

Abstract
The cure rate for chronic neurodegenerative diseases remains low, creating an urgent need for improved intervention methods. Recent studies have shown that enhancing mitochondrial function can mitigate the effects of these diseases. This paper comprehensively reviews the relationship between mitochondrial dysfunction and chronic neurodegenerative diseases, aiming to uncover the potential use of targeted mitochondrial interventions as viable therapeutic options. We detail five targeted mitochondrial intervention strategies for chronic neurodegenerative diseases that act by promoting mitophagy, inhibiting mitochondrial fission, enhancing mitochondrial biogenesis, applying mitochondria-targeting antioxidants, and transplanting mitochondria. Each method has unique advantages and potential limitations, making them suitable for various therapeutic situations. Therapies that promote mitophagy or inhibit mitochondrial fission could be particularly effective in slowing disease progression, especially in the early stages. In contrast, those that enhance mitochondrial biogenesis and apply mitochondria-targeting antioxidants may offer great benefits during the middle stages of the disease by improving cellular antioxidant capacity and energy metabolism. Mitochondrial transplantation, while still experimental, holds great promise for restoring the function of damaged cells. Future research should focus on exploring the mechanisms and effects of these intervention strategies, particularly regarding their safety and efficacy in clinical settings. Additionally, the development of innovative mitochondria-targeting approaches, such as gene editing and nanotechnology, may provide new solutions for treating chronic neurodegenerative diseases. Implementing combined therapeutic strategies that integrate multiple intervention methods could also enhance treatment outcomes.
Keywords: Alzheimer’s disease, amyotrophic lateral sclerosis, calcium homeostasis, oxidative stress, Huntington’s disease, mitochondrial dysfunction, mitochondria, mitophagy, neurodegenerative diseases, Parkinson’s disease, targeted therapy
Introduction
Action of mitochondria
One of the most predominant organelles in the cell is the mitochondrion, commonly referred to as the “energy factory” (Adam et al., 2025; Xu et al., 2025). The primary function of mitochondria involves transforming raw materials into final products by modulating the interactions among two or more molecules, thereby forming an integrated system (Monzel et al., 2023; Parikh and Shah, 2025; Ye et al., 2025). Mitochondria serve as energy suppliers, maintainers of cellular homeostasis, and regulators of various metabolic pathways, including apoptosis and calcium signaling (Picard and Shirihai, 2022; Vercellino and Sazanov, 2022). The release of neurotransmitters and synaptic plasticity are crucial processes that help regulate calcium ion homeostasis (Giorgi et al., 2018). Under normal circumstances, cells require considerable energy to sustain themselves, particularly for processes such as signal transmission between neurons and the synthesis and release of neurotransmitters. This energy is primarily derived from adenosine triphosphate (ATP) produced by the mitochondria (Chen et al., 2025).
During their growth, development, and recovery from damage, neurons synthesize new proteins and nucleic acids in processes that require a high energy input. By producing ATP, mitochondria provide indispensable energy sources for these activities (Hilton et al., 2024). To ensure a continuous and stable supply of energy to the brain, the nervous system employs various strategies to protect and optimize mitochondrial function (Menacho and Prigione, 2020). Maintaining an adequate and healthy number of mitochondria is crucial for meeting this system’s high energy demands. This involves fine-tuning the balance between the formation of new mitochondria, the division of existing structures, and the degradation of old components (Theparambil et al., 2024). As harmful substances, such as free radicals, are produced during oxidative phosphorylation in mitochondria, enhancing the antioxidant defense system is particularly important. For example, glutathione is a critical molecule that effectively removes reactive oxygen species (ROS), helping to prevent cellular damage (Li et al., 2022a; Houldsworth, 2024). Additionally, the overall efficiency of the energy conversion process within mitochondria can be further improved by adjusting specific enzyme activity levels or altering gene expression patterns (Wang et al., 2021). Certain gene products may also play roles in regulating the construction and function of mitochondria (Tan et al., 2024a).
Basic concepts and classification
Mitochondrial dysfunction refers to the impairment of mitochondrial oxidative phosphorylation, which reduces ATP production and leads to cellular dysfunction (Behl et al., 2023). This dysfunction not only disrupts energy metabolism but also triggers a cascade of reactions, including the increased production of ROS, induction of apoptosis, and activation of inflammatory pathways. These changes are critical factors in many diseases, particularly age-related degenerative disorders (Amorim et al., 2022).
Parkinson’s disease (PD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD) are classified as cognitive neurodegenerative diseases (CNDs), characterized by cognitive or sensory abnormalities resulting from the gradual loss of neuronal function or cell death. Despite their varied clinical presentations, these diseases share common pathological mechanisms. Key features include synaptic and neural network dysfunction, abnormal protein aggregation, protein homeostasis imbalances, and abnormalities in the cytoskeleton (Wilson et al., 2023).
Relationship between neurodegenerative diseases and mitochondrial dysfunction
First, because mitochondria are the primary ATP producers in cells, their dysfunction directly leads to an insufficient energy supply for neurons, negatively affecting the synthesis and release of neurotransmitters and impairing neuronal signal transduction (Popov, 2023). Second, mitochondrial dysfunction is closely linked to the inflammatory response and can cause damage to neuronal tissue by activating cells that release pro-inflammatory factors (Suski et al., 2018; Picca et al., 2020). This inflammatory response then further damages mitochondria and other cellular structures, creating a vicious cycle (Agirman et al., 2021; Qin et al., 2024).
Neuroinflammation and mitochondrial dysfunction are critical pathological features of neurodegenerative diseases. Recovering mitochondrial function can reduce neuroinflammation, which in turn creates a favorable microenvironment for nerve regeneration (Peruzzotti-Jametti et al., 2024). For example, mitochondria-targeted antioxidant peptides, such as elamipretide, can decrease neuroinflammation, inhibit neural cell apoptosis, and enhance survival pathways in nerve cells, thereby preventing the progression of neurodegenerative diseases (Nhu et al., 2021). As an innovative therapeutic strategy, mitochondrial transplantation can deliver healthy mitochondria to damaged nerve cells, effectively restoring their cellular energy metabolism, reducing oxidative stress, alleviating neuroinflammation, and promoting nerve regeneration (Eo et al., 2024). Additionally, the regulation of mitophagy, such as via the application of the drug rapamycin or activation of the PINK1/Parkin pathway, has shown potential in the treatment of AD and PD (Antico et al., 2025). The key event axis for targeted interventions of mitochondrial dysfunction in AD, PD, HD, and ALS is illustrated in Figure 1. This review focuses on the benefits of mitochondria as targets for neuroinflammation interventions, which can offer novel approaches that transcend the limitations of traditional neuroinflammation therapies that primarily address cytokine and immune regulation (Cai et al., 2024). Mitochondrial anti-neuroinflammation processes are closely associated with nerve regeneration, and research is providing new methods and techniques to achieve this goal. The ultimate aim of these studies is to develop more effective treatments to improve the prognosis of neurodegenerative diseases and enhance the quality of life of patients.
Figure 1.

Key event axis for targeted mitochondrial dysfunction interventions in AD, PD, HD, and ALS.
Created with BioRender.com. AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; Aβ: amyloid-β plaques; CHCHD10: coiled-coil-helix-coiled-coil-helix domain containing 10; HD: Huntington’s disease; MDVs: mitochondrial DNA variants; mHTT: mutant huntingtin; NAD+: nicotinamide adenine-positive; PD: Parkinson’s disease; PINK1: PTEN-induced putative kinase 1; Parkin: a gene related to autosomal recessive juvenile Parkinsonism.
Purpose
The purpose of this review is to comprehensively elucidate the mechanisms of mitochondrial function in chronic neurodegenerative diseases, with a particular focus on how mitochondrial dysfunction influences the occurrence and progression of these conditions. The author has closely followed the latest research, including new discoveries and molecular mechanisms related to mitochondrial dysfunction, emerging intervention strategies targeting mitochondria, and drugs designed to mitigate mitochondrial dysfunction. The goal is to provide new strategies and insights into potential interventions for chronic neurodegenerative diseases.
Search Strategy
Literature search strategy
To obtain the latest international advancements in research on neurodegenerative diseases and mitochondria, the following English literature search strategy was used:
Database selection: Web of Science, PubMed, Google Scholar, and Scopus database.
Search terms: Alzheimer’s disease; amyotrophic lateral sclerosis; apoptosis; ATP production; Huntington’s disease; inflammation; mitochondrial dysfunction; neurodegenerative disease; oxidative stress; Parkinson’s disease.
Time range for the search: 2010–2024.
Search method: Use keyword combinations along with subject terms and abstracts to conduct precise searches and facilitate effective screening.
Criteria for literature inclusion and exclusion
Select high-quality journal articles published within the last 5 years, prioritizing those with higher citation counts. Additionally, we focused on articles from top academic journals and those with high-impact factors.
Inclusion criteria
Articles must focus on neurodegenerative diseases and mitochondrial function, be published within the last 5 years in high-impact journals (e.g., Nature, Science), and be in English. Eligible articles may include reviews, basic research, case reports, or clinical studies.
Exclusion criteria
Exclude duplicate publications, articles with incomplete or inaccurate data, non-academic articles (e.g., press releases), and those that are irrelevant to the topic. This approach ensures the relevance, quality, and focus of the selected literature.
Mitochondrial Structure and Function
Mitochondrial structure
The outer and inner membranes of mitochondria boarder the intermembrane space. The outer membrane is smooth and highly permeable and features hydrophilic channels formed by porins that allow the passage of small molecules. In contrast, the inner membrane is folded into ridges, which increase its surface area and facilitate ATP synthesis. Cellular energy conversion is supported by the continual fusion and fission of the inner mitochondrial membrane structures. Defects in the fusion of the inner mitochondrial membrane can lead to a loss of mitochondrial DNA (mtDNA) and embryonic lethality in mice. Additionally, mutations in the human OPA1 gene are associated with genetic diseases such as optic atrophy (Yu et al., 2020a). The intermembrane space contains several key enzymes, including monoamine oxidase and superoxide dismutase (SOD), which play major roles in cellular metabolism.
The inner membrane and the cristae constitute the mitochondrial matrix, within which there is a large number of enzyme classes involved in biochemical reactions, as well as ribosomes, mtDNA, and other genetic material (Palma et al., 2020). Mitochondrial proteins are mostly encoded by the nuclear genome, although a few are encoded by mtDNA (Hu et al., 2024; Tábara et al., 2024).
Mitochondrial bioenergetics
The tricarboxylic acid (TCA) cycle mainly occurs in the mitochondrial matrix. This cycle operates under aerobic conditions, converting pyruvate into acetyl-CoA and generating reducing equivalents used for ATP synthesis (Wang et al., 2025).
