Information, Bethesda, Md.) and the Ribosomal Database Project II (Michigan State University, East Lansing). S-S-D.frap-327(D. frappieri)-a-A-19 is unique to the 16S rDNA sequences of the four D. frappieri strains (PCP-1, DP7, TCP-A, TCE1), of Desulfitobacterium chlororespirans, and of Desulfitobacterium hafniense but has at least two mismatches with other 16S rDNA of other Desulfitobacterium species. The S-S-D.frap-86(D. frappieri)-a-A-20 sequence is located in the 128-nt insertion characteristic of D. frappieri 16S sequences (3) and is unique to the 16S rDNA sequences of D. frappieri strains PCP-1, TCE1, and TPA but different from D. frappieri DP7 and other bacteria. S-S-D.frap-576(D. frappieri)-a-A-19 is unique to the 16S rDNA sequences of the four D. frappieri strains and of Desulfitobacterium hafniense but has at least two mismatches with 16S rDNA sequences of other Desulfitobacterium species. S-S-D.frap-327(D. frappieri)-a-A-19 and S-S-D.frap-86(D. frappieri)-a-A-20 were both labeled with Cy3, while S-S-D.frap-576(D. frappieri)-a-A-19 was labeled with Cy5. Fluorescent probes were synthesized by Medicorps (Montreal, Quebec, Canada). Oligonucleotide helpers (13) S-S-D.frap-555(D. frappieri)-a-A-21 and S-S-D.frap-595(D. frappieri)-a-A-18, synthesized by Hukabel Scientific LTD (Montreal, Quebec, Canada), were added to the hybridization solution to raise the fluorescent signal of S-S-D.frap-576(D. frappieri)-a-A-19.
TABLE 1.
Sequences of oligonucleotide probes targeting 16S rDNA sequencesc
| Probe | Target organisms | Sequence (5′-3′) | Positiona |
|---|---|---|---|
| ARCH915 | Archaea | GTGCTCCCCCGCCAATTCCT | |
| EUB338 | Eubacteria | GCTGCCTCCCGTAGGAGT | |
| S-S-D.frap-327 (D. frappieri)-a-A-19b | All strains of D. frappieri, Desulflitobacterium hafniense, and Desulfitobacterium chlororespirans | GCGGATCCATCTACTAACG | 345-327 |
| S-S-D.frap-86 (D. frappieri)-a-A-20b | All strains of D. frappieri except strain DP7 | ATCCACTTATCTGCTCCTTA | 105-86 |
| S-S-D.frap-576 (D. frappieri)-a-A-19b | All strains of D. frappieri and Desulfitobacterium hafniense | CCGTCATGTAAGTACATTA | 594-576 |
| S-S-D.frap-555 (D. frappieri)-a-A-21 | Helpers for the S-S-D.frap-576(D. frappieri)-a-A-19 probe | TTTACATACTTACCGTTCGTC | 575-555 |
| S-S-D.frap-595 (D. frappieri)-a-A-18 | Helpers for the S-S-D.frap-576(D. frappieri)-a-A-19 probe | GGGCTTCCTCCTCAGGTA | 612-595 |
Position relative to strain PCP-1 16S rDNA sequence.
Sequence is identical to the 16S rDNA sequence of the mentioned desulfitobacteria.
All oligonucleotides were labeled with Cy3 except S-S-D.frap-576(D. frappieri)-a-A-19, which was labeled with Cy5 and two helpers (unlabeled).
FISH.
Granular biomass was washed three times for 5 min each time in ice-cold filtered phosphate-buffered saline (PBS) buffer (130 mM NaCl, 7 mM Na2HPO4, 3 mM NaH2PO4·H2O) and fixed for 3 h in paraformaldehyde 4%-PBS, pH 7.2. Then, the granules were dehydrated in a graded series of ethanol solutions of 50, 70, 95, and 100%; incubated in pure xylene; and then incubated overnight at room temperature in a 50:50 preheated (at 60°C) solution of Paraplast and xylene. Solidified samples were then incubated three times in pure paraplast wax at 60°C for 1 h and were transferred to plastic molds. Solidified blocks were cut in 7-μm slices, and the slices were mounted on poly-l-lysine slides (Polysciences, Niles, Ill.). Blocks and mounted samples were kept at room temperature (23).
