

Abstract
Insecticidal toxins produced by Bacillus thuringiensis interact with specific receptors located in the midguts of susceptible larvae, and the interaction is followed by a series of biochemical events that lead to the death of the insect. In order to elucidate the mechanism of action of B. thuringiensis toxins, receptor protein-encoding genes from many insect species have been cloned and characterized. In this paper we report the cloning, expression, and characterization of Cry toxin-interacting aminopeptidase N (APN) isolated from the midgut of a polyphagous pest, Spodoptera litura. The S. litura APN cDNA was expressed in the Sf21 insect cell line by using a baculovirus expression system. Immunofluorescence staining of the cells revealed that the expressed APN was located at the surface of Sf21 cells. Treatment of Sf21 cells expressing S. litura APN with phosphatidylinositol-specific phospholipase C demonstrated that the APN was anchored in the membrane by a glycosylphosphatidylinositol moiety. Interaction of the expressed receptor with different Cry toxins was examined by immunofluorescence toxin binding studies and ligand blot and immunoprecipitation analyses. By these experiments we showed that the bioactive toxin, Cry1C, binds to the recombinant APN, while the nonbioactive toxin, Cry1Ac, showed no interaction.
The insecticidal crystal proteins (ICPs) produced by Bacillus thuringiensis during sporulation have been used as biopesticides for many decades. Members of the Cry1A and Cry1C classes of ICPs are toxic to lepidopteran larvae (33). Upon ingestion by a susceptible insect larva, the ICPs are released from the crystals as 135-kDa protoxins in the alkaline and reducing environment of the insect midgut. The released protoxins are cleaved to an active toxin (65 kDa) by proteases present in the midgut (9). Activated toxin subsequently interacts with a protein located at the apical membrane of the insect midgut columnar cell, and the interaction at these binding molecules is a significant determinant for the specificity of ICPs (11, 43). Toxin-binding proteins from several insects have been identified by ligand blot analysis (36) of brush border membrane (BBM) vesicles (BBMVs) prepared from the midguts of the insects (6, 26, 31). Three types of molecules, i.e., aminopeptidase N (APN), cadherin-like protein, and anionic glycoconjugate (42), in the insect midgut have been found to serve as receptors for the Cry toxins. APN (EC 3.4.11.2) serves as a binding molecule for Cry1C in Manduca sexta (21), for Cry1Aa in Bombyx mori (12, 13, 46), and for Cry1Ac in M. sexta (7, 15, 16, 31), Lymantria dispar (19, 41), and Heliothis virescens (10, 22, 28). The aminopeptidase-encoding genes from many insects have been cloned, sequenced, and expressed in Escherichia coli (46) and insect cell culture (3, 23, 35). In addition to aminopeptidase, cadherin-like protein has also been identified as the receptor for the Cry1A class of toxins in M. sexta (14, 39, 40) and B. mori (13, 25). A cDNA encoding a 210-kDa BT-R1 (B. thuringiensis toxin receptor) of M. sexta has been cloned and expressed in insect and mammalian cell culture (14). Nagamatsu et al. (25) reported the cloning and expression of a B. mori Cry1Aa toxin-binding cadherin-like protein, BtR175, in Sf9 cells. Recently, Jenkins and Dean (13) have examined the binding specificity of Cry1Aa toxin for APN and cadherin-like receptor purified from B. mori BBMVs.
A significant part of the information about toxin-receptor interaction comes from tobacco hornworm M. sexta (7, 15, 21, 23) and the polyphagous pest H. virescens (10, 22, 28). The aminopeptidases used in those studies were purified by anion-exchange chromatography of CHAPS {3-[(3-chol-amidopropyl)-dimethylammonio]-1-propanesulfonate}-sol-ubilized BBMV proteins. Although those experiments have provided important insights into the toxin-receptor interaction mechanism, the availability of receptor protein in large amounts remains a limitation for elucidating this mechanism. The characterization of receptors in the economically important pests by studying their binding to different Cry toxins will help us in evaluating the molecular mechanism of action of Cry toxin and will lead to a better understanding of the mechanism of development of resistance to these proteins in insects.
The lepidopteran moth Spodoptera litura (common cutworm) is a polyphagous pest affecting various economically important crops in Asia. Toxins of the Cry1A group have been found to be totally ineffective against Spodoptera species (30), but the pest is susceptible to Cry1C toxin. Here we describe the isolation of a 2,856-bp gene, encoding a 108-kDa APN, from the midgut tissue of S. litura. We have expressed the protein in Sf21 insect cells by using a baculovirus expression system. The expressed protein was analyzed by Western blot analysis and immunolocalization studies. The recombinant APN protein is glycosylated, is attached to the membrane by a glycosylphosphatidylinositol (GPI) anchor, and, most importantly, retains aminopeptidase activity. The levels of expression obtained by us are comparable to those reported earlier (23). The S. litura APN protein expressed at the surface of Sf21 cells interacted with Cry1C toxin but showed no binding to Cry1Ac toxin.
MATERIALS AND METHODS
Insect rearing and midgut isolation.
Eggs of S. litura were obtained from the Entomology Department of the Indian Agricultural Research Institute. The insect culture was maintained in the insect rearing facility of our laboratory. The larvae were reared on an artificial diet at 25°C and 70% relative humidity with a photoperiod of 12 h light and 12 h dark. Early-fifth-instar S. litura larvae were chilled on ice for 15 min and dissected to isolate the midgut tissue. The midgut was slit longitudinally, the peritrophic membrane was removed, and the residual midgut contents were rinsed away with ice-cold buffer (300 mM mannitol, 5 mM EGTA, 17 mM Tris-HCl, pH 7.5). The midgut tissue used for isolating RNA was rinsed with diethyl pyrocarbonate-treated water. The tissue was either used immediately or snap frozen in liquid nitrogen and then stored at −80°C.
Cloning of S. litura apn.
Total RNA was isolated from the S. litura midgut by using the guanidine isothiocyanate method (29). cDNA was synthesized by using an oligo(dT) primer with 5 μg of total RNA as the template and reverse transcriptase (Superscript II; Life Technologies) according to the manufacturer's instructions. Primers for screening APN were designed on the basis of two conserved regions in the insect aminopeptidases (3, 10, 16), AFPCYDEP (RKB5 [GCT TTY CCY TGY TAY GAY GAR CC]) and WWDVLWL (RKB3 [AGC CAN ARN WYR TCC CAC CA]) (where Y is C or T; R is A or G; N is A, C, G, or T; and W is A or T). Using these degenerate primers, PCR was performed with midgut cDNA as the template. A 600-bp fragment was amplified, and this was cloned into the pGEM-T easy vector (Promega) and sequenced by the dideoxy termination method of Sanger et al. (32). A set of gene-specific forward (Sp1F, CGA CAC TCT TGG CCC GTC) and reverse (Sp1R, GAC CAA GTT GGT GTT AGC) primers was designed on the basis of the sequence of the cloned insert.