The TCA cycle is the hub of energy metabolism in organisms, with carbon skeleton degradation products from various nutrients ultimately entering this cycle, where they are broken down into CO2 for excretion (Wang et al., 2025). Additionally, intermediates of the TCA cycle are crucial for biological development and the maintenance of cellular homeostasis. When mammalian cells face environmental stress or physiological pathologies, they undergo metabolic reprogramming through multiple strategies to meet their metabolic demands. Among these strategies are critical heterogeneous changes in substrate preference and alterations in key enzyme activities within the TCA cycle. Selective regulatory mechanisms governing TCA cycle activity in mammalian cells play a decisive role in determining cell fate (Arnold et al., 2022; De Martino et al., 2024).
Respiration is the basis of cellular energy metabolism. After organisms take up organic matter, they produce water and energy and release carbon dioxide (Guan et al., 2022). The mitochondrial respiratory chain is located in the inner membrane of the mitochondria. It includes four membrane protein complexes, complex I (NADH dehydrogenase), complex II (succinate dehydrogenase), complex III (cytochrome c reductase), and complex IV (cytochrome c oxidase), as well as electron transfer ligands (Guan et al., 2022). As the mitochondrial respiratory chain transfers electrons to oxygen, it pumps protons into the intermembrane space of the mitochondria, generating a proton gradient that drives ATP synthase’s (complex V) synthesis of ATP (Vercellino and Sazanov, 2022). As protons traverse the inner membrane, the difference in membrane potential can be used for the catalytic phosphorylation of ADP to form ATP (Lai et al., 2023).
Role of mitochondria in cell cycle regulation and cell death
Mitochondria are crucial for cell cycle regulation by providing energy and participating in various regulatory processes. They interact with cell cycle checkpoint mechanisms to ensure the normal progression of the cell cycle (Ryu et al., 2024). Apoptosis is closely associated with mitochondrial functions involving membrane permeability regulation by Bcl-2 family proteins and p53. Bcl-2 proteins are proapoptotic proteins that cleave the outer mitochondrial membrane. Necrosis is a passive form of cell death related to apoptosis. During necrosis, mitochondrial dysfunction results in decreased ATP production, ionic imbalance, and cell swelling, eventually leading to inflammation (Newton et al., 2024). Research has found that pyroptosis, a form of inflammatory cell death activated by gasdermin D (GSDMD), also leads to mitochondrial damage. Mitochondrial damage occurs once GSDMD is cleaved, and the pores formed by the N-terminal fragment of GSDMD may rapidly damage the outer mitochondrial membrane. These events suggest that mitochondrial damage plays a crucial role in pyroptosis (Miao et al., 2023).
Pathological Mechanisms of Mitochondrial Dysfunction
Mitochondrial dysfunction triggers a series of cascading reactions that profoundly affect neurons. First, it leads to reduced ATP production, resulting in an insufficient cellular energy supply, which in turn affects the normal function and survival of neurons. Second, during energy metabolism, mitochondria produce ROS and other free radicals that can accumulate excessively and damage cell membranes, proteins, and DNA, thereby accelerating neuronal degeneration (Swerdlow, 2018). The dysfunction of these organelles may also cause abnormal calcium efflux or influx, leading to an intracellular calcium overload that further damages the neuron (Chen et al., 2021). Even more seriously, mitochondrial dysfunction activates inflammasomes, which release inflammatory factors, exacerbating neuronal injury (Shimada et al., 2012). It also triggers apoptotic signaling pathways (Pihán et al., 2017), increasing cell apoptosis and necrosis, which lead to a reduction in the number of neurons and intensify neurodegenerative diseases. Finally, it affects the synthesis, storage, and release of neurotransmitters (Raefsky and Mattson, 2017). For example, mitochondrial dysfunction in dopaminergic neurons has been associated with dopamine deficiency in PD (Kim et al., 2016; Rodrigues et al., 2024).
Immune response of inflammation
Neuroinflammation is commonly referred to as an inflammatory response within the central nervous system (CNS) that stimulates various CNS diseases. This phenomenon results not only from pathogen invasion but also from the damage caused by trauma, ischemia, toxins, or other factors. Such damage is especially a risk when the blood-brain barrier is compromised, which makes persistent neuroinflammation more likely to occur (Kuźniar et al., 2024; Magnusson et al., 2024). A study by Onisiforou and Zanos (2024) revealed a notable association between HSV-1, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and AD. The coronavirus disease 2019 pandemic has provided new evidence regarding the role of viruses in the development of AD (Xia et al., 2021). The presence of SARS-CoV-2 not only poses a direct threat to the nervous system but also indirectly promotes local and systemic inflammation by activating inflammasomes and triggering cytokine storms, thereby affecting the function of the CNS (Kryńska et al., 2024). Additionally, brain tissue from individuals with HD was reported to exhibit a notable increase in microglia and astrocyte counts. These glial cells become overactive and secrete substantial amounts of pro-inflammatory factors, creating an environment unfavorable for neuronal survival and exacerbating disease progression.
Recent research has uncovered evidence that mitochondria are pivotal in modulating neuroimmune inflammation. The progression of neuroimmune inflammation can be effectively managed and controlled by modulating mitochondrial function and related signaling pathways. Mitochondria possess a unique signaling system that includes, but is not limited to, key components such as mtDNA and ROS. Upon pathogen invasion, mtDNA is released from the cytoplasm, activating the NLRP3 inflammasome or its CpG island sequence. This is recognized by Toll-like receptor 9 and causes it to activate the nuclear factor-kappa B (NF-κB) signaling pathway, ultimately leading to the production of excessive pro-inflammatory mediators (Figure 2).
Figure 2.

Mitochondrial dysfunction and immune inflammation.
Mitochondria regulate immune responses to maintain intracellular homeostasis. The extent and duration of mitochondrial damage can influence the intensity and duration of inflammatory responses. Mildly damaged or stressed mitochondria initiate a series of immune cascade reactions by releasing various damage-associated molecular patterns, such as mtDNA, mitochondrial-derived ROS, and cardiolipin. mtDNA promotes the release of pro-inflammatory cytokines such as TNF-α and IL-1β, as well as anti-inflammatory cytokines such as IL-10 from microglia, through activating the cGAS-STING signaling pathway, recognizing Toll-like receptor nine and activating the NF-κB signaling pathway, and directly activating the NLRP3 inflammasome. This helps to clear pathogens and repair tissue damage. Additionally, mitochondria achieve self-clearance of damaged mitochondria through PINK1/Parkin-mediated autophagy. However, in cases of severe mitochondrial dysfunction, the immunomodulatory capacity is also affected. Mitochondrial membrane depolarization and excessive ROS release lead to the massive production of pro-inflammatory cytokines, causing an overexuberant inflammatory response. The opening of the mitochondrial permeability transition pore, the release of cytochrome c into the cytoplasm, and the activation of downstream Caspase-3 initiate apoptosis, directly eliminating damaged neurons and resulting in neural tissue damage and dysfunction. Created with BioRender.com. cGAS: Cyclic GMP-AMP synthase; IL-10: interleukin-10; IL-1β: interleukin-1β; IL-6: interleukin-6; MAVS: mitochondrial antiviral signaling protein; mPTP: mitochondrial permeability transition pore; mtDNA: mitochondrial DNA; NF-κB: nuclear factor kappa-B; NLRP3: NOD-like receptor family pyrin domain containing 3; p62: sequestosome-1; ROS: reactive oxygen species; STING: stimulator of interferon genes; TLR9: Toll-like receptor 9; TNF-α: tumor necrosis factor-alpha.
PINK1/Parkin is a key protein in mitochondrial autophagy that regulates immune responses by inhibiting mitochondrial antigen presentation (Matheoud et al., 2016). Cossu et al. (2021) reported that the absence of PINK1/Parkin exacerbates the neuroinflammation caused by experimental autoimmune encephalomyelitis. Additionally, a lack of PINK1 was shown to alter the innate immune response of glial cells and increase nitric oxide (NO) production (Sun et al., 2018). This evidence suggests that defects in mitochondrial quality control impair the organelle’s ability to regulate immunity, and a loss of mitochondrial autophagy proteins further exacerbates the neuronal damage caused by the existing immune inflammation.
Intestinal flora imbalance
The bacteria, fungi, and parasites present in the gut have a close relationship with neurodegenerative diseases. The gut microbiota affects the CNS through the microbiota–gut–brain axis (MGBA), which in turn regulates the gut biome. Research has shown that amyloid-beta (Aβ) and tau proteins in the colon can exacerbate AD symptoms via the vagus nerve, with symptom relief observed after vagotomy (Zhan et al., 2023). In addition to the MGBA, other inter-organ communication systems include the lung–brain axis, which has attracted considerable attention in the field of biology in recent years. These discoveries offer new hope for the treatment of neurodegenerative diseases (Park and Lee, 2024).
In patients with AD, the concentration of short-chain fatty acids (SCFAs) is reduced in their cerebrospinal fluid, while their levels of trimethylamine N-oxide are elevated (Choi and Mook-Jung, 2023). SCFAs play a key role in the MGBA by regulating immune function, epigenetics, and neuroplasticity, either directly or indirectly, through their effects on neuronal survival, inflammatory cascades, and endocrine signaling. Additionally, SCFAs have shown antioxidant and anti-inflammatory properties, inhibiting amyloid deposition and improving microglial function in AD model mice (Jiang et al., 2021).
Transplanting the gut microbiota of patients with PD into wild-type mice reportedly leads to intestinal inflammation and alpha-synuclein (α-syn) accumulation in these mice, which subsequently developed systemic inflammation and intestinal barrier damage. The level of α-syn accumulation in the terminal ileum could serve as an early biomarker for PD (Munoz-Pinto et al., 2024). Changes in the ratios of dominant bacterial populations in the gut have been observed in HD mouse models with mutant huntingtin (mHTT) deposits. These changes were significantly improved through fecal microbiota transplantation and targeted antibiotic intervention, which helped reduce intestinal mHTT levels (Burtscher et al., 2024). An imbalance in intestinal flora can decrease beneficial metabolites while increasing harmful substances, such as trimethylamine N-oxide, which can damage the gut and blood–brain barrier, increasing the risk of chronic neurodegenerative disease-related pathogen infections. Pathogenic microbes and pro-inflammatory cytokines originating from the gut further exacerbate CNS disorders, increasing the likelihood of developing chronic neurodegenerative diseases (Wu et al., 2021; Brown and Heneka, 2024; Figure 3).
Figure 3.

Mitochondrial dysfunction and pathogenic microorganism.