To eliminate the paraplast wax and permeabilize the cells, slices were soaked two times for 10 min each time in pure xylene and then 1 time for 10 min in 100% ethanol (18). One milliliter of acetylation solution (100 mM triethanolamine, 0.25% acetic anhydride, 0.09% NaCl; pH 7.2) was added to each slide. The slides were incubated for 10 min at room temperature and rinsed with deionized water. The acetylation step helped to lower nonspecific binding of the probes to proteins (10). Then, 10 μl of hybridization solution (35% deionized formamide, 0.9 M NaCl, 0.02 M Tris-HCl, 0.01% sodium dodecyl sulfate; final pH, 7.2) containing 25 ng of fluorescent probe was directly added to each slice. Autofluorescence was verified by applying the hybridization buffer to a slice in the absence of a probe (23). To improve fluorescence signal with the S-S-D.frap-576(D. frappieri)-a-A-19 probe (labeled with Cy5), the two nonfluorescent helper oligonucleotides (25 ng each) were added to the hybridization buffer (13). After addition of the hybridization solution, the slides were separately incubated in chambers humidified by paper towels saturated with 1 M NaCl (27). Incubation was carried out in the dark at 46°C for 2 h. Following hybridization, the slides were washed twice at 48°C in an agitated bath containing washing buffer (0.04 M NaCl; 0.02 M Tris-HCl, pH 7.2; 0.01% sodium dodecyl sulfate; 0.005 M EDTA) for 20 min each (18). The slides were then rinsed with deionized water or PBS and stained with DAPI (4′,6-diamino-2-phenylindole dihydrochloride; 10 μg ml−1) for 10 min at room temperature and in the dark. Subsequently, slides were rinsed with deionized water or PBS, and a coverslip was added and sealed with nail varnish. Slides were examined at magnifications of ×100, ×250, and ×400 with an epifluorescence microscope (Laborlux S; E. Leitz, Wetzlar, Germany) equipped with filters for DAPI, Cy3, and Cy5 (Chroma Technology Corp., Brattleboro, Vt.) and a mercury short-arc photo optic lamp (HBO 103 W/2; OSRAM Sylvania Ltd., Mississauga, Ontario, Canada). Antifade was not used since initial experiments showed no significant difference between samples prepared with and without the antifade.
Images of granule cross sections were acquired using a charge-coupled device camera (Photometrics Coolsnap; Roper Scientific, Trenton, N.J.) with a filter and Coolsnap software (version 1.0.0; Roper Scientific). The exposure time was 2 s for the DAPI and Cy3 probes and 8 s for the Cy5 probe. Adobe Photoshop (version 5.0; Adobe Systems Inc., San Jose, Calif.) software was used to construct two-color images of stained granule sections.
The hybridization procedure was repeated three to eight times using different samples for each probe and for each sampling time. For each probe, three representative hybridized granules, each obtained in a separate hybridization procedure, were used to analyze distribution of fluorescent areas. The analysis of images was carried out using the NIH Image program (version 1.62; U.S. National Institutes of Health) according to the following sequence. The format of the images was converted to black and white by using a preset threshold value of signal intensity, i.e., white areas on the image represented hybridized cells. Then, the rectangular segments of the image were chosen. Each segment was typically 60 μm high and extended from an outer layer of a granule to its center. The segments were analyzed to determine the number of white pixels at each radial position. The resulting values were proportional to the specific area of hybridized cells. For each granule, at least five segments were selected, and the results were averaged. The results obtained from three different granules were also averaged.
Multispecies biofilm model.
The multispecies biofilm model consisted of partial differential equations representing material balances on key substrates and bacterial trophic groups. The bacterial trophic groups of the model corresponded to those of the oligonucleotide probes used in this study: PCP-degrading bacteria (x1), acidogenic bacteria (x2), and methanogens (x3). The substrates were PCP (s1) and sucrose (s2). For the sake of simplicity, intermediates of the anaerobic degradation of sucrose such as pyruvate, volatile fatty acids (VFAs), and acetate were grouped as a single entity, denoted as s3. Other simplifying assumptions used in the model are common in modeling of biofilms and can be found elsewhere (29, 31).
The overall interaction of substrates and bacterial species in the model is described by the following sequence of transformations:
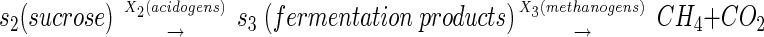 |
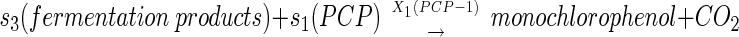 |
Bacterial growth and substrate transformations were modeled using nonlinear kinetic equations that accounted for substrate limitation of bacterial growth (e.g., sucrose-limited growth of acidogens and VFA-limited growth of methanogens, etc) as well as PCP toxicity. Substrate limitation was described using a Monod-like nonlinear dependence of the form L(s) = s/(ks + s), while the substrate inhibition dependence I(s) was described by the equation I(s) = kI/(kI + s), where ks and kI are the limitation and the inhibition constants, respectively and s is the substrate concentration.
Using these definitions, growth rates of PCP-degrading bacteria (μ1), acidogenic bacteria (μ2), and methanogenic bacteria (μ3) were defined as follows:
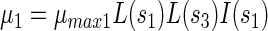 |
(1) |
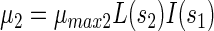 |
(2) |
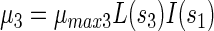 |
(3) |
where μmax1, μmax2, and μmax3 denote the specific growth rates of strain PCP-1, acidogenic, and methanogenic bacteria, respectively.
Specific consumption rates of PCP (q1), sucrose (q2), fermentation products by x3 (q3), fermentation products by x1 (q4), and production rate of fermentation products from sucrose (q5) were defined as follows:
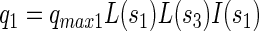 |
(4) |
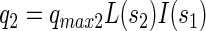 |
(5) |
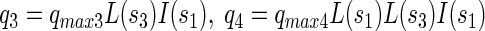 |
(6) |
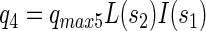 |
(7) |
where qmax denotes the maximal specific transformation rate.