The 3′ end of the gene was amplified with the Sp1F and oligo(dT) primers with cDNA as the template. Conditions for the PCR included a hot start of 1 min followed by 32 cycles each of 94°C for 1 min, 48°C for 40 s, and 72°C for 2 min. The 5′ end of the gene was amplified by using a nested PCR approach with Sp3R (GCG TGT TGG TCT GTC GGC TC) and oligo(dG) primers according to the protocol described in the 5′ rapid amplification of cDNA ends (5′ RACE) kit manual (Life Technologies). The 3′ RACE and 5′ RACE DNA fragments were cloned in the pGEM-Te vector and sequenced. Seven independent clones obtained after 3′ and 5′ RACE were sequenced. Identical sequences were obtained in all of the clones, establishing the fidelity of the RACE products. Primers SpolitF (CGA GTC TAG AAT GGG TAC CAA AAT GTT GGT TCC) and SpolitR (CGA CGC GGC CGC TAA CTC CAC AGG AAT CTC), corresponding to sequences of the 5′ and 3′ ends of the apn cDNA, amplified a product of 2.856 kb which was cloned in the pGEM-Te vector. All of the primers were synthesized from Integrated DNA Technologies.
Expression of S. litura APN in E. coli and production of polyclonal antibodies.
A 1,420-bp fragment from bp 1161 to 2580 (amino acids 387 to 860) was amplified by using the Sp4F (CTG CGC ACT GGG CTG ATC) and Sp13R (AGC TTC ACC ACC ATA TGT AG) primers and cloned into the pGEM-Te vector. This fragment was excised from pGEM-Te with SphI and PstI restriction enzymes and cloned into SphI- and PstI-digested E. coli expression vector pQE30 (Qiagen). To express the truncated apn gene, pQE30 carrying the 1.42-kb fragment was transformed into E. coli strain M15. Expression of the protein was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) after IPTG (isopropyl-β-d-thiogalactopyranoside) induction. A 51-kDa protein was produced in the inclusion bodies. The inclusion bodies were solubilized in SDS loading buffer containing β-mercaptoethanol and resolved by SDS-10% PAGE. The 51-kDa protein band was excised from the gel after staining and destaining and was macerated with a pestle and mortar. Antiserum was raised by immunization of a New Zealand White rabbit with 0.2 mg of truncated APN administered in Freund's complete adjuvant (Sigma). The rabbit was boosted two times with truncated APN protein administered in Freund's incomplete adjuvant (Sigma). The rabbit serum was collected 10 days after the second boost. The reactivity of the rabbit serum was assessed by Western blotting.
Preparation and purification of recombinant B. thuringiensis δ-endotoxins.
E. coli strain JM103 expressing the recombinant protoxin Cry1Ac was kindly provided by D. H. Dean, Ohio State University, Columbus. The gene for this protoxin has been cloned in the pKK 223-3 expression vector. The Cry1C-expressing vector pTZ19R in E. coli strain DH5α was obtained from the Bacillus Genetic Stock Center, Ohio State University. δ-endotoxins were purified from E. coli by a modification of the procedure described by Lee et al. (20). The crystal protein was solubilized in 50 mM sodium carbonate buffer (pH 10.5) containing 10 mM dithiothreitol at 37°C for 2 h with vigorous shaking. The unsolubilized proteins were removed by centrifugation at 15,000 × g at 4°C for 10 min. The solubilized crystal protein was digested with trypsin (United States Biochemical) at a trypsin/protoxin ratio of 1:50 (by mass) at 37°C. Another dose of trypsin was added after 1 h of incubation and incubated further for 30 min. Solubilized Cry1C protoxin was activated with trypsin at a trypsin/protoxin ratio of 1:10. The activated Cry1Ac and Cry1C toxins were purified by ion-exchange liquid chromatography using a Q-Sepharose anion exchanger (Pharmacia Biotech Inc.) equilibrated with 50 mM sodium carbonate buffer (pH 10.5). Bound toxin was eluted with a 50 to 500 mM NaCl linear gradient prepared in the sodium carbonate buffer, and individual fractions were analyzed by SDS-PAGE. Fractions containing the purified toxin were pooled and dialyzed against 10 mM HEPES (pH 7.4)-150 mM NaCl-3.4 mM EDTA) and stored at −20°C. After activation and purification, the E. coli-expressed Cry1C toxin was obtained as a near-homogenous 62-kDa protein (34).
Expression of S. litura apn in Sf21 cells.
Sf21 cells were grown and maintained at 27°C in TNM-FH medium (Grace's basic medium with yeastolate, lactalbumin hydrolysate, and l-glutamine) supplemented with 10% fetal bovine serum and 50 μg of gentamicin per ml (Pharmingen). Cells were grown as a monolayer in tissue culture flasks. Cell viability was determined by the trypan blue exclusion method.
To clone S. litura apn into the baculovirus transfer vector pBacPAK (Clontech), the gene was amplified with gene-specific primers containing XbaI and NotI sites and cloned into the pGEM-Te vector. The gene was excised from the vector and cloned into XbaI- and NotI-digested pBacPAK vector by standard recombinant DNA techniques (29). The reverse primer also contained a His tag-encoding sequence. The recombinant transfer vector carrying the S. litura apn gene was termed pBacPAK8-SlApn.
To generate recombinant Autographa californica nuclear polyhedrosis virus carrying the S. litura apn gene, 106 Sf21 cells were cotransfected with 500 ng of sterile pBacPAK8-SlApn DNA and BacPAK6 linearized viral DNA (Bsu36 I digest) according to the instructions given in the Clontech manual. After 5 days, the supernatant of the cotransfection plate containing the recombinant virus, BV-SlApn (for baculovirus-S. litura APN), was harvested and stored at 4°C. Plaque assay was carried out to purify the virus, and different plaque-purified viruses were amplified. The amplified viruses were analyzed by PCR for the aminopeptidase gene by using gene specific primers. The PCR-positive viruses were used to infect Sf21 cells to determine the expression of the recombinant protein. The expression of the protein was analyzed by SDS-PAGE and Western blotting. A time course study was carried out to determine the level of expression of the recombinant aminopeptidase as a function of time.
Immunofluorescence localization.
Sf21 cells were seeded onto a sterile microscope coverslip (18-mm diameter) in a 12-well tissue culture plate. The cells were then infected with BV-SlApn at a multiplicity of infection of 2 for 36 h at 27°C. After the indicated incubation time, the infected cells were washed three times with insect phosphate-buffered saline (PBS) (1 mM Na2HPO4, 10.5 mM KH2PO4, 140 mM NaCl, 40 mM KCl, pH 6.2). The cells in each well were fixed with 1 ml of 2% formaldehyde in PBS. The fixed cells were washed three times with standard PBS and blocked with 3% bovine serum albumin (BSA) in PBS at room temperature for 1 h. Following three washes with 1× PBS, the cells were incubated with anti-APN polyclonal serum (1:5,000) in PBS containing 0.5% BSA at room temperature for 1 h. In parallel, cells in another well were incubated with preimmune serum as a control. After incubation, cells were washed three times with PBS and incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit immunoglobulin G (IgG) secondary antibody (1:500) in PBS containing 0.5% BSA for 1 h at room temperature. Finally, cells were washed with 1× PBS, mounted in buffered glycerol under a coverslip, observed under a fluorescence microscope, and photographed.
Assay of APN and PIPLC treatment.
Uninfected Sf21 cells and BV-SlApn-infected Sf21 cells were harvested at 72 h postinfection and washed with cold 1× PBS. The cells were counted with a hemocytometer. Nearly 106 cells were resuspended in 500 μl of PBS and mixed with 500 μl of substrate solution (5 mM leucine-p-nitroanilide in 67 mM Na2HPO4-67 mM KH2PO4, pH 7.4) (18). The reaction was carried out at 37°C for 15 min. The amount of product (p-nitroaniline) released was measured spectrophotometrically at 405 nm.