Intestinal microflora can produce a variety of neurotransmitter precursors or directly synthesize certain neurotransmitters. For example, bacteria such as Escherichia coli, Streptococcus spp., and Staphylococcus spp. found in the intestinal tract can secrete dopamine. Additionally, some probiotics, including Lactobacillus and Bifidobacterium, can produce tryptophan, a precursor to serotonin, which subsequently promotes serotonin synthesis. The metabolic products of intestinal flora, particularly SCFAs, provide energy for intestinal epithelial cells, help maintain intestinal barrier function, and influence the release and signaling of neurotransmitters. Furthermore, SCFAs can affect neural tissue metabolism and mitochondrial function through blood circulation and enteric nerves. However, dysbiosis in the intestinal flora can trigger abnormal activation of the intestinal immune system. This leads to the release of inflammatory factors such as tumor necrosis factor-α and interleukin-6, which increase oxidative stress and further damage mitochondria. Additionally, a reduction in SCFA production or an imbalance in SCFA ratios can compromise the intestinal barrier, allowing an increase in harmful bacteria and their metabolites, such as TMAO and lipopolysaccharides. These toxic substances can cross the intestinal barrier and the blood–brain barrier, exacerbating inflammatory damage to the nervous system. Created with BioRender.com. 5-HT: 5-Hydroxytryptamine; Aβ: amyloid-β; CytC: cytochrome c oxidase; DA: dopamine; GABA: gamma-aminobutyric acid; Glu: glutamic acid; IL-6: interleukin-6; LPS: lipopolysaccharide; mPTP: mitochondrial permeability transition pore; SCFAs: short-chain fatty acids; TMAO: trimethylamine N-oxide; TNF-α: tumor necrosis factor-alpha.
Dysfunctional mitochondria can disrupt the gut microbiota and induce chronic inflammation throughout the body, leading to inflammatory diseases in the gut and other bodily systems. Urbauer et al. (2024) knocked out the gene responsible for producing the Hsp60 protein in mouse intestinal epithelial cells, creating a model of mitochondrial dysfunction. Their findings suggested that mitochondrial damage is associated with intestinal tissue damage and changes in the microbiome. Furthermore, based on the theory that mitochondria originated as bacteria, the dysbiosis of gut microbiota may lead to neurodegenerative changes that specifically target mitochondria. Bacterial pathogen-associated molecular patterns and neuronal mitochondrial damage-associated molecular patterns can stimulate the innate immune system and exacerbate chronic inflammation (Cardoso and Empadinhas, 2018). Collectively, these studies suggest that developing drugs targeting certain mitochondrial pathways or interactions between the microbiome and mitochondria may represent novel and effective intervention strategies for the treatment of chronic neurodegenerative diseases.
Oxidative stress
Mitochondrial oxidative stress is a complex pathophysiological process characterized by an imbalance in the generation and clearance of ROS within dysfunctional mitochondria, and thus a disruption of cellular redox homeostasis (Wen et al., 2025b). ROS include superoxide anions (·O2–), hydrogen peroxide (H2O2), and hydroxyl radicals (·OH). Elevated levels of ROS are closely associated with oxidative stress and chronic neurodegenerative diseases (Dash et al., 2024). Under normal physiological conditions, ROS play crucial roles as signaling molecules in cell signaling and immune regulation. However, when ROS levels become abnormally elevated, they can trigger oxidative damage and exacerbate the body’s inflammatory response. As shown in Figure 3, ROS can activate various inflammatory signaling pathways, such as the NF-κB and mitogen-activated protein kinase (MAPK) pathways, leading to the accelerated release of pro-inflammatory cytokines (Wen et al., 2025b). Excessive ROS can also damage the mitochondrial respiratory chain, resulting in insufficient ATP production, reduced membrane potential, altered membrane permeability, and calcium overload, all of which contribute to mitochondrial dysfunction. MtDNA is particularly vulnerable to ROS damage due to its proximity to the respiratory chain and lack of histone protection. Mutations in mtDNA can further increase ROS production, creating a vicious cycle of damage (Long et al., 2024).
Notably, the brain, an organ characterized by high energy expenditure and oxygen demand, possesses relatively few antioxidant defense mechanisms, making it particularly vulnerable to oxidative stress (Borković-Mitić et al., 2021). Research indicates that oxidative stress not only induces tau phosphorylation modifications but also promotes the formation of neurofibrillary tangles. Under pathological conditions, a cascade of reactions eventually leads to tau hyperphosphorylation, which accelerates microtubule depolymerization and disrupts neuronal signaling (da Cunha Germano et al., 2023; Figure 4).
Figure 4.

Mitochondrial dysfunction and oxidative stress.
Mitochondria, when functioning normally, produce small amounts of ROS and reactive nitrogen species (RNS) that act as signaling molecules. These molecules play a role in intracellular signal transduction and immune regulation by activating pathways such as Nrf2/ARE and MAPK, as well as stimulating cells such as microglia and astrocytes. The antioxidative defense system of mitochondria, which includes superoxide dismutase, vitamin E, and alpha-linolenic acid, helps maintain a balance between oxidation and antioxidation in the body. However, when mitochondrial dysfunction occurs, the function of the respiratory chain complexes becomes impaired, hindering electron transport and leading to an increase in mitochondrial-derived ROS production. Excessive ROS can attack lipids, proteins, and DNA within the mitochondria, further exacerbating mitochondrial damage and creating a vicious cycle. Oxidative stress can exacerbate mitochondrial dysfunction, as the large amounts of ROS generated during this process can damage mitochondrial structure and function. This damage can affect the permeability of the mitochondrial membrane, leading to mitochondrial swelling and cristae rupture. Consequently, these changes disrupt the energy metabolism and signal transduction functions of mitochondria while also promoting neuroinflammation. Created with BioRender.com. ALA: Alpha-lipoic acid; ARE: antioxidant response element; ATP: adenosine triphosphate; Aβ: amyloid-β; GST: glutathione S-transferase; HO-1: heme oxygenase 1; IL-1β: interleukin-1β; IL-6: interleukin-6; MAPK: mitogen-activated protein kinase; mPTP: mitochondrial permeability transition pore; NQO-1: quinone oxidoreductase 1; Nrf2: nuclear factor erythroid 2-related factor 2; RNS: reactive nitrogen species; ROS: reactive oxygen species; TNF-α: tumor necrosis factor-alpha; VitC: vitamin C; VitE: vitamin E.
Dysfunction of neurotransmitters
Neurotransmitters are the chemical substances responsible for facilitating communication between neural cells or between neural cells and effector cells. They play a crucial role in regulating synaptic plasticity, as well as in learning, memory, and cognitive function.
Glutamate is the primary excitatory neurotransmitter in the CNS. When its concentration significantly increases, it can lead to excitotoxic effects that are closely associated with neuronal damage and various CNDs (Verma et al., 2022; Yang et al., 2023). There is a correlation between neuronal cell death in patients with HD and the excitotoxicity induced by elevated glutamate levels (Jiang et al., 2023). In the brains of patients with AD, Aβ can bind to the N-methyl-D-aspartate receptor, disrupting its normal signaling activity and inducing the neurotoxic effects of glutamate (Yang et al., 2023; Figure 5).
Figure 5.

Mitochondrial dysfunction and excitotoxicity.
When extracellular glutamate levels are excessively high, glutamate receptors on neurons become overactivated, disrupting normal neurotransmission and cognitive function. This overactivation simultaneously increases Ca2+ influx, triggering calcium overload, which in turn promotes the production of reactive nitrogen species and mitochondrial fission. These processes lead to oxidative stress and further exacerbate mitochondrial damage. Collectively, these toxic effects can result in neuronal injury or even cell death. Mitochondrial dysfunction, in turn, impairs cell’s ability to uptake and metabolize glutamate, leading to increased extracellular glutamate levels and the initiation of excitotoxicity. Created with BioRender.com. ATP: Adenosine triphosphate; CytC: cytochrome c oxidase; GSH: glutathione; MFN2: mitochondrial fusion factor 2; MMP: mitochondrial membrane potential; mPTP: mitochondrial permeability transition pore; NMDAR: N-methyl-D-aspartate receptor; NO: nitric oxide; NOS: nitric oxide synthase; ROS: reactive oxygen species; SLC7A11: solute carrier family 7 member 11.
Evidence has been reported linking the neurotoxicity of glutamate and mitochondrial function (Vaglio-Garro et al., 2024). Researchers found that the mitochondria of glutamate-treated neurons were shortened and fragmented, indicating that glutamate can promote mitochondrial division. This process was typically accompanied by a reduction in mitochondrial membrane potential and an overproduction of ROS, which are associated with neurotoxic effects. The Weidinger team confirmed that a deficiency in key enzymes of the TCA cycle, such as the 2-oxoglutarate dehydrogenase complex, impairs the mitochondrial uptake of exogenous glutamate, thereby exacerbating its toxic effects. Additionally, a reduction in 2-oxoglutarate dehydrogenase complex activity may be linked to increased neuroinflammation and oxidative stress (Weidinger et al., 2023).
Other neurotransmitters, such as acetylcholine, dopamine, and gamma-aminobutyric acid, also play important roles in the evolution of chronic neurodegenerative diseases. The cognitive and memory impairments exhibited by patients with AD are closely related to defects in cholinergic neurotransmission within the CNS, which are characterized by the reduced synthesis, transport, and release of acetylcholine in the brain, as well as a decrease in the expression levels of cholinergic receptors. The progressive loss of dopaminergic neurons is a hallmark pathological feature of PD (Teleanu et al., 2022). In the brains of patients with PD, there is an increase in the levels of toxic metabolites of tryptophan, likely due to the secondary effects of mitochondrial complex I inhibition, which further exacerbates mitochondrial damage and ultimately leads to the death of dopaminergic neurons (Liang et al., 2021). NO is an inhibitory neurotransmitter released by nitric oxide synthase (NOS) neurons within the enteric nervous system. It plays a crucial role in regulating smooth muscle function, ensuring the normalization of colon movement and assisting in the defecation process. NOS, as the core rate-limiting enzyme for NO generation, is essential for this function (Idrizaj et al., 2021). In animal models of PD, a lack of NOS negatively affects the expression levels of antioxidant genes, leading to decreased NO synthesis in the enteric nervous system and exacerbating the abnormal accumulation of α-syn. This phenomenon significantly inhibits the autonomous rhythmic contractions of gastrointestinal smooth muscle, resulting in impaired gastrointestinal motility. In clinical practice, this pathological process often manifests as constipation, which is a common non-motor symptom experienced by patients with PD.
Disorder of mitochondrial energy metabolism
Studies have shown that ATP levels are significantly reduced in patients with AD, PD, and HD (Cai et al., 2019; Castora et al., 2023). This energy deficit, resulting from impaired mitochondrial function, hampers neurons’ ability to maintain normal physiological functions, ultimately leading to cell death and the progression of neurodegenerative diseases.
As the primary energy supply for the brain, glucose undergoes a series of complex mitochondrial energy conversion processes, such as the TCA cycle and oxidative phosphorylation/electron transfer system (OXPHOS/ETC). Under normal physiological conditions, microglia in patients with AD primarily rely on the oxidation of glucose to obtain energy. Glucose is first transported into the cytoplasm via the glucose transporter and then converted into pyruvate through glycolysis. Acetoacetate is further metabolized into acetyl-CoA within the mitochondria; acetyl-CoA then participates in the TCA cycle, releasing a small amount of energy. The ETC uses electron transfer to create chemical potential energy, which is essential for the synthesis of ATP (Figure 6). When stimulated by factors such as Aβ and phosphorylated tau protein, or during periods of stress, the energy demands of microglia increase sharply. At this point, microglia demonstrate metabolic adaptability by shifting from OXPHOS to glycolytic pathways to meet their energy needs (Li et al., 2022b).