Also, to account for cell transport in the biofilm, a diffusive flux of solids (i.e., bacterial cells) was introduced in agreement with the model formulation proposed by Wanner and Reichert (31).
To describe material balance on each bacterial and chemical species, a set of partial differential equations was defined as follows:
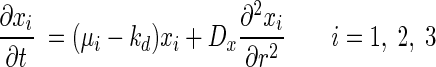 |
(8) |
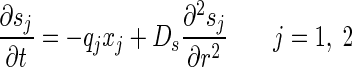 |
(9) |
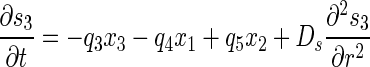 |
(10) |
with the boundary conditions
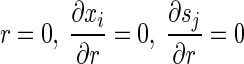 |
(11) |
 |
(12) |
where xb and sb are the bulk concentrations of bacterial species and substrates, respectively.
The set of equations (equations 1 to 12) was transformed to a dimensionless form and solved numerically in Matlab (Mathworks, Inc. [Natick, Mass.] by using a finite-difference method. The source code for the model equations (equations 1 to 12) along with a graphical interface written in Matlab can be accessed at http://www.bri.nrc.ca/envbioeng/PCPmodel.html.
In the simulations, bulk substrate concentrations were chosen with respect to experimental data obtained during reactor operation. The following values were used (in milligrams liter−1): s1 = 5, s2 = 200, and s3 = 30. Bulk concentrations of bacterial species were (in grams of VSS liter−1) x1 = 1, x2 = 5, and x3 = 5. Maximal density of biomass in the biofilm was set to 80 g of VSS liter−1. No biofilm growth was allowed if total density of all bacterial species exceeded this value. Also, a constant granule radius of 1.5 mm, which corresponded to an average radius of biomass granules by the end of the reactor experiment, was assumed. Other model parameters are listed in Table 2.
TABLE 2.
Model parameters
| Parameter(s) | Value | Dimension |
|---|---|---|
| Ds1, Ds2, Ds3 | 5 · 10−4 | dm2 day−1 |
| Dx1 | 10−7-10−6 | dm2 day−1 |
| Dx2, Dx3 | 10−7 | dm2 day−1 |
| qmax1 | 400 | mg g of VSS−1 |
| qmax2 | 1,500 | mg g of VSS−1 |
| qmax3 | 250 | mg g of VSS−1 |
| qmax4 | 400 | mg g of VSS−1 |
| qmax5 | 200 | mg g of VSS−1 |
| μmax1 | 0.3-2.2 | day−1 |
| μmax2 | 1.0 | day−1 |
| μmax3 | 0.6 | day−1 |
| ks1 | 0.2 | mg liter−1 |
| ks2 | 25 | mg liter−1 |
| ks3 | 50 | mg liter−1 |
| kl | 10 | mg liter−1 |
| xmax | 80 | g liter−1 |
RESULTS
FISH.
The anaerobic granules analyzed by FISH originated from a PCP-treating UASB reactor. As described in the introduction, successful augmentation of this reactor was evidenced by an increase in the strain PCP-1 population from 106 to 1010 cell g of VSS−1 within a period of 70 days (28). Samples were taken from the lowest port of the UASB reactor corresponding to the lower portion of the sludge bed where the PCP concentration would be highest. Samples were withdrawn after 24 h and 1, 2, 3, 4, 5, and 9 weeks of reactor operation. Negative controls consisted of Champlain sludge granules not exposed to PCP and strain PCP-1 and of granules taken from a lab-scale UASB reactor never exposed to PCP and not inoculated with strain PCP-1.
Distribution of Eubacteria and Archaea in the granules was studied using the Cy3-EUB338 and Cy3-ARC915 probes, respectively. Analysis of transects across the granule centers demonstrated a homogeneous distribution of Eubacteria and Archaea in the anaerobic granules used to inoculate the reactor. In the 24-h samples, a homogeneous distribution of Archaea was observed, but a very thin (<10-μm-thick) and noncontinuous outer layer of Eubacteria was noted (results not shown). Growth of the outer eubacterial layer was visible in samples taken at week 1, 2, and 3, and by the fourth week of reactor operation a distinct outer layer of Eubacteria was formed (Fig. 1B). This layer had a thickness of 50 to 100 μm and was observed in all subsequent samples. The eubacterial layer was followed by a region colonized by Archaea (Fig. 1A) with some eubacterial colonies (Fig 1B). The hybridization experiments were carried out in triplicates yielding similar distribution of species.
FIG. 1.