Infected Sf21 cells were harvested at 72 h postinfection and washed with PBS. Nearly 106 cells were resuspended into 500 μl of PBS and incubated with 0.5 U of phosphatidylinositol-specific phospholipase C (PIPLC). Following incubation at 37°C for 1 h, the cells were centrifuged at 16,000 × g for 10 min. The supernatant and the pellet resuspended in 500 μl of PBS were mixed with 500 μl of substrate solution and assayed for APN activity.
Preparation of BBMVs and Sf21 cell membranes.
BBMVs were prepared from fifth-instar larval midguts by the differential MgCl2 precipitation method of Wolfersberger et al. (45) and stored in buffer (300 mM mannitol, 5 mM EGTA, 17 mM Tris-HCl, pH 7.5) at −80°C until use.
To isolate the membranes of Sf21 cells, the infected cells were washed two times with PBS and resuspended in buffer A (20 mM Tris-HCl [pH 7.5], 0.5 mM EDTA, 10 mM NaCl, 10 μg of leupeptin per ml, 10 μg of aprotinin per ml, and 1 mM phenylmethylsulfonyl fluoride). The cell suspension was sonicated on ice (four 15-s pulses) and observed under the microscope for complete lysis. The homogenate was centrifuged at low speed (700 × g at 4°C for 5 min) to pellet heavier cellular debris, and the supernatant was subjected to ultracentrifugation (100,000 × g for 60 min at 4°C) in a 50 Ti rotor (Beckman). The high-speed pellet comprising membranes was washed two times with buffer A and suspended in the same buffer prior to snap freezing in liquid N2 and storage at −80°C.
Solubilization of expressed APN.
Membrane extracts prepared from Sf21 cells expressing S. litura APN and uninfected cells were solubilized at 4°C for 2 h in solubilization buffer (20 mM Tris-HCl [pH 7.5], 200 mM NaCl, 0.5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10 μg of leupeptin per ml, and 10 μg of aprotinin per ml) containing 1% (wt/vol) nonionic detergent (CHAPS; United States Biochemical). Undissolved material was removed by ultracentrifugation (100,000 × g for 60 min) at 4°C in a 50 Ti rotor (Beckman). The supernatant containing the solubilized recombinant APN protein was used for immunoprecipitation studies.
Ligand blotting.
Membrane extracts of Sf21 cells expressing S. litura APN were heated in SDS sample buffer containing β-mercaptoethanol, resolved by SDS-7.5% PAGE, and electrotransferred to nitrocellulose membrane (38). After being blocked with 3% BSA in Tris-buffered saline (50 mM Tris-HCl [pH 7.5], 150 mM NaCl) containing 0.1% Tween 20 (TBST), the membrane was incubated with 1 μg of purified Cry toxins per ml in TBST containing 0.2% BSA at room temperature for 1 h. The unbound toxins were removed by three washes with TBST. The membrane was then incubated with antibodies against the respective overlaid toxins at room temperature for 1 h. After three washes with TBST, the blots were incubated with alkaline phosphatase-conjugated goat anti-rabbit IgG antibodies (Calbiochem) for 1 h at room temperature. The immunoblots were washed again with TBST and developed using p-nitroblue tetrazolium chloride (NBT)-5-bromo-4-chloro-3-indolyl phosphate toluidine (BCIP) substrate (Sigma) as recommended by the manufacturer.
The above-described procedure for ligand blotting was used with S. litura BBMV proteins. Twenty-five micrograms of protein was resolved by SDS-7.5% PAGE and electrotransferred to nitrocellulose membrane. The membrane was overlaid with 2 μg of purified Cry1C toxin per ml, followed by incubation with anti-Cry1C toxin antiserum, and as a control the membrane was probed directly with anti-Cry1C toxin antiserum without preincubation with Cry1C toxin.
The electroblotted BBMV proteins were also probed with anti-APN antibodies (1:50,000). The blots were then incubated with alkaline phosphatase-conjugated goat anti-rabbit IgG and NBT-BCIP substrate.
The glycosylation status of the expressed APN was examined by transferring the membrane extracts from infected and uninfected cells onto nitrocellulose membrane and incubating with biotin-labeled concanavalin A (Sigma) (5 μg/ml in PBS) for 1 h. The membrane was probed with a 1:5,000 streptavidin-alkaline phosphatase complex (Sigma) and developed with NBT-BCIP.
Immunoprecipitation of Cry1C and APN.
Purified Cry1C toxin (5 μg) was incubated with 100 μg of CHAPS-solubilized membrane proteins (prepared from both infected and uninfected cells) in a binding buffer (50 mM sodium phosphate [pH 7.5], 50 mM NaCl, and 3 mM MgCl2) at 4°C on an end-over-end mixer for 3 h. To this mixture anti-Cry1C antiserum (5 μl) was added and then incubated further at 4°C. After 3 h of incubation, 100 μl of protein A-Sepharose 4B equilibrated with binding buffer was added. The reaction mixture was incubated at 4°C for 2 h with rotation. The protein A-Sepharose beads were pelleted at room temperature and washed once with wash buffer (10 mM Tris-HCl [pH 7.5], 2 mM EDTA, and 0.2% NP-40) containing 150 mM NaCl, once with wash buffer containing 300 mM NaCl, and twice with the same buffer (without NaCl and NP-40). The beads were resuspended in SDS sample buffer containing β-mercaptoethanol and heated at 100°C for 5 min to dissociate the bound protein-toxin-antitoxin antiserum complex. After centrifugation at 12,000 × g for 5 min, the supernatant was resolved by SDS-7.5% PAGE and electrotransferred to nitrocellulose membrane. The membrane was incubated with anti-APN antibodies followed by alkaline phosphatase conjugated goat anti-rabbit IgG antibodies and developed with NBT-BCIP.
Immunofluorescence toxin binding studies.
Sf21 cells were seeded, infected with recombinant virus, fixed, and blocked as described above. Following blocking, the cells were overlaid with 1 μg purified Cry1Ac and Cry1C toxins per ml in 1× PBS containing 0.2% BSA at room temperature for 1 h. Cells were then washed with PBS and incubated at room temperature for 1 h with respective antitoxin antiserum (1:2,500). Following three washes with PBS, the cells were incubated with FITC-conjugated goat anti-rabbit IgG secondary antibody (1:500) at room temperature for 1 h. Finally, the cells were washed three times with PBS, mounted in buffered glycerol, observed under a fluorescence microscope, and photographed.
Protein concentration determination.
Protein concentrations in samples without detergent were determined by the dye binding method of Bradford (1), and those in samples containing detergent were determined with a detergent-compatible protein assay kit (Bio-Rad) with BSA (Sigma) as the standard.
Nucleotide sequence accession number.
The GenBank accession number for the S. litura APN sequence is AF320764.
RESULTS
Cloning, sequencing, and characterization of S. litura APN.