Figure 6.

Mitochondrial dysfunction and energy metabolism.
Mitochondria are the primary sites for ATP production within cells, converting energy from nutrients into ATP through oxidative phosphorylation. However, mitochondrial dysfunction can disrupt glucose energy metabolism. For instance, mitochondrial DNA damage, abnormalities in respiratory chain complexes, and altered mitochondrial membrane permeability can impede the TCA cycle and the oxidative phosphorylation process, leading to reduced ATP production. This results in insufficient directly usable energy for cells, which affects various energy-requiring cellular activities. Meanwhile, fatty acid oxidation is enhanced to produce acetyl-CoA, and the uptake of glutamine by mitochondria increases, thereby compensating for the reduced energy production and strengthening the capacity of the TCA cycle. Created with BioRender.com. ATP: Adenosine triphosphate; Aβ: amyloid-β; ETC: electron transport chain; GLUT-1: glucose transporter; GLUT-3: glucose transporter 3; GST: glutathione S-transferase; LDH: lactate dehydrogenase; OXPHOS: oxidative phosphorylation; ROS: reactive oxygen species; TCA: tricarboxylic acid; TNF-α: tumor necrosis factor-alpha; α-KG: alpha-ketoglutarate.
In the early stages of AD, the efficiency of glucose utilization decreases, leading to increased energy expenditure. Additionally, the expression levels of glucose transporters GLUT1 and GLUT3 are significantly downregulated in the brains of patients (Dewanjee et al., 2022). In patients with PD, ATP concentrations decrease in the midbrain and putamen, along with reductions in high-energy phosphate compounds in the basal ganglia and frontal regions. Additionally, there is decreased glucose metabolism activity in the occipital and parietal areas (Cai et al., 2019). Memory functional decline has been associated with decreased [18F]fluorodeoxyglucose (18F-FDG) metabolism in the posterior parietal and temporal regions. Conversely, attention-related behavioral performance correlates with metabolic FDG levels in the frontal areas (Borsche et al., 2023).
Wang et al. (2024a) demonstrated the occurrence of significantly reduced glucose consumption in the basal ganglia region, along with elevated lactate concentrations in the basal ganglia and occipital cortex of patients with HD. The diminished glucose uptake in the brains of patients with HD leads to an increasing reliance on the glycolytic pathway’s consumption of glucose, while the proportion of glucose metabolized through the pentose phosphate pathway decreases correspondingly. This reduction limits the production of NADPH and glutathione, which are essential for subsequential antioxidant protection. As a result, key enzymes in glycolysis and the TCA cycle become susceptible to oxidative damage, further restricting energy generation and creating a vicious cycle (Jurcau and Jurcau, 2023).
The maintenance of calcium homeostasis is a requisite for cellular activity, and mitochondria are involved in regulating intracellular calcium concentrations. A disruption in calcium homeostasis leads to the formation of Aβ plaques, the accumulation of neurofibrillary tangles, and the dysfunction of synaptic plasticity, leading to delayed neurodegenerative changes in the brain, excitotoxicity, and glial cell-induced neuroinflammation (Chanaday et al., 2021).
In chronic neurodegenerative diseases, mitochondrial calcium uptake increases due to abnormal changes in calcium channels. The excessive accumulation of calcium in mitochondria can lead to the opening of mitochondrial permeability transition pores (mPTPs) and the release of proapoptotic factors, such as cytochrome c, triggering apoptosis, as well as affect the cytoplasmic calcium buffer system (Calvo-Rodriguez and Bacskai, 2021; Griffioen, 2023).
Under physiological conditions, reduced amounts of calcium can lead to the leakage of ROS from mitochondrial respiratory chain complexes I and III. However, under pathological conditions, ROS production increases. The disruption to the pH gradient across the mitochondrial membrane promotes ROS production by interrupting the mitochondrial membrane potential associated with calcium uptake and thereby influencing the spatial organization of ROS-formation sites and the rate of mitochondrial respiration (Palma et al., 2024). Notably, the mitochondrial Ca2+ uniporter has been shown to trigger ROS production by affecting the structural load of mitochondria and the concentration of Ca2+ (Princen et al., 2024).
Mutation of genes
Mitochondrial heterogeneity refers to the simultaneous presence of both wild-type and mutant mitochondrial DNA (mtDNA) during cell development, disease, and aging. It is characterized by the variability of mtDNA under conditions of cell division, across different tissues, and in varying environmental contexts. Various factors, including mtDNA mutations, environmental influences, and lifestyle choices, can lead to variations in mitochondrial functions and status (Mareckova et al., 2025). For example, studies have demonstrated the occurrence of mitochondrial heterogeneity in dopaminergic neurons in the substantia nigra of patients with PD. Within these neurons, some mitochondria are damaged or functionally abnormal, leading to neuronal death, while others possess relatively normal mitochondria that can maintain proper neuronal function (Trinh et al., 2023; Kong et al., 2024). This level of heterogeneity may complicate and hinder interventions for PD. Additionally, key subunits of the respiratory chain complex are encoded by mtDNA, and mutations in mtDNA can impair mitochondrial function, resulting in decreased ATP production and increased ROS generation (Kotrys et al., 2024). Mitochondrial function can also be affected by mutations in the nuclear genome, such as within the genes MFN2, OPA1, and C9orf72 (Heuer, 2023; Figure 7).
Figure 7.

Mitochondrial dysfunction and gene mutation.
Mutations affect mitochondrial function in both mitochondrial and nuclear genomes, including genes such as MFN2 and OPA1. The protein encoded by the OPA1 gene is located on the inner mitochondrial membrane, where it facilitates the fusion of the inner mitochondrial membranes and regulates the remodeling of mitochondrial cristae. Mutations in the OPA1 gene can lead to mitochondrial fragmentation, resulting in abnormal mitochondrial morphology and impaired function. This fragmentation increases mitochondrial membrane permeability, reduces ATP production, and leads to excessive generation of reactive oxygen species. Created with BioRender.com. MFN2: Mitofusin 2; OPA1: optic atrophy 1; PINK1: PTEN-induced putative kinase 1; Parkin: E3 ubiquitin-protein ligase Parkin.
Although mtDNA fragments may be inserted into target sites during genome editing, genome editing process can also trigger mtDNA instability, leading to mtDNA insertion into the nuclear genome. These genes encode proteins that are involved in the regulation of mitophagy, and mutations in these genes can lead to the accumulation of damaged mitochondria.
Changes of dynamics
Mitochondrial fragments dynamically undergo fusion and fission to form a highly interconnected tubular network in the cell (Burté et al., 2015). Key regulatory proteins involved in fusion, such as OPA1, MFN1, and MFN2, have a balanced coexistence with proteins that regulate fission, including FIS1, GDAP1, OPA3, and DRP, under normal mitochondrial conditions.
In neurodegenerative diseases, there are significant changes in mitochondrial morphology (Figure 8). These morphological changes influence the distribution of mitochondria, potentially leading to an uneven distribution of energy supply and localized ROS accumulation. Mitochondria form a dynamic network structure through fusion and fission. In neurodegenerative diseases, the network-remodeling process is often unbalanced, with a lack of fusion-related protein activity and an elevation of fission-related protein activity. Excessive fission can lead to a loss of mitochondrial function. This imbalance affects the overall function of mitochondria and cellular health (Bustamante-Barrientos et al., 2023; Grel et al., 2023; Monzel et al., 2023), and thereby exacerbates neurodegenerative pathologies (Burté et al., 2015; Wu et al., 2024b).
Figure 8.

Mitochondrial dysfunction and kinetic changes.
Mitochondria are in a dynamic state of fusion and fission, regulated by fusion-related proteins such as OPA1 and MFN1/2 on the mitochondrial membrane, as well as fission-related proteins such as FIS1, DRP1, and GDAP1. This regulation maintains a dynamic balance in mitochondrial morphology. Changes in mitochondrial morphology can affect mitochondrial dynamics, resulting in an uneven distribution of mitochondria in neurons, which can lead to an imbalance in energy supply and affect normal neuronal activity. When mitochondrial fission is dominant, the increase in non-functional mitochondria promotes the initiation of mitophagy while causing mitochondrial damage and dysfunction. Created with BioRender.com. DRP1: Dynamin-related protein 1; FIS1: mitochondrial fission protein 1; GDAP1: ganglioside-induced differentiation-associated protein 1; MFN1/2: mitofusin 1/2; OPA1: optic atrophy 1; OPA3: outer mitochondrial membrane lipid metabolism regulator OPA3.
Process of mitophagy
Mitochondrial autophagy is a selective process aimed at clearing damaged or dysfunctional mitochondria. This process is one of the key mechanisms in mitochondrial quality control and is associated with neurodegenerative diseases, cancer, aging, and other physiological and pathological conditions (Antico et al., 2025).
Multiple mammalian mitophagy pathways have been identified (Antico et al., 2025). For example, the primary regulatory pathway for mitophagy, the PINK1/Parkin signaling pathway, mediates the autophagic clearance of depolarized mitochondria associated with PD (Marchesan et al., 2024). In healthy mitochondria, PINK1 enters the inner membrane and is degraded by proteasomes. In patients with PD, oxidative stress, mtDNA mutations, and abnormal aggregations of α-syn lead to a reduction in mitochondrial membrane potential (ΔΨm) and an increase in ROS, resulting in functional abnormalities in PINK1 and Parkin. Under these conditions, PINK1 phosphorylates various proteins, including Parkin. Phosphorylated Parkin is then transported from the cytoplasm to the outer mitochondrial membrane, further promoting mitochondrial degradation (Guardia-Laguarta et al., 2019). Other proteins such as MFN1/2, OPA1, and BCL2 also participate in the regulation of mitophagy (Narendra and Youle, 2024). The receptors BNIP3, NIX, and FUNDC1 on the outer mitochondrial membrane induce mitochondrial autophagy under hypoxic conditions, with NIX specifically responsible for clearing mitochondria during the maturation stage of red blood cells (Cao et al., 2023).
The main pathways mediating mitophagy are the ubiquitin-dependent pathway and the non-ubiquitin-dependent pathway. In the ubiquitin-mediated pathway, the PINK1 protein cannot penetrate the inner mitochondrial membrane but instead activates Parkin on the outer mitochondrial membrane, transforming it into an active E3 ubiquitin-protein ligase. This enzyme subsequently catalyzes ubiquitination modifications on several outer membrane components (Heo et al., 2015). Following ubiquitination, autophagic proteins, such as p62, OPTN, and NDP52, accumulate on the outer mitochondrial membrane. TANK-binding kinase 1 phosphorylates these autophagic proteins, enhancing their binding affinity for ubiquitin and LC3, and thereby facilitating the recruitment of ubiquitinated proteins to autophagosomes through their interactions with LC3. Mature autophagosomes then fuse with lysosomes to form autolysosomes, where the mitochondria are ultimately degraded (Antico et al., 2025). In contrast, in the non-ubiquitin-mediated pathway, mitochondrial outer membrane proteins such as NIX, BNIP3, and FUNDC1 can directly bind to LC3 without undergoing ubiquitination (Tang and Dong, 2025; Figure 9).