Images of anaerobic granules taken from a UASB reactor augmented with D. frappieri PCP-1 and hybridized with fluorescent probes coupled to Cy3 (red) or Cy5 (red) and counterstained with DAPI (blue). Shown are sections of granules hybridized with probe specific to Archaea, after 4 weeks of reactor operation (A); probe specific to Eubacteria, after 4 weeks of reactor operation (B); and species-specific probe S-S-D.frap-327(D. frappieri)-a-A-19 (labeled with Cy3), 3, 4, 5 and 9 weeks of reactor operation, respectively (C, D, E, F). Bars correspond to 200 μm. Pink coloration results from the combination of red and blue when both signals are superimposed within the 7-μm-thick cross section of the granule. The white square on each panel indicates the granule center.
Distribution of strain PCP-1 cells was studied using three species-specific probes (Table 1). In the 24-h granules and in samples taken 1 and 2 weeks after reactor startup, no or low nonspecific signal was observed following FISH with all three species-specific fluorescent probes (results not shown). In samples taken at weeks 3 and 4, strain PCP-1 microcolonies were visible at the surface of the granules (Fig. 1C and D). In the 5-week granules (Fig. 1E), a large number of microcolonies were found in the outer layer. Finally, in the 9-week granules strain PCP-1 microcolonies densely colonized the outer layer, which attained a thickness of 30 to 60 μm (Fig. 1F).
Throughout the experiment, the validity of hybridization results was confirmed using both negative controls and hybridization with two species-specific probes (double hybridization). Negative controls (sludge granules without strain PCP-1 added and not exposed to PCP) showed some random nonspecific hybridization signals. The intensity of nonspecific signals with all three probes was low. These signals were not caused by Desulfitobacterium indigenous to the initial granular sludge, since PCR carried out with total DNA extracted from both negative control samples and primers specific to the Desulfitobacterium genus (19, 28) showed no positive signal (results not shown).
Statistical analysis of fluorescent areas provided one-dimensional representation of signal distribution on the images, which confirmed visual observations. After 1 week of reactor operation only a weak fluorescent background was observed in granule samples (Fig. 2A), but at week 5, regions at the edges of the granules were more fluorescent than the background (Fig. 2B). By the end of the experiment (week 9), up to 35% of the outer layer of the granule hybridized with PCP-1-specific probes (Fig. 2C), while hybridized area was below 2% in the biofilm interior.
FIG. 2.

Profiles of specific areas hybridized with S-S-D.frap-327(D. frappieri)-a-A-19 probe after 1 (A), 5 (B), and 9 (C) weeks of reactor operation. Broken lines show standard deviation of the area.
Fluorescent signals on samples obtained at weeks 3 through 9 with double hybridization (S-S-D.frap-327(D. frappieri)-a-A-19 and S-S-D.frap-576(D. frappieri)-a-A-19 or S-S-D.frap-86(D. frappieri)-a-A-20 and S-S-D.frap-576(D. frappieri)-a-A-19 confirmed that the fluorescent outer layer consisted of strain PCP-1 cells (Fig. 3), as both combinations gave the same hybridization pattern in triplicate experiments. Simultaneously, a double hybridization using negative control samples showed no overlapping of the nonspecific signals.
FIG. 3.

Images of anaerobic granules taken from a UASB reactor augmented with D. frappieri PCP-1, after 9 weeks of operation. Shown are granule cross sections counterstained with DAPI (blue) (A and C) and hybridized either with S-S-D.frap-86(D. frappieri)-a-A-20 (Cy3) and S-S-D.frap-576(D. frappieri)-a-A-19 (Cy5) (B) or with S-S-D.frap-327(D. frappieri)-a-A-19 (Cy3) and S-S-D.frap-576(D. frappieri)-a-A-19 (D). Overlapping fluorescent signal from both probes was converted to yellow.
Modeling of biofilm colonization by D. frappieri PCP-1.
A biofilm model outlined in the Materials and Methods section was used to simulate population dynamics and distribution of bacterial species in the biofilm. Based on the experimental results, the simulation was started with a homogeneous initial distribution of acidogenic and methanogenic bacteria in the granular biofilm. Reactor inoculation with strain PCP-1 was simulated by maintaining a constant concentration of strain PCP-1 in the bulk liquid for a period of 3 days while setting the concentration of strain PCP-1 in the biofilm to zero. Then, the concentration of strain PCP-1 in the bulk was set to a near zero value to simulate a continuous mode of reactor operation, which led to a washout of free cells from the bulk liquid.
Biofilm colonization by strain PCP-1 was simulated for a period of 70 days. By the end of the integration period, the predicted distribution of bacterial trophic groups resembled the granule structure demonstrated by FISH. Acidogenic bacteria colonized the outer layer, while the maximal density of methanogenic bacteria was predicted in the interior region of the biofilm (Fig. 4A). The distribution of acidogenic bacteria and methanogens mirrored that of corresponding substrates, sucrose and fermentation products (Fig. 4B). Also, at a granule diameter of 3 mm the existence of a carbon source-limited granule core was predicted (Fig. 4B).
FIG. 4.

Distribution of strain PCP-1 (x1), acidogenic (x2), and methanogenic (x3) bacteria (A) and of substrates and intermediates (B) predicted by the biofilm model. Bacterial and substrate concentrations were normalized to concentrations in bulk liquid. Simulation parameters: integration time, 70 days; granule diameter, 3 mm. Other parameters are given in Table 2. Zero on the x axis corresponds to the granule center, and 1 corresponds to the granule surface.