Based on the published sequences of aminopeptidase genes from different insect species, degenerate primers were designed to clone the APN gene from S. litura. A 600-bp DNA fragment was amplified with the degenerate primers. The amplified product was cloned in the pGEM-Te vector and sequenced. A search for the sequence of cloned fragment in the National Center for Biotechnology Information nucleic acid data bank revealed homology with aminopeptidase-coding sequences of other insects. By using the RACE technique, the 5′ and 3′ ends of the gene were amplified and sequenced. Gene-specific primers corresponding to the 5′ and 3′ ends of the apn gene with cDNA as the template amplified the full-length gene of 2.856 kb. Analysis of the gene sequence with PC-GENE revealed an open reading frame corresponding to 952 amino acids with a molecular mass of 108,675 Da, an N-terminal cleavable signal peptide (residues 1 to 20), a C-terminal GPI modification site (residues 930 to 952), four N-glycosylation sites, and zinc-binding signature sequence characteristic of zinc metallopeptidases (Fig. 1). The amino acid sequence also contains four Cys residues which are highly conserved among APN molecules of higher eukaryotes (rat, rabbit, pig, and human).
FIG. 1.

(a) Deduced amino acid sequence of APN from S. litura. The 2,856-bp cDNA clone encodes a 952-amino-acid polypeptide. The putative NH2-terminal cleavable peptide and the GPI signal peptide at the COOH terminus are double underlined. The zinc metallopeptidase signature is indicated with a dotted line. The putative N-glycosylation sites are underlined. The four Cys residues that are conserved among eukaryotic aminopeptidases are in boldface. (b) Schematic representation and putative signature domains of S. litura APN.
By using the ClustalW alignment program (Mac Vector version 7), the deduced amino acid sequence of S. litura APN was aligned with those previously reported for 16 insect aminopeptidases. The multiple and pairwise alignments were carried out using following parameters: similarity matrix-blosum, open gap penalty of 10.0, and extended gap penalty of 0.1. The pairwise alignment showed that S. litura APN exhibited 67% identity to APN2 isolated from Helicoverpa punctigera (5) but only 25 to 35% identity to other APN sequences. The dendrogram constructed on the basis of this alignment by using the neighbor-joining method and Best Tree mode revealed that the reported APNs can be clustered into five homology groups (Fig. 2). Four homology groups were described earlier by Oltean et al. (28); the fifth group contains S. litura APN and H. punctigera APN2.
FIG. 2.

Phylogenetic tree describing the sequence similarity of known insect aminopeptidases. The tree was constructed by the neighbor-joining method with the Best Tree mode. A value of 0.1 corresponds to a difference of 10% between two sequences. GenBank accession numbers are as follows: Plutella xylostella APN3 (PxAPN3), AJ222699 (P. Denholf, unpublished data); S. litura APN (SlAPN), AF320764 (this study); H. punctigera APN2 (HpAPN2), AF217249 (5); Epiphyas postvittana APN (EpAPN), AF276241 (35); L. dispar APN1 (LdAPN1), AF126442 (8); H. virescens 120-kDa protein (Hv120kDa), U35096 (10); H. punctigera APN3 (HpAPN3), AF217250 (5); Plodia interpunctella APN1 (PiAPN1), AF034483 (47); P. xylostella APNA (PxAPNA), AF020389 (2); H. virescens 170-kDa protein (Hv170kDa), AF173552 (28); H. punctigera APN1 (HpAPN1), AF217248 (5); M. sexta APN1 (MsAPN1), X89081 (16); B. mori APN1 (BmAPN1), AF084257 (46); P. xylostella APN1 (PxAPN1), X97878 (3); L. dispar APN2 (LdAPN2), AF126443 (8); M. sexta APN2 (MsAPN2), X97877 (3); and B. mori APN2 (BmAPN2), AB011497 (12).
Expression of S. litura APN in Sf21 cells.
The full-length apn cDNA with a His tag-encoding sequence at the 3′ end was cloned in the baculovirus transfer vector. Recombinant viruses (BV-SlApn) were identified by plaque assays and screened by PCR and Western blot analysis. The Coomassie blue-stained gel (Fig. 3, lanes 2 and 4) showed the expression of a 108-kDa protein in the infected-cell lysate and membranes, which was not detected in the uninfected-cell lysate and membranes (Fig. 3, lanes 1 and 3). The 108-kDa protein reacted with anti-APN antibodies, confirming the expression of S. litura APN driven off the polyhedrin promoter in the insect cells with the baculovirus expression system. The level of APN expression in Sf21 cells was investigated at different time points after infection. Synthesis of the recombinant APN began within 24 h postinfection and continued through the time period (96 h) examined. No appreciable increase in the expression levels of APN was found after 72 h of infection. Therefore, cells harvested at 3 days postinfection were considered to be the most suitable for protein isolation and characterization. An interesting difference was observed in the blots developed with anti-APN and anti-His antibodies. Two bands, upper band A and lower band B, were highlighted with anti-APN antibodies, whereas only the lower band B was highlighted with anti-His antibodies (Fig. 4). The synthesis of two forms of APN in Sf21 cells is probably a consequence of differential sorting of nascent APN, as illustrated diagrammatically in Fig. 5.
FIG. 3.

Expression of S. litura APN in Sf21 cells. (A) One million uninfected and 72-h-infected cells were harvested, and total cell extract was prepared by resuspending and boiling the cells in SDS sample buffer containing β-mercaptoethanol. (B) Membranes were isolated from both uninfected and infected cells by the procedure described in Materials and Methods. Cell extracts and equal amounts of membrane proteins were resolved by SDS-7.5% PAGE and stained with Coomassie brilliant blue. Lanes 1 and 3, uninfected Sf21 cells; lanes 2 and 4, BV-SlApn-infected cells. The positions of molecular mass markers (in kilodaltons) are indicated on the left.
FIG. 4.

Time course of S. litura APN expression in Sf21 insect cell culture. U, total cell extract of uninfected cells. BV-SlApn-infected Sf21 cells (106) were harvested at the indicated times. Total cell extract was resolved by SDS-7.5% PAGE and electrotransferred to nitrocellulose membrane. Western blot analysis was carried out with (i) polyclonal antibodies raised against APN and (ii) monoclonal anti-histidine tag antibodies. Bands A and B represent the two forms of APN expressed in the Sf21 cells. The positions of molecular mass markers (in kilodaltons) are indicated on the left.
FIG. 5.

Flow diagram illustrating the occurrence of a heterogenous population of APN molecules expressed in Sf21 cells as shown in Fig. 4. Population A consists of molecules that have undergone transamidation at the COOH terminus and are transported to plasma membrane; these molecules lose the COOH-terminal His tag and are not visualized with anti-His antibodies. Population B consists of molecules that remain unprocessed and are retained in the endoplasmic reticulum (ER) with the His tag. These molecules are visualized with anti-His antibodies. Details are given in Discussion.
Characterization of the recombinant APN expressed in BV-SlApn-infected insect cells.
The expressed APN was characterized for its catalytic efficiency, specificity, and localization. The catalytic competence of the expressed protein was examined by assaying for the aminopeptidase activity. Preliminary analyses revealed the presence of low levels of endogenous aminopeptidase activity associated with Sf21 cells. The values reported here have taken into account this endogenous activity. A significant difference of 17,000 nmol/mg/h in the specific activity of APN-expressing cells over the uninfected host cells was observed. The specific inhibitors amastatin and bestatin at a 1 mM concentration inhibited aminopeptidase and reduced the activity displayed by APN-expressing cells to 4% (data not shown).