Figure 9.
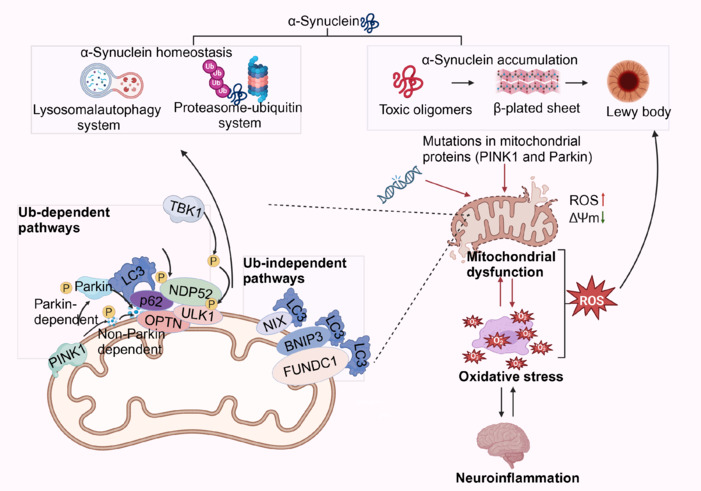
Mitochondrial dysfunction and mitophagy.
In normal mitochondria, PINK1 enters the inner mitochondrial membrane and is degraded by the proteasome without triggering autophagy. However, when mitochondria are damaged, a decrease in mitochondrial membrane potential, mutations in mitochondrial DNA, and oxidative stress lead to an excessive accumulation of PINK1 on the inner mitochondrial membrane. This accumulation results in the phosphorylation of Parkin protein. The phosphorylated Parkin then translocates from the cytoplasm to the outer mitochondrial membrane, where it binds with PINK1 and is subsequently converted into an active ubiquitin ligase. This active ubiquitin ligase ubiquitinates the outer mitochondrial membrane, attracting autophagy proteins with ubiquitin-binding receptors, such as p62, OPTN, and NDP52, which aggregate on the outer mitochondrial membrane. These autophagy proteins are phosphorylated to facilitate the interaction between the ubiquitinated mitochondria and the autophagosome marker LC3, forming an autophagosome. The autophagosome then fuses with a lysosome to form an autolysosome, ultimately leading to the degradation of mitochondrial components by lysosomal hydrolases. Created with BioRender.com. BNIP3: BCL2 interacting protein 3; FUNDC1: Fission 1; NDP52: nucleotide-binding oligomerization domain-like receptor; NIX: nucleophosmin/nucleoplasmin X-associated protein; OPTN: optineurin; PINK1: PTEN-induced putative kinase 1; ROS: reactive oxygen species; TBK1: TANK-binding kinase 1; ULK1: Unc-51 like autophagy activating kinase 1; α-synuclein: alpha-synuclein; ΔΨm: mitochondrial membrane potential.
The dysfunction of PINK1 and Parkin in patients with PD can lead to the accumulation of damaged mitochondria. This phenomenon may be associated with oxidative stress, damage to mitochondrial DNA, and the abnormal accumulation of α-syn (Moskal et al., 2020). In AD, an increase in the LC3-II/I ratio indicates enhanced autophagic activity, although the specific mechanisms involved are not yet fully understood. This increase may be related to the clearance of Aβ and could contribute to neuronal damage and death (Wang et al., 2024b).
The occurrence of autophagy in chronic neurodegenerative diseases is complex. Moderate autophagy can help to clear damaged mitochondria and other cellular components, thereby maintaining cellular health (Wu et al., 2024a). However, abnormal autophagy can lead to increased cell damage and cell death. For instance, in AD, moderately enhanced autophagy activity may aid in the clearance of Aβ (Xie et al., 2022). Additionally, there may be obstacles to the initiation of autophagy in some neurodegenerative diseases, leading to increased damage to mitochondria and cellular stress (López-Otín et al., 2023).
Relationship Between Mitochondrial Dysfunction and Chronic Neurodegenerative Diseases
Mitochondrial dysfunction and chronic neurodegenerative diseases: Unraveling the connection
Chronic neurodegenerative diseases are a group of neurological disorders that pose considerable risks to human health. Their pathogenesis is complex, and existing treatment measures are often ineffective. Chronic neurodegenerative diseases include conditions such as AD, PD, HD, and ALS. They share common features, including neuronal injury, degeneration leading to cell death, and a decline in neurological function (Ozgen et al., 2022). Researching effective treatment strategies for chronic neurodegenerative diseases is a major challenge in modern neurology, as current therapies are limited, and traditional medications typically only alleviate symptoms without providing a complete cure. This limitation exacerbates the social and economic burdens of these conditions, particularly as the aging population grows (Abramov and Bachurin, 2021). Fundamentally, the occurrence and progression of chronic neurodegenerative diseases result from the interactions of multiple underlying mechanisms, necessitating multifaceted and multi-targeted therapeutic approaches.
The neuronal cells of patients with chronic neurodegenerative diseases exhibit physiological dysfunction and various damaged organelles, particularly mitochondria. Morphologically and functionally abnormal mitochondria are major factors in the development of CNDs, manifesting mainly as ATP depletion, oxidative stress, and Ca2+ homeostasis imbalances. Studies have shown that abnormal mitochondria and impaired energy metabolism in the brains of those affected by AD often precede the clinical pathological changes associated with the disease (Hui and Just, 2022; Bhatti et al., 2023). Electron microscopy results from brain tissue biopsies of patients with PD revealed evidence of mitochondrial structural damage, primarily disruptions in mitochondrial cristae and partial or complete loss of internal structure (Samanta et al., 2024). In brains affected by AD and PD, levels of a key regulator of mitochondrial biogenesis, peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), are decreased (Rojo et al., 2017; Chen et al., 2020). In HD, there is abnormal oxidative stress, ROS accumulation, mitochondrial morphological changes, increased mitochondrial fragmentation, and a decrease in the activity of respiratory chain complexes II, III, and IV (Cherubini et al., 2020). In ALS, mitochondrial dysfunction in astrocytes leads to altered mitochondrial glutamate metabolism, impairing the cells’ ability to clear synaptic glutamate. This results in excessive glutamate accumulation, triggering neuronal excitotoxicity (Granatiero and Manfredi, 2019). Given that mitochondrial dysfunction significantly impacts CNDs, improving mitochondrial function may provide a therapeutic avenue for mitigating the progression of these diseases (Figure 10).
Figure 10.

Relationship between mitochondrial dysfunction and chronic neurodegenerative diseases.
Mitochondrial dysfunction is primarily characterized by the obstruction of oxidative phosphorylation, resulting in reduced ATP synthesis, excessive production of ROS, and disturbances in calcium homeostasis. Additionally, it involves mitochondrial fission, enhanced autophagy, mutations in mitochondrial DNA, dysregulation of immune responses leading to chronic inflammation and damage, and abnormal apoptosis mediated by mitochondria, all contributing to cell death. These impaired mitochondrial functions not only disrupt cellular processes but also act as intracellular foreign bodies, triggering excessive inflammatory responses. This, in turn, directly contributes to and accelerates the onset and progression of neurodegenerative diseases such as AD, PD, HD, and ALS. The characteristic pathological changes of AD, including Aβ protein deposition and neurofibrillary tangles, are closely associated with chronic neuroinflammation and neuronal apoptosis mediated by dysfunctional mitochondria. These factors play a significant role in the pathogenesis of both PD and HD. Created with BioRender.com. AD: Alzheimer’s disease; Aβ: amyloid-β; ALS: amyotrophic lateral sclerosis; BH3-only: Bcl-2 homology domain only proteins; CytC: cytochrome c oxidase; HD: Huntington’s disease; IL-6: interleukin-6; INF: interferon; NLRP3: NOD-like receptor family pyrin domain containing 3; OXPHOS: oxidative phosphorylation; PD: Parkinson’s disease; ROS: reactive oxygen species; TNF-α: tumor necrosis factor-alpha.
Mitochondrial dysfunction and Alzheimer’s disease
The primary manifestations of AD are the deposition of Aβ and tau protein fibrils, which are accompanied by neuronal loss. The main symptoms include cognitive impairment and memory decline, making AD the most common CND, accounting for 80% of dementia diagnoses (Pszczołowska et al., 2024). Although the exact mechanism of AD remains unclear, mitochondrial dysfunction, along with the resulting oxidative stress and neuroinflammation, is considered a major contributing factor. Previous studies have reported that mitochondrial dysfunction promotes the generation of Aβ and the phosphorylation of tau protein (Yan and Baolu, 2013; Mei and Yanling, 2018). In patients with AD, the activity of the oxidative phosphorylation complexes is reduced, leading to observable pathological changes, such as mitochondrial swelling, fragmentation, and neurite degeneration, which can occur before clinical symptoms appear (Pszczołowska et al., 2024). Additionally, decreased glucose metabolism in brain tissue contributes to memory impairment in patients with AD. In an AD model Caenorhabditis elegans that overexpressed Aβ and tau proteins, memory impairment was ameliorated by targeting mitophagy through the PINK1/Parkin-dependent pathway, providing new insights for potential interventions in AD (Fang et al., 2019).
Under normal physiological conditions, ROS are produced by the mitochondria. However, aging disrupts the dynamic balance of free radicals, leading to the excessive accumulation of ROS, DNA sequence damage, and altered gene expression, and ultimately resulting in cell apoptosis, necrosis, or even carcinogenesis. CNDs are closely associated with oxidative stress. In the early stages of these diseases, before the formation of neurofibrillary tangles, increased levels of markers of DNA and RNA oxidative damage can be observed in neurons (Zhou et al., 2022). Several drugs targeting mitochondrial dysfunction in AD are currently at various stages of clinical trial. Dhapola et al. (2022) found that calcium channel blockers such as nifedipine, as well as antioxidants such as MitoQ, curcumin, and apigenin, alleviated the memory dysfunction and cognitive deficits in AD mouse models by intervening in mitochondrial dysfunction. Additionally, using small-molecule compounds to target voltage-dependent anion channels and maintain intracellular calcium homeostasis presents a potential method for intervening in AD (Chen et al., 2023; Yang et al., 2024). Therapeutics that target mitochondrial dysfunction to enhance mitochondrial protein homeostasis hold considerable potential for intervening in the onset and progression of AD (Sorrentino et al., 2017; Xie et al., 2022; Cummings et al., 2024).