It should be kept in mind that the deterministic one-dimensional biofilm model presented in this work does not distinguish between uniform cell growth and growth in the form of scattered microcolonies, as in the observations presented above for samples taken at weeks 3, 4, and 5. Consequently, the model prediction, while appearing as a continuous function of the biofilm distance, can apply for both a uniform cell distribution and a multiplicity of microcolonies.
Sensitivity analysis of strain PCP-1 distribution to variations in the model parameters showed that the specific growth rate of strain PCP-1 and bacterial diffusive transport governed the distribution. A high specific growth rate combined with a low value of bacterial diffusion kept the bacterial growth predominantly at the surface (Fig. 5, line A). Understandably, at a given specific growth rate, setting the model with a larger value for bacterial diffusion (e.g., 10−6 dm2 day−1; Fig. 5, line B) was required to obtain a higher cell density in the biofilm core. A low value of specific growth rate (0.3 day−1) also led to an almost homogeneous distribution (Fig. 5, line C). A distribution similar to that observed experimentally in Fig. 2B and C was obtained by using a value of 2.2 day−1 for the specific growth rate and a bacterial diffusion of 10−7 dm2 day−1, i.e., high growth rate and low bacterial transport. This specific growth rate was similar to that cited in the literature for a pure culture of strain PCP-1 grown on a rich medium (3).
FIG. 5.

The effect of specific growth rate and bacterial diffusion on the distribution of strain PCP-1 in the biofilm. (A) Fast growth, slow transport. Parameters: μmax1 = 2.2 day−1, Dx1 = 10−7 dm2 day−1, xmax1 = 0.11 g/liter. (B) Fast transport and growth. Parameters: μmax1 = 2.2 day−1, Dx1 = 10−6 dm2 day−1, xmax1 = 0.05 g/liter. (C) Slow transport and growth. Parameters: μmax1 = 0.3 day−1, Dx1 = 10−7 dm2 day−1, xmax1 = 0.02 g/liter. In all equations, μmax1 is the maximal specific growth rate, Dx1 is the effective bacterial diffusivity, and xmax1 is the maximal density of strain PCP-1. Zero on the x axis corresponds to the granule center, and 1 corresponds to the granule surface.
DISCUSSION
Hybridization results of the present study demonstrated that by the 4th week of reactor operation, bacterial distribution had become stratified, with Eubacteria dominating mostly in the peripheral portion of the granule and Archaea in the middle and inner parts of the granule (Fig. 1A and B). The Eub338 probe detects 97% of all Eubacteria. Even though it fails to detect some groups of bacteria such as Planctomycetales and Verrucomicrobia (8), which could be present in the granules, the overall distribution of Eubacteria was correctly represented.
The outermost Eubacteria most likely corresponded to fermentative bacteria which transform sucrose and pyruvate to acetate and/or other VFAs, as well as hydrogen and CO2. Eubacteria colonies observed in the inner region of the granule (Fig. 1B) can be attributed to acetogenic bacteria. Such a layered distribution of Eubacteria and Archaea is well established for anaerobic granules fed with carbohydrates (12, 15, 16, 21, 25) as the initial step of carbohydrate degradation is notably faster than subsequent steps. In contrast, at the time of inoculation both Eubacteria and Archaea were homogeneously distributed throughout the granules (results not shown). This random distribution was similar to that published previously for granules of the same origin (23), i.e., an industrial reactor treating protein-rich effluents. When proteolysis and acetogenesis from amino acids are the limiting steps in the overall degradation process, a uniform microstructure develops in granules with a random distribution of Methanosaeta populations (11, 12, 14). In comparison, the results shown in Fig. 1A and B reflect the shift in the carbon source that the granules underwent from the industrial reactor to the laboratory-scale operation. This is a clear example of how the architecture of the granule is changed in response to a change in the carbon source composition.
As shown in our previous study (28), proliferation of strain PCP-1 allowed for an increase in the volumetric PCP load from 5 to 80 mg/liter of reactor volume (LR)/day. Throughout the experiment, a constant methane yield was observed, suggesting that the dechlorination activity of strain PCP-1 protected other members of the anaerobic consortium, in particular methanogens, from PCP toxicity. Although the competitive-PCR-based enumeration did not provide information on the distribution of strain PCP-1 in the biofilm, it was postulated that strain PCP-1 essentially colonized the outer granule layer. This was inferred from the fact that such a distribution was a requisite for fast consumption of PCP combined with high methanogenic activity. The methanogens were expected to occupy the granule core, which was shielded from the PCP (28). The results of FISH presented above proved this assumption was correct. Three weeks after reactor augmentation, strain PCP-1 microcolonies were detected in the outer layer of the granules (Fig. 1C). The area of cells hybridized with the PCP-1-specific probes progressively increased, and by the end of the experiment a dense outer layer of strain PCP-1 was observed (Fig. 1F). It should be noted that since the FISH technique only detects cells with a high ribosomal content, it is theoretically possible to have some PCP-1 cells with a low metabolic state inside the granule that would not be detected.