When recombinant APN-expressing Sf21 cells were lysed and subjected to high-speed ultracentrifugation, S. litura APN was completely retained in the pellet fraction, indicating that APN was expressed as a membrane-anchored protein. Since the amino acid sequence of the nascent APN showed the presence of a GPI anchor addition site at the carboxyl terminus and a cleavable signal peptide sequence at the amino terminus, we investigated the location of APN on the surface of infected Sf21 cells. Immunofluorescence studies on immobilized infected Sf21 cells with anti-APN antibodies showed an intense cell surface fluorescence, indicating that the expressed APN was located on the surface of Sf21 cells (Fig. 6B). No cell surface fluorescence was observed in uninfected cells stained with anti-APN antibodies (data not shown) or in infected cells stained with preimmune serum (Fig. 6A).
FIG. 6.

Immunolocalization of expressed S. litura APN. Surface expression of the APN was examined at 36 h postinfection by immunofluorescence staining with rabbit polyclonal antibodies raised against APN and FITC-conjugated goat anti-rabbit IgG. (A) Cells overlaid with preimmune serum. (B) Cells overlaid with anti-APN antibodies.
The nature of membrane anchorage of recombinant S. litura APN expressed in Sf21 cells was determined by treating the Sf21 cells with PIPLC. PIPLC treatment released a fraction of the expressed APN population into the supernatant, as only 68% of the APN activity was retained in the pellet fraction, showing that S. litura APN is attached to Sf21 cells by means of a GPI anchor.
Visualization of biotin-labeled concanavalin A blots of uninfected and infected Sf21 cell membrane proteins with the streptavidin-alkaline phosphatase complex showed a reaction with most of the proteins, including the expressed APN (Fig. 7, lanes 1 and 2, respectively). This indicates that S. litura APN expressed in Sf21 cells is glycosylated. The E. coli-expressed truncated APN protein of 51 kDa showed no reaction with the streptavidin-alkaline phosphatase complex and served as a negative control (Fig. 7, lane 3).
FIG. 7.

Concanavalin A lectin blot detection of glycosylated proteins in Sf21 cell membranes. Lane 1, uninfected cell membranes. Lane 2, infected cell membranes. Lane 3, E. coli-expressed 51-kDa APN. The positions of molecular mass markers (in kilodaltons) are indicated on the left.
Identification of a S. litura BBM protein that binds to the Cry1C toxin.
We investigated the binding of Cry1C toxin to S. litura BBM proteins by a protein blot technique (36). On overlaying of the electroblotted S. litura BBMV proteins with the Cry1C toxin followed by anti-Cry1C antiserum and secondary antibodies as described in Materials and Methods, multiple bands were highlighted (Fig. 8A, lane 2). One of these bands was also observed in the control blot in which the toxin was omitted, indicating that it could be a nonspecific signal (Fig. 8A, lane 1). When the electroblotted S. litura BBMVs were probed with anti-APN antibodies, three bands were highlighted (Fig. 8A, lane 3). These interacting proteins did not react with preimmune serum (data not shown). Among the multiple proteins visualized with the Cry1C overlay, only one protein, corresponding to 108 kDa, reacted with anti-APN antibodies and hence tentatively was identified as aminopeptidase. This protein also corresponded to the Sf21 cell-expressed APN protein (Fig. 8B). Together, these results indicate that the APN highlighted at 108 kDa in the BBMVs might serve as a receptor for Cry1C toxin in the insect midgut.
FIG. 8.

Identification of Cry1C toxin-binding protein in S. litura BBMV. (A) Twenty-five micrograms of BBMV proteins was resolved by SDS-PAGE and transferred to nitrocellulose membrane. The membrane strips were incubated with anti-Cry1C antiserum without prior overlaying with Cry1C toxin (lane 1), with Cry1C toxin and anti-Cry1C antiserum (lane 2), and with anti-APN antibodies (lane 3). (B) Sf21-expressed S. litura APN was transferred to the membrane and probed with anti-APN antibodies. The membranes were then incubated with alkaline phosphatase-conjugated secondary antibody and NBT-BCIP substrate as described in Materials and Methods. The positions of molecular mass markers (in kilodaltons) are indicated on the left.
Interaction of the surface-expressed APN with different Cry toxins.
The toxin-binding characteristics of the expressed S. litura APN were analyzed by ligand blotting experiments. For ligand blotting, identical amounts of membrane proteins prepared from uninfected and infected Sf21 cells were resolved by SDS-PAGE, electroblotted onto nitrocellulose membrane, and overlaid with Cry toxins. The interaction was detected by incubating the blot with the respective antitoxin antibodies. A band corresponding to the expressed APN was highlighted in the infected membranes incubated with Cry1C toxin (Fig. 9, lane 2) and was not observed in Cry1C-overlaid uninfected membranes (Fig. 9, lane 1). This corroborates the interaction of S. litura APN with Cry1C toxin. No such band was highlighted in the infected membranes incubated with Cry1Ac toxin, indicating the lack of interaction between the two (Fig. 9, lane 3).
FIG. 9.

Ligand blot analysis of insect cells expressing S. litura APN. Membrane proteins (2.5 μg) prepared from uninfected Sf21 cells (lane 1) and BV-SlApn-infected cells (lane 2 and 3) were solubilized in SDS sample buffer containing β-mercaptoethanol, separated by SDS-7.5% PAGE, and electrotransferred to nitrocellulose membrane. The nitrocellulose membrane was probed with Cry1C (1 μg/ml) (lanes 1 and 2) and Cry1Ac (1 μg/ml) (lane 3) δ-endotoxins. Binding of the toxin was detected by using antibodies against the toxin followed by alkaline phosphatase-conjugated goat anti-rabbit IgG. The positions of molecular mass markers (in kilodaltons) are indicated on the left.
The S. litura APN-Cry1C interaction was also examined by immunoprecipitation experiments. When CHAPS-solubilized membranes were incubated with Cry1C toxin, the toxin-protein complex was pulled down with anti-Cry1C antibodies by using protein A-Sepharose beads. Western analysis of the immunoprecipitated protein with anti-APN antibodies was carried out as described in Materials and Methods. Both forms of expressed APN were immunoprecipitated from the CHAPS-solubilized infected membranes, thus indicating that both interact with Cry1C toxin (Fig. 10, lane 2). No interacting protein was visualized when the solubilized uninfected membranes were used for immunoprecipitation (Fig. 10, lane 1). The control lacking Cry1C toxin did not exhibit any interacting protein on probing with anti-APN antibodies (data not shown).
FIG. 10.
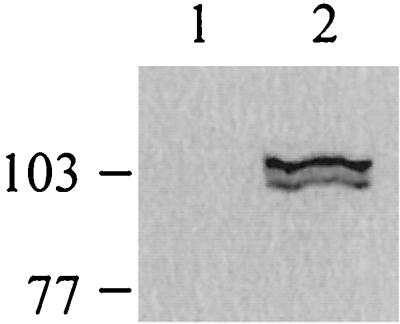
Immunoprecipitation of APN with Cry1C toxin. CHAPS-solubilized membrane proteins from uninfected cells (lane 1) and infected cells (lane 2) were incubated with purified Cry1C toxin. Using anti-Cry1C antiserum, the protein-toxin complex was pulled down with protein A-Sepharose beads. The beads were boiled in SDS sample buffer containing β-mercaptoethanol, resolved by SDS-PAGE, and electrotransferred to nitrocellulose membrane. The membrane was probed with anti-APN antibodies. The positions of molecular mass markers (in kilodaltons) are indicated on the left.