Mitochondrial dysfunction and Parkinson’s disease
PD is characterized by the accumulation of α-syn aggregates in dopaminergic neurons, leading to the formation of Lewy bodies and subsequent neuronal loss that primarily affects the substantia nigra. Symptoms of PD include resting tremors, increased muscle tone, cognitive impairment, and depression. PD was among the first CNDs to be linked to mitochondrial dysfunction. Dopaminergic neurons are particularly sensitive to mitochondrial dysfunction (Blagov et al., 2024). Mitochondrial dysfunction, primarily caused by respiratory chain damage due to complex I inhibition, contributes to increased oxidative stress and is a major factor in the progressive degeneration of dopaminergic neurons (Moradi Vastegani et al., 2023). This dysfunction can lead to abnormal apoptosis, and impaired mitophagy may exacerbate cell death (Venderova and Park, 2012). Studies have shown that knocking out caspase 3 can slow the manifestation of mPTP-induced Parkinsonian symptoms in mice, indicating there is a strong connection between mitochondria-induced apoptosis and PD. The PINK1/Parkin pathway mediates mitophagy, and inhibition of these pathway proteins can result in the impaired clearance of damaged mitochondria, increased ROS production, and the excessive accumulation of α-syn. Additionally, the increased expression of dynamin-related protein 1 (Drp1) has been observed in PD models induced by mPTP and rotenone. Inhibiting mitochondrial fission has been shown to improve PD symptoms in mice, suggesting that mitochondrial dynamics play a major role in the development of PD (García-Revilla et al., 2024; Yao et al., 2024). Haque et al. (2022) found that inhibiting mitochondrial fission can reduce α-syn expression, providing valuable insights into potential interventions in PD.
Zheng et al. (2024) proposed various methods to improve mitochondrial dysfunction and intervene in PD, and subsequent studies have resulted in notable progress. These include methods for clearing mitochondrial ROS, enhancing autophagy, promoting mitochondrial biogenesis, transplanting healthy mitochondria, utilizing photobiological regulation, and implementing exercise interventions. For instance, targeting PINK1 with triphosphate agonists and their derivatives, as well as chloride and its analogs, can increase PINK1 stability or accumulation within mitochondria. Additionally, ROCK inhibitors, such as fasudil, SR3677, and SR3677 hydrochloride, have been shown to enhance Parkin-mediated mitophagy and improve PD phenotypes, and are potential targets for intervention in PD (Moskal et al., 2020). LRRK2 kinase inhibitors, including DNL201 and DNL151, along with pyrazolopyrimidine LRRK2 small-molecule inhibitors, suppress LRRK2 activity, neuronal α-syn aggregation, and neurodegeneration, demonstrating their potential for PD intervention (Moskal et al., 2020; Ntetsika et al., 2021). Moreover, Alda-1, a mitochondria-targeted antioxidant activator, as well as acetaldehyde dehydrogenase 2, voltage-dependent anion channel-targeted cholesterol oxime, PPAR-γ agonists, melatonin, amphetamine, α-lipoic acid, and other medications such as Mito Q10 can all play protective roles in mitochondrial function and contribute to anti-PD effects (McFarthing et al., 2024).
Mitochondrial dysfunction and Huntington’s disease
HD is an inherited neurodegenerative disorder caused by abnormal CAG repeat sequences in the huntingtin (HTT) gene, leading to the accumulation of mutant HTT (mHTT) proteins in the brain. HD primarily affects the striatum and involves brain-derived neurotrophic factor (BDNF) deficiency and damage to GABAergic neurons. Clinically, it manifests as cognitive impairment, psychiatric symptoms, choreiform movements, and abnormal muscle tone (Lin et al., 2025). The pathological process of HD also involves mitochondrial dysfunction, neuroinflammation, and oxidative stress (Burtscher et al., 2024). Tong et al. (2024) reported that patients with HD exhibit low energy metabolism, elevated lactate levels, and muscle fiber atrophy, resulting in weight loss. The mHTT protein plays an important role in mitochondrial energy metabolism, as its interaction with the mitochondrial membrane leading to Ca2+ release and abnormal mitochondrial morphology and dynamics. Alternatively, mHTT can indirectly inhibit mitochondrial function through abnormal transcriptional regulation. PGC-1α may be a key player linking transcriptional abnormalities in HD with mitochondrial dysfunction. In the striatum of patients with HD, reduced expression of PGC-1α inhibits mitochondrial biogenesis and suppresses the neurotrophic effects of BDNF. In transgenic mouse models, overexpression of PGC-1α was observed to reduce mHTT aggregation and promote BDNF expression, thereby improving neuronal degeneration and motor deficits (Boulos et al., 2024). Furthermore, it has been reported that time-restricted feeding may improve mitochondrial function, maintain metabolic balance, upregulate autophagy to clear mHTT, and restore striatal BDNF levels, offering potential intervention effects for neurodegenerative diseases such as HD (Wells et al., 2024). Naia et al. (2017) used resveratrol (a SIRT1 activator) to activate deacetylase activity in an HD model, resulting in improved HD-related brain atrophy and motor dysfunction symptom relief. These improvements may have been attributable to the promotion of mitochondrial PGC-1α mRNA transcription and protein expression (Naia et al., 2017). mHTT aggregation can induce ROS production; the expression of downstream antioxidant factors, such as Nrf2, SOD, and GPx-1, is inhibited by downregulating PGC-1α expression, leading to oxidative stress that further exacerbates neuronal damage. An insufficient mitochondrial energy supply and calcium overload result in glutamate accumulation outside of neuron, causing sustained excitotoxicity, which is a major cause of neuronal death in patients with HD (Dai et al., 2023). Mitochondria isolated from lymphoblastoid cells of patients with HD showed reduced ETC complex activity and mitochondrial membrane depolarization, and underwent gradual destruction as the polyglutamine repeat length increased (Dai et al., 2023).
In their report, Bajpai et al. (2024) discuss in detail several HD intervention strategies targeting oxidative stress, including drug interventions aimed at reducing protein misfolding and the HTT aggregation caused by HTT gene CAG repeat expansions. They used gene editing technologies, such as CRISPR-Cas9, to demonstrate the effectiveness of antioxidant interventions in HD (Bajpai et al., 2024). Chakraborty et al. (2024) constructed an in vitro cell culture system to simulate the oxidative stress environment of HD and found that F2,6BP reduced oxidative stress markers and enhanced mitochondrial respiratory function. Administering F2,6BP to HD transgenic mouse models significantly improved their motor functions and extended their survival, while reducing the aggregation of pathological proteins in the brain. In summary, mitochondrial dysfunction is a crucial factor in the development of HD. The above studies indicate that methods for intervening in mitochondrial oxidative stress have potential therapeutic efficacy for CNDs and could drive the development of new drugs.
Mitochondrial dysfunction and amyotrophic lateral sclerosis
ALS is a condition involving the disintegration of neuromuscular junctions and muscle atrophy. The main pathogenic mechanisms of ALS currently include mitochondrial dysfunction, neuroinflammation, glutamate excitotoxicity, SOD1 gene mutations, and abnormal protein aggregation (Kubat and Picone, 2024). Mitochondrial swelling and vacuolization have been observed in motor neurons. Abnormal mitochondrial transport leads to mitochondria clusters in axons and an insufficient supply of energy at the neuromuscular junction, impairing synaptic transmission and resulting in muscle atrophy and motor dysfunction (Kubat and Picone, 2024). Researchers have reported finding a deficiency in cytochrome c oxidase I in patients with ALS, along with the abnormal accumulation of mitochondria at the axon terminals of their spinal anterior horn motor neurons. Rizea et al. (2024) reported that several ALS-related pathogenic genes, such as SOD1 and TDP-43, can cause mitochondrial dysfunction. TDP-43 protein aggregations in the cytoplasm of motor neurons in patients with ALS increase cellular sensitivity to oxidative stress. The aggregated TDP-43 protein enters the mitochondrial matrix, inhibiting mitochondrial protein translation, leading to structural and functional abnormalities and exacerbating oxidative stress (Rizea et al., 2024). Mitochondrial abnormalities directly stimulate neurons, causing damage that leads to the expansion of inflammation, calcium overload, and increased glutamate toxicity, and indirectly resulting in motor neuron degeneration and death. The entry of TDP-43 protein into mitochondria triggers mtDNA release, thereby activating the cGAS/STING pathway, which may contribute to neuronal death and could represent a common mechanism of neuronal death in patients with ALS. A disruption of calcium homeostasis is a crucial part of ALS pathogenesis. An imbalance in calcium homeostasis primarily arises from the increase in calcium ions permeating the cell membrane and a reduction in their clearance from cells. In patients with ALS, intracellular calcium buffering systems are dysfunctional, and the ability of mitochondria to absorb calcium ions is diminished, making it difficult for neurons to restore calcium levels after excitation. The sustained high calcium levels ultimately damage neurons (Yu et al., 2020b).
Tan et al. (2024b) focused on the role of BRD7 in the survival of ALS motor neurons, considering its function as a regulator associated with cellular aging. Knocking out the BRD7 gene inhibited the mitochondrial translocation of p21 and p53, reducing apoptosis and senescence. Guo et al. (2024) demonstrated that FUNDC1-mediated mitophagy improved motor function and extended survival rates in ALS mice given intrathecal injections of AAV9 vectors expressing FUNDC1 or EGFP, providing a scientific basis for new intervention strategies. Improving mitochondrial function has not only become a potential intervention target for ALS but also provides hope for the development of new drugs.
Mitochondria as Targets for Chronic Neurodegenerative Disease Intervention
Mitochondrial damage and dysfunction are involved in many stages of CNDs, suggesting that mitochondria are a potential intervention target. Mitochondrial dysfunction, an important cause of CNDs, is primarily characterized by TCA cycle disorders that lead to insufficient ATP production, excessive ROS generation, calcium overload, and abnormal apoptotic pathways. Additionally, abnormalities in mitochondrial quality control, dynamics, and biogenesis have been observed in these conditions (Klemmensen et al., 2024), and mitochondria are key targets in the intervention of these diseases (Figure 11).
Figure 11.

Mechanistic pathways of mitochondria-targeted drugs.
The main mechanisms of mitochondrial-targeted therapy include regulating mitochondrial dynamics—specifically, promoting mitochondrial fusion and inhibiting mitochondrial fission; regulating the mitochondrial apoptosis pathway, which involves promoting the expression of anti-apoptotic proteins such as Bcl-2 and Bcl-xL while inhibiting the expression of pro-apoptotic proteins such as Bax and Bak; inducing mitophagy to eliminate abnormal mitochondria by promoting the AMPK/ULK1 pathway and inhibiting the PI3K/AKT/mTOR pathway to activate the autophagy process; and promoting mitochondrial biogenesis by targeting factors that regulate the expression of genes related to mitochondrial biogenesis, such as PGC-1α, PPARγ, and SIRT3. Created with BioRender.com. AMPK: aMP-activated protein kinase; BH3-only: bcl-2 homology domain only proteins; Beclin1: recombinant human Beclin 1 protein; DRP1: dynamin-related protein 1; LC3: microtubule-associated protein light chain 3; mTOR: mammalian target of rapamycin; MFF: mitochondrial fission factor; Mfn1/2: mitochondrial fusion factor 1/2; mPTP: mitochondrial permeability transition pore; P62: sequestosome-1; PPARγ: peroxisome proliferator-activated receptor γ; PGC1-α: peroxisome proliferator-activated receptor γ coactivator 1α; SIRT3: Sirtuin-3; SOD1: superoxide dismutase 1; TFAM: transcription factor A, mitochondrial; ULK1: Unc-51 like autophagy activating kinase 1.