The observations of a dense outer layer of strain PCP-1 are not entirely in agreement with previously published results, which showed that dehalogenating strains in anaerobic granules were distributed in both the exterior and interior part of the biofilm. By immunofluorescence assay, Ahring et al. (1) demonstrated that Desulfomonile tiedjei formed microcolonies at the subsurface and surface of granules of a 3-chlorobenzoate-degrading UASB bioreactor. Also, inoculation of Desulfitobacterium hafniense DCB-2 in a PCP-degrading UASB reactor containing sterilized granules led to the generation of a net structure by this strain (7). Finally, Horber et al. (17) showed that Dehalospirillum multivorans built a net-like structure at the surface of PCE-fed live granules but formed microcolonies principally in the center of autoclaved granules.
In an attempt to clarify these differences in the distribution of newly introduced strains in a biofilm (Table 3), the granule colonization by strain PCP-1 was simulated using a dynamic multispecies model, which accounted not only for substrate diffusion but also for bacterial cell transport. Cell transport would have to be accounted for if newly added cells were found in the biofilm interior in a relatively short time. The anaerobic granule has a porous structure, with numerous channels and wells (20) that can easily serve as entry and transport routes for the cells. Cell transport can be mediated by bacterial motility, convection and/or diffusion. Strain PCP-1 is nonmotile (3). Convection is unlikely within a solid matrix, although one may imagine the gas microbubble release to displace the liquid at countercurrent, and thus to cause local convective motion in the pits and channels. Finally diffusive transport remains probably the most prominent mechanism by which the cell can enter and travel across the inner granule space. In diffusive transport, bacteria exhibit a Brownian motion, at an average speed of 40 μm h−1, which is responsible for crossing any quiescent liquid layer (30). Thus, bacterial diffusion was accounted for in the model. When the model was set with a large value for the specific growth rate and a low value for the bacterial diffusive transport term for strain PCP-1, computer simulations (Fig. 5A) were in agreement with tendencies displayed by FISH results. Most of the strain PCP-1 growth was predicted to occur in the outer section of the biofilm. When the model is set with either a large value for both the specific growth rate and the bacterial diffusive transport term or a much lower value for the specific growth rate and low bacterial diffusive transport term, the distribution is more uniform (Fig. 5, lines B and C). The latter set might explain the deep penetration of Desulfomonile tiedjei. Like D. frappieri, this strain is large and nonmotile, but in contrast it has a low growth rate (Table 3). The deeper penetration and more uniform distribution of Dehalospirillum multivorans and Desulfitobacterium hafniense in the granules most likely results from the fact that the granules were autoclaved. This means that there was no competition for space and substrate with other bacteria, which would promote a more uniform growth. The motility of both Desulfitobacterium hafniense and Dehalospirillum multivorans and the smaller size of the latter are other factors contribution to a deeper penetration.
TABLE 3.
Comparison of UASB granules bioaugmented with dehalogenating anaerobic strains
| Added strain | Pure strain characteristicsb
|
Reference | Initial granules | Distribution of added strain in granules (reference) | Chloroorganics used | |||
|---|---|---|---|---|---|---|---|---|
| Size (μm2)a | Motility | μmax (d−1) | Temp (°C) | |||||
| Desulfitobacterium frappieri PCP-1 | 1.5-3 | − | 5.5 | 29 | 3 | Live | Outermost layer (this study) | PCP |
| Desulfomonile tiedjei | 4-10 | − | 0.1 | 37 | 9 | Live | Inner and surface microcolonies (1) | CB |
| Dehalospirillum multivorans | 0.8-2.5 | + | 6.6 | 30 | 24 | Live | Surface microcolonies (17) | PCE |
| Autoclaved | Microcolonies in the center (17) | |||||||
| Desulfitobacterium hafniense | 2-4 | + | 0.3 | 30 | 6 | Autoclaved | Uniform distribution (7) | PCP |
Cross-sectional surface calculated from the dimensions given in the corresponding reference.
Growth substrate for all strains was pyruvate.
The model also predicted the methanogens (Archaea) to proliferate in the interior section of the biofilm, and acidogenic bacteria (Eubacteria) together with most of the strain PCP-1 to grow in the outer section of the biofilm. This is explained by the fact that the outer region contained the highest concentration of sucrose (s2, Fig. 4B), a primary substrate for fermenters, and of pyruvate (s3, Fig. 4B), a cosubstrate required for dehalogenating PCP to monochlorophenol. Presence of strain PCP-1 in the outer layer of the granules resulted in a fast decrease of PCP concentration with the depth of biofilm. Thus, PCP toxicity was brought below a threshold value which would affect metabolism of other members of the consortium, in particular methanogens, which are present in the granule core (Fig. 1A).
In conclusion, FISH of anaerobic granules augmented in D. frappieri PCP-1 revealed a predominant growth of this strain at the periphery of the biofilm in the form of microcolonies. These microcolonies appeared to proliferate over time to form a dense outer layer of strain PCP-1. Mathematical simulation of the colonization process using a dynamic multispecies biofilm model agreed with the experimentally observed granule structure, provided that the model is set with a large value of specific growth rate and a low value for the bacterial diffusive transport term.