Since we demonstrated that the recombinant APN is expressed at the surface of Sf21 cells, a binding assay was also performed with intact insect cells. The interaction between the two molecules was detected by using antitoxin antibodies and FITC-conjugated secondary antibody. The fluorescing cells indicate the binding of the expressed APN with Cry1C toxin (Fig. 11B), whereas faint fluorescence was observed in Cry1Ac-overlaid cells (Fig. 11A). This indicates that the recombinant APN interacts with Cry1C toxin but not with Cry1Ac toxin. Controls in which cells were directly incubated with antitoxin antibodies without prior overlaying with toxin showed no fluorescence. The uninfected Sf21 cells showed no fluorescence when overlaid with any of the toxins (data not shown).
FIG. 11.

Differential toxin binding to surface-expressed S. litura APN. Sf21 cells infected for 36 h were overlaid with Cry1Ac (A) or Cry1C (B) toxin (1 μg/ml). Binding of the toxin was detected by using antibodies against the toxin followed by FITC-conjugated goat anti-rabbit IgG.
DISCUSSION
S. litura (Lepidotera: Noctuidae) is a polyphagous pest known to attack many economically important crops such as groundnut, castor, maize, safflower, banana, and many vegetables. B. thuringiensis formulations containing Cry1C are active against Spodoptera spp. and have been deployed for commercial use. The most recent application of Cry proteins for the control of Spodoptera larvae is the development of transgenic crops that express a synthetic cry1Ca gene.
Bioassays conducted in our laboratory to determine the relative sensitivities of S. litura to different Cry toxins revealed that the second-instar larvae were susceptible to Cry1C toxin (50% lethal dose, 200 ng/3.8 cm2) and were not affected by Cry1Ac toxin even at up to 2,000 ng/cm2. Our results are in agreement with earlier reports that demonstrate the tolerance of larvae of Spodoptera species to Cry1A toxins and their susceptibility to Cry1C toxin (24, 30).
Our attempts to express the complete S. litura apn cDNA in E. coli failed; hence, we attempted the expression of apn in insect cells by using a baculovirus expression system. The full-length cDNA was successfully expressed in the S. frugiperda (Sf21) cell culture. The levels of S. litura APN expression were satisfactory and comparable to those in an earlier report (23). The specific activity of Sf21 cells expressing S. litura APN was significantly higher than the values observed when uninfected cells were used for the assay, thus indicating that the expressed APN is catalytically active. The aminopeptidase activity of expressed protein was found to be sensitive to the specific APN inhibitors amastatin and bestatin, with the percentage of inhibition being 96% with both inhibitors. The expressed APN was localized at the surface of insect cells as predicted from the presence of a cleavable N-terminal signal peptide (44) and C-terminal GPI anchor addition signal sequence (predicted from a sequence-based algorithm developed by Eisenhaber et al. [4]). Immunofluorescence staining of whole cells with anti-APN antibodies confirmed the surface localization of recombinant APN produced in insect cells. Partial release of aminopeptidase activity from the S. litura APN-expressing Sf21 cells after PIPLC treatment indicated that at least a fraction of the expressed APN population was anchored in the membrane by a GPI anchor. Some studies have expressed doubts about using the polyhedrin promoter for the expression of aminopeptidase in S. frugiperda insect cell culture (23). However, in the present study the expressed S. litura APN protein not only was catalytically active but was processed faithfully to be anchored in the plasma membrane of culture cells.
Resolving the expressed APN protein and probing separately with anti-APN antibodies and C terminus-associated His tag antibodies revealed the presence of two forms of APN. Both forms reacted with anti-APN antibodies, whereas only one of the forms reacted with anti-His antibodies. We believe that the appearance of these two protein forms could be a consequence of following different biochemical events: (i) transamidation reaction limitation due to overburdening of posttranslational machinery and (ii) inefficient processing due to the presence of serine at the ω+2 site (17). Thus, the APN molecules in upper band A are the ones which are processed at their carboxyl termini by transamidase, in turn losing the His tag, and the ones in lower band B are unprocessed and retained in the endoplasmic reticulum with the His tag intact.
Several Cry toxin-binding molecules have been identified by performing ligand blot analysis of BBMVs prepared from insect midguts. Aminopeptidases, proteins belonging to the cadherin superfamily (37), and anionic glycoconjugates are the three types of molecules present in the midguts of insect that have been employed by Cry toxins to exert their toxicity towards the susceptible insects. The binding of Cry toxins either to total proteins extracted from BBMVs (6, 30, 43) or to APN/cadherin-like receptor purified from BBMVs (13, 15, 19, 22, 35) has been evaluated. The divergence in structure within aminopeptidases and cadherin-like receptor molecules is significant. The multiple aminopeptidase isoforms known to exist in the insects are believed to have arisen as a consequence of gene duplication (2, 8). To date several aminopeptidase-encoding genes have been isolated from different insects. Oltean et al. (28) carried out a dendrogram analysis of the reported insect aminopeptidases that have been described as Cry1A toxin-binding proteins and clustered them in four homology groups. S. litura APN and H. punctigera APN2 exhibited 67% identity to each other and formed a fifth group. These two APNs showed less identity to other APNs and thus appear to interact with a different class of Cry toxins. To date there have been three reports of studies in which aminopeptidase-encoding genes have been cloned and expressed in heterologous insect cell cultures (3, 23, 35). In two of these studies (23, 35) the protein was expressed at the surface of the cells. In these reports toxin-receptor interaction was demonstrated by ligand blot experiments, but they could not demonstrate binding to intact APN-expressing cells.
Cry1C toxin has been shown to bind to multiple BBM proteins in three Spodoptera spp. by ligand blotting (27). In another independent report, 40- and 110-kDa proteins in Spodoptera littoralis BBMVs were found to interact with Cry1C toxin (30). None of these proteins has been characterized further. We also investigated the binding of Cry1C toxin to S. litura BBMVs and to the expressed APN by using the ligand blot technique. Among the proteins that were highlighted on overlaying of the S. litura BBMVs with Cry1C toxin, a 108-kDa protein, which not only interacted with the anti-APN antibodies but also corresponded to the expressed S. litura APN, was found to be the specific Cry1C-binding protein in BBMVs. Thus, it might be the aminopeptidase in the insect midgut that serves as a receptor for the Cry1C toxin. Cry1C also interacted with the 108-kDa recombinant APN in the infected-cell membranes, with which Cry1Ac did not show any interaction. The specific precipitation of the expressed S. litura APN from the solubilized membrane proteins by Cry1C toxin in the immunoprecipitation experiment also showed that the two proteins interact with each other.