Promotion of mitochondrial autophagy
The promotion of mitochondrial autophagy is a prominent research topic. Mitophagy is a self-protective mechanism in cells whereby damaged or stressed mitochondria are targeted for degradation. Via the timely clearing of damaged or aged mitochondria, this process helps avoid inflammation and stress-induced damage, thereby maintaining the cell’s healthy state (Lin et al., 2019). When selective mitochondrial autophagy is impaired, dysfunctional mitochondria accumulate continuously. In the CNS, mitochondrial dysfunction or the absence of related proteins can disrupt the normal transport and distribution of mitochondria, leading to a reduced number of mitochondria in neuronal axons, dendrites, and synaptosomes. This affects the formation of dendritic spines and synapses as well as the conduction of signals in neuronal axons and communication between neurons (Gu et al., 2024). Beclin 1 initiates autophagy by connecting with LC3 to form autophagosomes, completing the autophagic process (Liu et al., 2019). The target of rapamycin (mTOR) also regulates mitophagy and controls autophagy by regulating the activity of the ULK1 complex. Under normal conditions, mTOR signaling is suppressed, maintaining basal levels of autophagy within cells. However, the enhanced inhibition of ULK1 by mTOR occurs through the mTOR signaling pathway, which reduces autophagy. This disruption of intracellular homeostasis can ultimately lead to cell death, making it an effective target for intervention in CNDs (Liu et al., 2023). Experimental studies have shown that ATP can enhance PINK1 activity, while introducing the deubiquitinase USP30 inhibitor can increase ubiquitination levels in neuronal models (Blagov et al., 2024; Narendra and Youle, 2024). Both approaches promote PINK1/Parkin pathway-mediated autophagy and ameliorate neuronal damage. This further indicates that inhibiting the mTOR signaling pathway could be an important strategy for indirectly activating autophagy to intervene in diseases. Enhancing the function of PINK1 or Parkin through gene therapy has been shown to effectively promote the clearance of damaged mitochondria and improve neuropathological features in PD models (Antico et al., 2025). Targeted gene therapies are being developed for specific genetic mutations, such as SOD1 gene mutations in ALS (Wen et al., 2025b). CRISPR/Cas9 technology can be utilized to repair or knock out mutations in genes associated with mitochondrial dysfunction, helping to restore normal mitophagy function (Eghbalsaied et al., 2024). Natural products often offer high safety and multiple targets, but their mechanisms of action can be complex, and quality control can be challenging. In contrast, CRISPR/Cas9 gene editing technology presents the advantages of high efficiency and flexibility. Despite the challenges related to its delivery and ethical considerations, this technology shows great potential for the treatment of CNDs. The activation of mitochondrial autophagy has emerged as an important breakthrough in therapeutic approaches for CNDs. With its role in clearing toxic proteins, mitochondrial autophagy offers promise as a target of effective interventions for various conditions, including AD, PD, HD, and ALS.
Inhibition of mitochondrial fission
Normally, the dynamic equilibrium of mitochondria is maintained by various proteins that ensure the stability of organelle processes. However, under stress conditions, mitochondria tend to divide more frequently, disrupting this equilibrium. One of the key proteins involved in membrane cleavage is Drp1, which plays a critical role in regulating mitochondrial fission and, consequently, mitochondrial dynamics and autophagy. Studies have shown that Mdivi-1 can limit Aβ accumulation and improve mitochondrial function by reducing hydrogen peroxide production and lipid peroxidation, thereby positively impacting symptom improvement in AD (Marx et al., 2024; Pszczołowska et al., 2024). Additionally, research indicates that melatonin positively influences mitochondrial dynamics by modulating Drp1. However, the underlying mechanisms by which melatonin regulates mitochondrial dynamics remain to be fully explored, and this could represent a potential intervening strategy in CNDs.
The inhibition of Drp1 results in the mitigation of damage to dopaminergic neurons in PD models. For example, P110, a heptapeptide inhibitor designed to inhibit the Drp1–Fis1 interaction, was shown to reduce pathological features in a variety of chronic degenerative disease models (Rios et al., 2023). In addition, the knockdown or inhibition of Drp1 expression by CRISPR/Cas 9 technology effectively reduced mitochondrial division and improved the pathological characteristics of neurodegenerative diseases (Gómez-Deza et al., 2024). P110 and Mdivi-1 have broad and straightforward advantages as small-molecule inhibitors, but have potential side effects. They are suitable for short-term interventions and the rapid regulation of mitochondrial dynamics, but require further safety assessments through clinical experiments. In contrast, CRISPR/Cas9 knockout of Drp1 has high specificity and basic research value, making it suitable for diseases that require long-term gene regulation; however, the potential risks should be carefully evaluated.
Promotion of mitochondrial biogenesis
Mitochondrial biosynthesis refers to the involvement of nuclear encoding and mitochondrial gene expression in generating new mitochondria. It serves as a prerequisite and foundation for the normal functioning of mitochondria and participates in mitochondrial quality control. The regulation of gene expression during mitochondrial biogenesis involves peroxisome proliferator-activated receptor γ (PPARγ) and sirtuins. Additionally, peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), which is directly related to NRF-1, primarily regulates mitochondrial biogenesis and increases the transcription of NRF-1-regulated genes (Wen et al., 2025a). The PPARγ/PGC-1α axis is activated by rosiglitazone and bezafibrate, while quercetin and resveratrol can activate sirtuins and AMPK and thereby promote mitochondrial biogenesis. Bezafibrate is known to increase mitochondrial protein and ATP production, providing neuroprotective effects in mitochondrial encephalopathy mice (Wang et al., 2019).
It has also been shown that dietary supplements, such as resveratrol and coenzyme Q10, can promote mitochondrial biogenesis by activating key regulators such as PGC-1α (Ghosh and Kumar, 2024). Among the current drug options, bezafibrate is an ideal choice in terms of its safety, effect on multiple targets, and promotion of mitochondrial biogenesis. It can not only efficiently promote mitochondrial biogenesis but also achieve multi-target regulation through the activation of PPARα and PPARγ, improving lipid metabolism and insulin resistance while demonstrating excellent overall efficacy (Douiev et al., 2020). Additionally, its high safety profile and low cardiovascular risk make it particularly advantageous for clinical applications. Meanwhile, coenzyme Q10 is a natural antioxidant with a high safety profile, making it suitable for adjuvant therapy, especially for improving long-term mitochondrial function and reducing oxidative stress. By reducing oxidative stress, it indirectly promotes mitochondrial biogenesis, providing continuous mitochondrial support for patients and further optimizing treatment outcomes.
Mitochondrial-targeted antioxidation
Studies show that the function of mitochondrial oxidative respiratory complex I is significantly over-active in PD. Additionally, enhanced lipid peroxidation and the reduced consumption of SOD and glutathione have been observed in the densest part of the substantia nigra, indicating the occurrence of oxidative stress damage (Cenini et al., 2019). An important strategy for intervening in CNDs is to target mitochondria with drug interventions to correct mitochondrial dysfunction and reverse the resulting neuronal oxidative stress damage. For instance, when AD rats were treated with ginsenosides, glutathione consumption was reduced, SOD and glutathione peroxidase activities were restored, while ROS levels were decreased in the brain tissue. Mito-Q may protect mitochondrial function by reducing mitochondrial fission and inhibiting ROS formation (Wen et al., 2025b). Monoamine oxidase B inhibitors interfere with PD by reducing the production of ROS. Mito Vit E and Mito TEMPO, derivatives of vitamin E, have been shown to have anti-lipid peroxidation effects and can effectively protect mitochondria from oxidation. In vitro experimental results have also demonstrated their high efficiency in combating mitochondrial oxidative stress damage (Pszczołowska et al., 2024). Another study indicated that curcumin exerts antioxidant and anti-inflammatory effects by reducing cytochrome c release and activating caspase 3, increasing the mitochondrial membrane potential and decreasing the expression level of Bcl-2 protein (Nicoliche et al., 2024).
Overall, each of these drugs has advantages in targeted mitochondrial antioxidant therapy. Most of the studied drugs can ameliorate mitochondrial dysfunction and cellular oxidative stress damage; however, no systematic clinical applications have been reported. This suggests that further research attention should be given to these drugs to promote their application in clinical settings (Zong et al., 2024).
Transplantation of mitochondria
In clinical trials, several mitochondria-related diseases have been alleviated through mitochondrial replacement therapy, leading to the widespread development of mitochondrial transplantation methods in recent years (Chang et al., 2021). A previous study in MPTP-induced PD rats confirmed that the intravenous administration of mitochondria can reduce ROS and inhibit neuronal cell necrosis and apoptosis, ultimately halting the progression of PD. The experiment demonstrated that, in the PD rat model induced by neurotoxins, bare mitochondria isolated from healthy cells can be successfully transplanted into diseased cells via in situ or intravenous injection, restoring their mitochondrial function and subsequently improving their activity (Shi et al., 2017).
Exosome-mediated mitochondrial transplantation is considered a promising modality due to its low immunogenicity and high stability (Cao et al., 2024). In a 6-hydroxydopamine-induced PD rat model, the injection of similar and heterogeneous mitochondria into the medial forebrain tract reduced the deterioration of dopaminergic neurons. In an AD mouse model, transplantation of intact mitochondria significantly improved cognitive function, reduced neuronal loss, and enhanced mitochondrial function in the brain and liver, with no toxicity observed (Zong et al., 2024).
The effectiveness of mitotic transplantation is influenced by many factors, including the different administration routes, the rate at which cells absorb mitochondria, and the intrinsic state of the recipient cells. Therefore, further in-depth research is required in the field of mitochondrial transplantation. Nevertheless, this emerging treatment method has shown great potential in the treatment of many diseases, including myocardial ischemia, reperfusion injury, and neurological disorders (Kim et al., 2023). However, it still faces challenges related to mitochondrial origin, preservation, transplantation efficiency, and safety in clinical applications. Future studies are needed to further optimize the technical processes involved in mitochondrial transplantation and to explore more efficient preservation methods and targeted delivery strategies to promote its widespread use in clinical practice.