The present study has broad implications for decontamination of chemical-polluted waters as it confirms the possibility to design granules for specific bioremediation needs. It shows that the de novo biodegradation abilities can be incorporated in the granule, since the added strain PCP-1 cells are intimately associated with the biofilm structure. The anaerobic granule seems to be an adequate matrix for efficient attachment of exogenous microorganisms.
Moreover, sensitivity analysis of the model parameters suggested that an inward propagation of strain PCP-1 can be expected at lower specific growth rates and/or higher bacterial transport. A higher bacterial transport could be achieved during initial colonization of solid matrices, such as sterilized granules or inert porous supports. Consequently, the added cells colonizing a deeper part of the biofilm are expected to be shielded from any major attrition. This might be of particular concern if such reactors are subjected to various shearing factors, such as the relatively high liquid superficial velocity and possibly high production and release of gas bubbles. This may guarantee that such a reactor could operate at a high dilution rate, and thus a low hydraulic residence time, which is instrumental in biosystems that have to be used at large scale in an economical manner. On the other hand, the association of new specific strains with live anaerobic granules offers a relatively easy way of engineering stable multispecies consortia that are capable of addressing target compounds. Multiple functions (e.g., cosubstrate fermentation or intermediates mineralization) and mutualistic ties are essential for completeness of degradation. Those assets are also instrumental for cost-effective specialized biosystems on a large scale.
Acknowledgments
We greatly appreciate the expert technical assistance of M.-J. Lévesque.
This study was funded partially (M.L. and R.V.) by the Natural Science and Engineering Research Council (NSERC grant no. OGP0155558) and partially (B.T., G.D.L., and S.R.G.) by the National Research Council of Canada (NRC grant no. 44648).
REFERENCES
- 1.Ahring, K. B. K., N. Christiansen, I. Mathrani, H. Hendriksen, A. J. L. Macario, and E. Conway De Macario. 1992. Introduction of a de novo bioremediation ability, aryl reductive dechlorination, into anaerobic granular sludge by inoculation of sludge with Desulfomonile tiedjei. Appl. Environ. Microbiol. 58:3677-3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amann, R. I., B. J. Binder, R. J. Olson, S. W. Chisholm, R. Devereux, and D. A. Stahl. 1990. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial population. Appl. Environ. Microbiol. 56:1919-1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouchard, B., R. Beaudet, R. Villemur, G. McSween, F. Lépine, and J.-G. Bisaillon. 1996. Isolation and characterization of Desulfitobacterium frappieri sp. nov., an anaerobic bacterium which reductively dechlorinates pentachlorophenol to 3-chlorophenol. Int. J. Syst. Bacteriol. 46:1010-1015. [DOI] [PubMed] [Google Scholar]
- 4.Bouchez, T., D. Patureau, P. Dabert, S. Juretschko, J. Dore, P. Delgenes, R. Moletta, and M. Wagner 2000. Ecological study of a bioaugmentation failure. Environ. Microbiol. 2:179-190. [DOI] [PubMed] [Google Scholar]
- 5.Bouchez, T., D. Patureau, P. Dabert, M. Wagner, J. P. Delgenes, and R. Moletta 2000. Successful and unsuccessful bioaugmentation experiments monitored by fluorescent in situ hybridization. Water Sci. Technol. 41:61-68.11382009 [Google Scholar]
- 6.Christiansen, N., and B. K. Ahring 1996. Desulfitobacterium hafniense sp. nov., an anaerobic, reductively dechlorinating bacterium. Int. J. Syst. Bacteriol. 46:442-448. [Google Scholar]
- 7.Christiansen, N., and K. Ahring 1996. Introduction of a de novo bioremediation activity into anaerobic granular sludge using the dechlorinating bacterium DCB-2. Antonie Leeuwenhoek 69:61-66. [DOI] [PubMed] [Google Scholar]
- 8.Daims, H., A. Brühl, R. Amman, K.-H. Schleifer, and M. Wagner. 1999. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: Development and evaluation of a more comprehensive nprobe set. Syst. Appl. Microbiol. 22:434-444. [DOI] [PubMed] [Google Scholar]
- 9.De Weerd, K. A., L. Mandelco, R. S. Tanner, C. R. Woese, and J. M. Suflita. 1990. Desulfomonile tiedje gen. nov. and sp. nov., a novel anaerobic, dehalogenating, sulfate-reducing bacterium. Arch. Microbiol. 154:23-30. [Google Scholar]
- 10.Emson, P. C., and M. J. Gait. 1992. In situ hybridization with biotinylated probes, p. 45-59. In D. G. Wilkinson (ed.), In situ hybridization: a practical approach. IRL Press, New York, N.Y.