Surface localization of the expressed APN prompted us to use whole cells for studying the toxin-binding characteristics of the recombinant S. litura APN. Immunofluorescence toxin binding experiments with whole cells showed that the expressed APN recognized the bioactive Cry1C toxin and, critically, did not show any binding towards biologically inactive Cry1Ac toxin. For the first time, we are able to demonstrate by immunofluorescence the binding of a Cry toxin to recombinant APN expressed at the surface of Sf21 cells. These results demonstrate that the expression of S. litura APN in insect cell culture confers specific binding of the lepidopteran-specific Cry1C toxin to intact cells as well as membranes prepared from these cells. These binding experiments with S. litura APN indicate that the expressed aminopeptidase exhibits a degree of specificity that renders it a candidate receptor for Cry1C toxin in the S. litura midgut and is responsible for Cry1C toxin sensitivity in the larvae. Luo et al. (21) isolated a 106-kDa soluble form of APN from M. sexta that was shown to function as a Cry1C-binding protein. It did not bind to Cry1Ac toxin. They showed that the 106-kDa APN was functionally related but structurally different from a Cry1Ac-binding 115-kDa APN isolated from the same insect. They described the 106- and 115-kDa proteins as isoforms of aminopeptidase. Previous studies have shown that 120- and 170-kDa APNs serve as receptors for the Cry1A class of δ-endotoxins, whereas the 108-kDa APN isolated from S. litura in the present study and the 106-kDa APN from M. sexta (21) have been found to bind Cry1C toxin. These results reveal the complexity in the mode of action of B. thuringiensis toxins at the molecular level.
The results of the present study show that the Sf21 cells are suitable for expression of binding-competent receptor ligand in significant amounts. They also highlight the utility of cell culture-expressed APN for studying receptor-ligand interaction.
Acknowledgments
We thank Donald H. Dean (Ohio State University) for providing recombinant Cry1Ac.
N.A. gratefully acknowledges the financial support provided by the Center for Scientific and Industrial Research (CSIR), India, during this course of study.
REFERENCES
- 1.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 2.Chang, W. X. Z., L. J. Gahan, B. E. Tabashnik, and D. G. Heckel. 1999. A new aminopeptidase from diamondback moth provides evidence for a gene duplication event in Lepidoptera. Insect Mol. Biol. 8:171-177. [DOI] [PubMed] [Google Scholar]
- 3.Denolf, P., K. Hendrickx, J. Van Damme, S. Jansens, M. Peferoen, D. Degheele, and J. Van Rie. 1997. Cloning and characterization of M. sexta and P. xylostella midgut aminopeptidase N enzymes related to Bacillus thuringiensis toxin-binding proteins. Eur. J. Biochem. 248:748-761. [DOI] [PubMed] [Google Scholar]
- 4.Eisenhaber, B., P. Bork, and F. Eisenhaber. 1999. Prediction of potential GPI-modification sites in proprotein sequences. J. Mol. Biol. 292:741-758. [DOI] [PubMed] [Google Scholar]
- 5.Emmerling, M., D. Chandler, and M. Sandeman. 2001. Molecular cloning of three cDNAs encoding aminopeptidases from the midgut of Helicoverpa punctigera, the Australian native budworm. Insect Biochem. Mol. Biol. 31:899-907. [DOI] [PubMed] [Google Scholar]
- 6.Garczynski, S. F., J. W. Crim, and M. J. Adang. 1991. Identification of putative insect brush border membrane-binding molecules specific to Bacillus thuringiensis δ-endotoxin by protein blot analysis. Appl. Environ. Microbiol. 57:2816-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garczynski, S. F., and M. J. Adang. 1995. Bacillus thuringiensis CryIA(c) δ-endotoxin binding aminopeptidase in the Manduca sexta midgut has a glycosyl-phosphatidylinositol anchor. Insect Biochem. Mol. Biol. 25:409-415. [Google Scholar]
- 8.Garner, K. J., S. Hiremath, K. Lehtoma, and A. P. Valaitis. 1999. Cloning and complete sequence characterization of two gypsy moth aminopeptidase N cDNAs, including the receptor for Cry1Ac toxin. Insect Biochem. Mol. Biol. 29: 527-535. [DOI] [PubMed] [Google Scholar]
- 9.Gill, S. S., E. A. Cowles, and P. V. Pietrantonio. 1992. The mode of action of Bacillus thuringiensis endotoxins. Annu. Rev. Entomol. 37:615-636. [DOI] [PubMed] [Google Scholar]
- 10.Gill, S., E. Cowles, and V. Francis. 1995. Identification, isolation and cloning of a Bacillus thuringiensis CryIA(a) toxin-binding protein from the midgut of the lepidopteran insect Heliothis virescens. J. Biol. Chem. 270:27277-27282. [DOI] [PubMed] [Google Scholar]
- 11.Hofmann, C., H. Vanderbruggen, H. Hofte, J. Van Rie, S. Jansens, and H. Van Mellaert. 1988. Specificity of Bacillus thuringiensis δ-endotoxins is correlated with the presence of high-affinity binding sites in the brush border membrane of target insect midguts. Proc. Natl. Acad. Sci. USA 85:7844-7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hua, G., K. Tsukamoto, and H. Ikezawa. 1998. Cloning and sequence analysis of the aminopeptidase N isozyme (APN2) from Bombyx mori midgut. Comp. Biochem. Physiol. B 121:213-222. [DOI] [PubMed] [Google Scholar]
- 13.Jenkins, J. L., and D. H. Dean. 2001. Binding specificity of Bacillus thuringiensis Cry1Aa for purified, native Bombyx mori aminopeptidase N and cadherin-like receptors. BMC Biochem. 2:12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keeton, T. P., and L. A. Bulla, Jr. 1997. Ligand specificity and affinity of BT-R1, the Bacillus thuringiensis Cry1A toxin receptor from Manduca sexta, expressed in mammalian and insect cell cultures. Appl. Environ. Microbiol. 63:3419-3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knight, P. J. K., N. Crickmore, and D. J. Ellar. 1994. The receptor for Bacillus thuringiensis CryIA(c) delta-endotoxin in the brush border membrane of the lepidopteran Manduca sexta is aminopeptidase N. Mol. Microbiol. 11:429-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knight, P. J. K., B. Knowles, and D. J. Ellar. 1995. Molecular cloning of an insect aminopeptidase N that serves as a receptor for Bacillus thuringiensis CryIA(a) toxin. J. Biol. Chem. 270:17765-17770. [DOI] [PubMed] [Google Scholar]
- 17.Kodukula, K., L. Gerber, R. Amthauer, L. Brink, and S. Udenfriend. 1993. Biosynthesis of glycosylphosphatidylinositol (GPI)-anchored membrane proteins in intact cells: specific amino acid requirements adjacent to the site of cleavage and glycosyl-PtdIns attachment. J. Cell Biol. 120:657-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunz, D., F. Buhling, H. J. Hutter, T. Aoyagi, and N. Ansorge. 1993. Aminopeptidase N (CD13, EC 3.4.11.2) occurs on the surface of resting and concanavalin A-stimulated lymphocytes. J. Biol. Chem. Hoppe-Seyler 374:291-296. [DOI] [PubMed] [Google Scholar]
- 19.Lee, M., T. You, B. Young, J. Cotrill, A. Valaitis, and D. H. Dean. 1996. Aminopeptidase N purified from gypsy moth brush border membrane vesicles is a specific receptor for Bacillus thuringiensis CryIA(c) toxin. Appl. Environ. Microbiol. 62:2845-2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee, M. K., R. E. Milne, A. Z. Ge, and D. H. Dean. 1992. Location of a Bombyx mori receptor binding region on a Bacillus thuringiensis delta-endotoxin. J. Biol. Chem. 267:3115-3121. [PubMed] [Google Scholar]
- 21.Luo, K., Y. J. Lu, and M. J. Adang. 1996. A 106-kDa form of aminopeptidase is a receptor for Bacillus thuringiensis Cry1C δ-endotoxin in the brush border membrane of Manduca sexta. Insect Biochem. Mol. Biol. 26:783-791. [Google Scholar]
- 22.Luo, K., S. Sangadala, L. Masson, A. Mazza, R. Brousseau, and M. J. Adang. 1997. The Heliothis virescens 170 kDa aminopeptidase functions as “Receptor A” by mediating specific Bacillus thuringiensis Cry1A δ-endotoxin binding and pore formation. Insect Biochem. Mol. Biol. 27:735-743. [DOI] [PubMed] [Google Scholar]
- 23.Luo, K., J. R. McLachlin, M. R. Brown, and M. J. Adang. 1999. Expression of a glycosylphosphatidylinositol-linked Manduca sexta aminopeptidase N in insect cells. Protein Exp. Purif. 17:113-122. [DOI] [PubMed] [Google Scholar]
- 24.Moar, W. J., L. Masson, R. Brousseau, and J. T. Trumble. 1990. Toxicity of Spodoptera exigua and Trichoplusia ni of individual P1 protoxins and sporulated cultures of Heliothis virescens subsp. kurstaki HD-1 and NRD-12. Appl. Environ. Microbiol. 56:2480-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagamatsu, Y., T. Koike, K. Sasaki, A. Yoshimoto, and Y. Furukawa. 1999. The cadherin-like protein is essential to specificity determination and cytotoxic action of the Bacillus thuringiensis insecticidal Cry1Aa toxin. FEBS Lett. 460:385-390. [DOI] [PubMed] [Google Scholar]
- 26.Oddou, P., H. Hartmann, and M. Geiser. 1991. Identification and characterization of Heliothis virescens midgut membrane proteins binding Bacillus thuringiensis delta-endotoxins. Eur. J. Biochem. 202:673-680. [DOI] [PubMed] [Google Scholar]
- 27.Oddou, P., H. Hartmann, F. Radecke, and M. Geiser. 1993. Immunologically unrelated Heliothis sp. and Spodoptera sp. midgut membrane proteins bind Bacillus thuringiensis Cry1A(b) delta-endotoxin. Eur. J. Biochem. 212:145-150. [DOI] [PubMed] [Google Scholar]
- 28.Oltean, D. I., A. K. Pullikuth, H.-K. Lee, and S. S. Gill. 1999. Partial purification and characterization of Bacillus thuringiensis Cry1A toxin receptor A from Heliothis virescens and cloning of the corresponding cDNA. Appl. Environ. Microbiol. 65:4760-4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 30.Sanchis, V., and D. J. Ellar. 1993. Identification and partial purification of a Bacillus thuringiensis Cry1C δ-endotoxin binding protein from Spodoptera littoralis gut membranes. FEBS Lett. 316:264-268. [DOI] [PubMed] [Google Scholar]
- 31.Sangadala, S., F. S. Walters, L. H. English, and M. J. Adang. 1994. A mixture of Manduca sexta aminopeptidases and phosphatase enhances Bacillus thuringiensis insecticidal CryIA(c) toxin binding and 86Rb-K+ efflux in vitro. J. Biol. Chem. 269:10088-10092. [PubMed] [Google Scholar]
- 32.Sanger, F., S. Nicklen, and A. R. Coulson, A. R. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnepf, E., N. Crickmore, J. Van Rie, D. Lereclus, J. Baum, J. Feitelson, D. R. Zeigler, and D. H. Dean. 1998. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol. Mol. Biol. Rev. 62:775-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwartz, J.-L., L. Garneau, D. Savaria, L. Masson, R. Brousseau, and E. Rousseau. 1993. Lepidopteran specific crystal toxins from Bacillus thuringiensis form cation- and anion-selective channels in planar lipid bilayers. J. Membr. Biol. 132:53-62. [DOI] [PubMed] [Google Scholar]
- 35.Simpson, R. M., and R. D. Newcomb. 2000. Binding of Bacillus thuringiensis δ-endotoxins Cry1Ac and Cry1Ba to a 120-kDa aminopeptidase N Epiphyas postvittana purified from both brush border membrane vesicles and baculovirus-infected Sf9 cells. Insect Biochem. Mol. Biol. 30:1069-1078. [DOI] [PubMed] [Google Scholar]
- 36.Soutar, A. K., and D. P. Wade. 1989. Ligand blotting, p. 55-70. In T. E. Creighton (ed.), Protein function: a practical approach. IRL Press, Oxford, United Kingdom.
- 37.Takeichi, M. 1991. Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251:1451-1455. [DOI] [PubMed] [Google Scholar]
- 38.Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vadlamudi, R. K., T. H. Ji, and L. A. Bulla. 1993. A specific binding protein from Manduca sexta for the insecticidal toxin of Bacillus thuringiensis subsp. berliner. J. Biol. Chem. 268:12334-12340. [PubMed] [Google Scholar]
- 40.Vadlamudi, R. K., B. Weber, I. Ji, T. H. Ji, and L. A. Bulla. 1995. Cloning and expression of a receptor for an insecticidal toxin of Bacillus thuringiensis. J. Biol. Chem. 270:5490-5494. [DOI] [PubMed] [Google Scholar]
- 41.Valaitis, A., M. K. Lee, F. Rajamohan, and D. H. Dean. 1995. Brush border membrane aminopeptidase N in the midgut of the gypsy moth serves as the receptor for the CryIAc δ-endotoxins of Bacillus thuringiensis. Insect Biochem. Mol. Biol. 25:1143-1151. [DOI] [PubMed] [Google Scholar]
- 42.Valaitis, A. P., J. L. Jenkins, M. K. Lee, D. H. Dean, and K. J. Garner. 2001. Isolation and partial characterization of gypsy moth BTR-270, an anionic brush border membrane glycoconjugate that binds Bacillus thuringiensis Cry1A toxins with high affinity. Arch. Insect Biochem. Physiol. 46:186-200. [DOI] [PubMed] [Google Scholar]
- 43.Van Rie, J., S. Jansens, H. Hofte, D. Degheele, and H. Van Mellaert. 1990. Receptors on the brush border membrane of the insect midgut as determinants of the specificity of Bacillus thuringiensis delta-endotoxins. Appl. Environ. Microbiol. 56:1378-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Heijne, G. 1986. A new method for predicting signal sequence cleavage sites. Nucleic Acids Res. 14:4683-4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolfersberger, M. G., P. Luethy, A. Maurer, P. Parenti, F. V. Sacchi, B. Giordana, and G. M. Honozet. 1987. Preparation and partial characterization of amino acid transporting brush border membrane vesicles from the larval midgut of the cabbage butterfly. Comp. Biochem. Physiol. A 86:301-308. [Google Scholar]
- 46.Yaoi, K., K. Nakanishi, T. Kadotani, M. Imamura, N. Koizumi, H. Iwahana, and R. Sato. 1999. cDNA cloning and expression of Bacillus thuringiensis Cry1Aa toxin binding 120-kDa aminopeptidase N from Bombyx mori. Biochim. Biophys. Acta 1444:131-137. [DOI] [PubMed] [Google Scholar]
- 47.Zhu, Y. C., K. J. Kramer, B. Oppert, and A. K. Dowdy. 2000. cDNAs of aminopeptidase-like protein genes from Plodia interpunctella strains with different susceptibilities to Bacillus thuringiensis toxins. Insect Biochem. Mol. Biol. 30:215-224. [DOI] [PubMed] [Google Scholar]