- 11.Fang, H. H. P., H. K. Chui, and Y. Y. Li. 1995. Effects of degradation kinetics on the microstructure of anaerobic biogranules. Water Sci. Technol. 32:165-172. [Google Scholar]
- 12.Fang, H. H. P., H. K. Chui, and Y. Y. Li. 1994. Microbial structure and activity of UASB granules treating different wastewaters. Water Sci. Technol. 30:87-96. [Google Scholar]
- 13.Fuchs, B. N., F. O. Glöckner, J. Wulf, and R. Amann. 2000. Unlabeled helper oligonucleotides increase the in situ accessibility to 16S rRNA of fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 66:3603-3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grotenhuis, J. T. C., M. Smit, C. M. Plugge, X. Yuansheng, A. A. M. Van Lammeren, A. J. M. Stams, and A. J. B. Zehnder. 1991. Bacteriological composition and structure of granular sludge adapted to different substrates. Appl. Environ. Microbiol. 57:1942-1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guiot, S. R., A. Pauss, and J. W. Costerton 1992. A structured model of the anaerobic granule consortium. Water Sci. Technol. 25:1-10. [Google Scholar]
- 16.Harmsen, H. J., and H. M. P. Kengen, A. D. L. Akkermans, A. J. M. Stams, and W. M. De Vos. 1996. Detection and localization of synthrophic propionate-oxidizing bacteria in granular sludge by in situ hybridization using 16S rRNA-based oligonucleotide probes. Appl. Environ. Microbiol. 62:1656-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horber, C., N. Christiansen, E. Arvin, and B. Ahring. 1998. Improved dechlorinating performance of upflow anaerobic sludge blanket reactors by incorporation of Dehalospirillum multivorans into granular sludge. Appl. Environ. Microbiol. 64:1860-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jansenn, K. 1996. In situ hybridization and immunochemistry, p. 14.0.1-14.2.8. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidma, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology. John Wiley and Sons, Inc., New York, N.Y.
- 19.Lanthier, M., R. Villemur, F. Lépine, J.-G. Bisaillon, and R. Beaudet. 2001. Geographic distribution of Desulfitobacterium frappieri PCP-1 and Desulfitobacterium spp. in soils from the province of Quebec, Canada. FEMS Microbiol. Ecol. 36:185-191. [DOI] [PubMed] [Google Scholar]
- 20.MacLeod, F. A., S. R. Guiot, and J. W. Costerton. 1990. Layered structure of bacterial aggregates produced in an upflow anaerobic sludge bed and filter reactor. Appl. Environ. Microbiol. 56:1598-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quarmby, J. F., and C. F. Forster. 1995. A comparative study of the internal architecture of anaerobic granular sludge. J. Chem. Tech. Biotechnol. 63:60-68. [Google Scholar]
- 22.Raskin, L., J. M. Stromley, B. E. Rittmann, and D. A. Stahl. 1994. Group-specific 16S rRNA hybridization probes to describe natural communities of methanogens. Appl. Environ. Microbiol. 60:1232-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rocheleau, S., C. W. Greer, J. Lawrence, C. Cantin, L. Laramée, and S. R. Guiot. 1999. Differentiation of Methanosaeta concilii and Methanosarcina barkeri in anaerobic mesophilic granular sludge by fluorescent in situ hybridization and confocal scanning laser microscopy. Appl. Environ. Microbiol. 65:2222-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schloz-Muramatsu, H., A. Neumann, M. Messner, E. Moore, and G. Diekert. 1995. Isolation and characterization of Dehalospirillum multivorans gen. nov., sp. nov., a tetrachloroethene-utilizing, strictly anaerobic bacterium. Arch. Microbiol. 163:48-56. [Google Scholar]
- 25.Sekiguchi, Y., Y. Kamagata, K. Nakamura, A. Ohashi, and H. Harada. 1999. Fluorescence in situ hybridization using 16S rRNA-targeted oligonucleotides reveals localization of methanogens and selected uncultured bacteria in mesophilic and thermophilic sludge granules. Appl. Environ. Microbiol. 65:1280-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Speece, R. E. 1996. Anaerobic biotechnology for industrial wastewaters. Archae Press, Nashville, Tenn.
- 27.Stahl, D. A., and R. Amann. 1991. Development and application of nucleic acid probes, p. 205-248. In M. G. E. Stackebrandt (ed.), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Inc., New York, N.Y.
- 28.Tartakovsky, B., M.-J. Lévesque, R. Dumortier, R. Beaudet, and S. R. Guiot. 1999. Biodegradation of pentachlorophenol in a continuous anaerobic reactor augmented with Desulfitobacterium frappieri PCP-1. Appl. Environ. Microbiol. 65:4357-4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tartakovsky, B., and S. R. Guiot. 1997. Modeling and analysis of layered stationary anaerobic granular biofilms. Biotechnol. Bioeng. 54:122-130. [DOI] [PubMed] [Google Scholar]
- 30.van Loosdrecht, M. C. M., J. Lyklema, W. Norde, and A. J. B. Zehnder. 1990. Influence of interfaces on microbial activity. Microbiol. Rev. 54:75-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wanner, O., and P. Reichert. 1996. Mathematical modeling of mixed-culture biofilm. Biotechnol. Bioeng. 49:172-184. [DOI] [PubMed] [Google Scholar]