To modify the gut microbiota and maintain mitochondrial health, lifestyle interventions, such as reducing carbohydrate intake, controlling caloric intake, and performing appropriate exercise, can also be beneficial (Zhang et al., 2023). New approaches, such as stem cell mitochondrial transfer and targeted microRNAs (miRNAs) are gradually being developed. Studies show that miRNA dysregulation drives ROS production and thus mitochondrial homeostasis dysfunction in PD (Ebadpour et al., 2024; Shaheen et al., 2024). Heightened oxidative stress damage is also caused by increased ROS formation due to electron leakage from the ETC (Moradi Vastegani et al., 2023).
Clinical Translation
CNDs are a class of diseases for which there is currently no cure, and they commonly manifest as significant cognitive and motor dysfunctions in the older adult population. Numerous studies have shown that mitochondrial dysfunction plays a central role in the pathogenesis of many age-related CNDs. The oxidative stress response and mtDNA mutations are considered important factors driving the aging process (Antico et al., 2025; Wang et al., 2025). The pathogenesis of CNDs is complex and intricate, and the potential efficacy of interventions targeting mitochondria has opened up new avenues for the development of therapeutic methods for CNDs.
As of December 14, 2024, a comprehensive search of databases identified 310 drugs aimed at treating patients with AD, PD, HD, or ALS that function by ameliorating mitochondrial dysfunction. Among these drugs, 122 developed for AD primarily reduce oxidative stress, accounting for 58.9% of the total. Additionally, drugs targeting mitochondrial biogenesis and energy metabolism each represented 7.1%. In terms of drug sources, proprietary drugs made up a major portion, at 75.8%, while traditional Chinese medicines, antibodies, and antibiotics collectively accounted for 10.7%. The clinical development stages were examined, and the highest number of drugs were in Phase II trials, comprising 33.0%, which indicates that many of these drugs have already demonstrated potential value in earlier stages of research (Figure 12).
Figure 12.

Schematic representation of all drugs targeting mitochondrial dysfunction for AD.
As of December 14, 2024, all drugs targeting mitochondrial dysfunction for Alzheimer’s disease intervention are illustrated in a circular chart. The outermost circle represents the stages of drug development, which include the research stage, preclinical stage, and Phases I, II, III, and IV clinical trials. Different colored blocks denote ten aspects of mitochondrial improvement. Various fonts indicate the classification of drugs, such as drugs developed by our research group (indicated in bold red), traditional Chinese medicines and their extracts (italicized and underlined), natural compounds extracted from plants and gut microbiota (italicized), small-molecule compounds (black font), substances produced in the human body, antibiotics and antibodies (underlined), and organic compounds that are self-developed or already on the market (without special formatting). Created with BioRender.com. AD: Alzheimer’s disease; Aβ: amyloid-beta; AZBAX4, AZBAX6: specific compounds; ASI: aSI-5MT, a monoamine oxidase inhibitor; ASI: aSI-1368464, a sigma-1 receptor agonist; Astragulus: a traditional Chinese medicine; BRV: Berberine; BIIB080: a monoclonal antibody targeting Aβ protofibrils; Bumetanide: a diuretic drug; Canagliflozin: a sodium-glucose cotransporter 2 (SGLT2) inhibitor; Ca2+: calcium ion; Cu2-xSe-TPPNPs: copper-deficient selenide-based transition metal phosphine complexes; DNP: 2,4-dinitrophenol, a protonophore; dexmedetomidine: an alpha-2 adrenergic agonist; DHA: docosahexaenoic acid; Dihuang Yinzi: a traditional Chinese medicine formula; Edaravone: a free radical scavenger; Fasudil: a Rho kinase inhibitor; Fosgonimeton: not a recognized drug name, might be a typo; Fisetin: a flavonoid; GSK4527225: a small molecule inhibitor of BACE1; Gastrodin: a component of the traditional Chinese herb Gastrodia elata; Givinostat: a histone deacetylase inhibitor; L-DOPA: Levodopa, a precursor of dopamine; liraglutide: a glucagon-like peptide-1 (GLP-1) analog; Mdivi-1: mitochondrial division inhibitor 1; Montelukast: a leukotriene receptor antagonist; NanoLithiumNP03: lithium nanoparticles; NMN: nicotinamide mononucleotide; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; Quercetin: a flavonoid; Qifu-yin: a traditional Chinese medicine formula; REM0046127: Rem Sleep Modulator; Rapamycin: also known as sirolimus, an immunosuppressive drug; Resveratrol: a natural polyphenol found in red wine; Rasagiline: a monoamine oxidase B inhibitor; sirtuin-NAD: sirtuin activator that works through NAD+; SS-31: shogaol analog; VH: vitamin H (Biotin); YA3D2, YA3D4, YA3D5: specific compounds.
Among the 103 drugs targeting mitochondria for PD intervention, their primary mechanism involves reducing oxidative stress, accounting for 41.7% of such drugs. However, drugs aimed at improving mitochondrial dynamics are rarely available in the current market. Regarding drug sources, the vast majority are self-developed, constituting 76.7%, followed by human-derived substances, antibodies, and antibiotics, which collectively account for 7.77% (Figure 13).
Figure 13.

Schematic representation of all drugs targeting mitochondrial dysfunction for PD.
As of December 14, 2024, all drugs targeting mitochondrial dysfunction for PD intervention are depicted in a circular chart. The outermost circle represents the stages of drug development, including the research stage, preclinical stage, and Phases I, II, III, and IV clinical trials. Different colored blocks represent ten aspects of mitochondrial improvement. Various fonts indicate the classification of drugs, such as drugs developed by our research group (indicated in bold red), traditional Chinese medicines and their extracts (italicized and underlined), natural compounds extracted from plants and gut microbiota (italicized), small molecule compounds (black font), substances produced in the human body, antibiotics and antibodies (underlined), and organic compounds that are self-developed or already on the market (without special formatting). Created with BioRender.com. AdipoRon: Adiponectin receptor agonist; Aureusidin: a natural product with potential neuroprotective effects; AAV-GAD: adeno-associated virus carrying glutamate decarboxylase gene; AAV2-GDNF: adeno-associated virus type 2 carrying glial cell line-derived neurotrophic factor; AGB101: a specific compound; Acteoside: a compound with potential anti-inflammatory properties; A-dopamine: a specific compound; Apomorphine: a dopamine agonist used in the treatment of Parkinson’s disease; BPNSS: Basal ganglia pacemaker activity; Bifenthrin: a synthetic pyrethroid insecticide; BIIB094: a monoclonal antibody developed by Biogen; Cardiolipin: a phospholipid found in the inner mitochondrial membrane; Ca2+: Calcium ion; Canagliflozin: a sodium-glucose cotransporter 2 (SGLT2) inhibitor; Cytarabine: an antineoplastic agent; Chrelin: Ghrelin, a hormone that stimulates appetite; Ceftriaxone: a third-generation cephalosporin antibiotic; Capsaicin: a compound found in chili peppers with analgesic properties; DR-Ab: Dopamine receptor antibody; Doxycycline: a tetracycline antibiotic; Dipraglurant: a specific compound; Ecologic BARRIER: a term that might refer to a specific treatment approach or compound; Exenatide: a glucagon-like peptide-1 (GLP-1) analog; Empagliflozin: a sodium-glucose cotransporter 2 (SGLT2) inhibitor; FTY720: a prodrug of fingolimod, a sphingosine-1-phosphate receptor modulator; Fasudil: a Rho kinase inhibitor; FMT: Fecal microbiota transplantation; Formoterol: a long-acting beta2-adrenergic agonist; Folic Acid: a B vitamin important for DNA synthesis and cell division; Ginsenoside Rg3: a compound found in ginseng with potential therapeutic effects; Gemfibrozil: a lipid-lowering drug; Hederagenin: a triterpenoid compound with anti-inflammatory properties; Hydroxypropyl beta: Hydroxypropyl beta-cyclodextrin, a drug carrier; Idebenone: an antioxidant used in the treatment of cognitive disorders; Istradefylline: a selective adenosine A2A receptor antagonist; Levodopa: a prodrug of dopamine; Lithium aspartate: a compound used as a mood stabilizer; Lactobacillus acidophilus: a probiotic bacterium; L-DOPA: Levodopa, a prodrug of dopamine; Levetiracetam: an antiepileptic drug; Metformin: a medication used to treat type 2 diabetes; Montelukast: a leukotriene receptor antagonist; MG149: a specific compound, details not widely recognized; NAC: N-acetylcysteine, an antioxidant; Nilotinib: a tyrosine kinase inhibitor; Nicergoline: a vasodilator used in the treatment of cerebrovascular diseases; Opicapone: a peripherally acting catechol-O-methyltransferase inhibitor; PTX: Pentoxifylline, a medication used to improve blood flow; Pimavanserin: a selective serotonin 5-HT2A receptor inverse agonist; Psilocybin: a psychoactive compound found in certain mushrooms; Pyridostigmine: an acetylcholinesterase inhibitor; Phillyrin: a compound extracted from the plant Phellodendron amurense with potential neuroprotective effects; Probucol: a lipid-lowering drug; Rifaximin: a minimally absorbed antibiotic; Radotinib: a specific compound; Rivastigimine: an acetylcholinesterase inhibitor; Rosiglitazone: a thiazolidinedione antidiabetic drug; SQJZ: Specific compound or code; Semaglutide: a glucagon-like peptide-1 (GLP-1) analog; Suvecaltamide: a specific compound; TT01001 derivatives: Derivatives of a specific compound, details not widely recognized; Talineuren: a specific compound; Transchalcone: a compound with potential anti-inflammatory properties; Tolcapone: a catechol-O-methyltransferase inhibitor; Uncaria rhynchophylla: a plant species used in traditional medicine.
Among the 62 drugs targeting mitochondria for HD intervention, their mechanistic focus is on improving energy metabolism and alleviating oxidative stress, with both approaches accounting for 30.6% of such drugs. Regarding drug sources, self-developed drugs had the highest proportion (62.9 %), followed by human-derived substances, antibiotics, and antibodies, collectively accounting for 12.9%. In terms of clinical stage distribution, phase IV trial drugs were the most numerous, constituting 62.9% (Figure 14).
Figure 14.

Schematic representation of all drugs targeting mitochondrial dysfunction for HD.
The figure shows the status of all drugs targeting mitochondrial dysfunction in HD as of December 14, 2024. The outermost circle represents the stages of drug development, including the research phase, preclinical stage, and clinical Phases I, II, III, and IV. Different colored blocks represent ten aspects of mitochondrial improvement. Different fonts indicate the classification of drugs, such as drugs developed by our research group (indicated in bold red), traditional Chinese medicine and its extracts (italicized and underlined), natural compounds extracted from plants and gut microbiota (italicized), small molecule compounds (indicated in black), substances produced by the human body, antibiotics and antibodies (underlined), and organic compounds independently developed or already marketed (without special formatting). Created with BioRender.com. ACP-204: A specific compound, details not widely recognized; Bacopa monnieri: a traditional Ayurvedic herb with potential cognitive benefits; Branaplam: a specific compound, details not widely recognized; Bezafibrate: a fibric acid derivative used in the treatment of hyperlipidemia; CoQ10: coenzyme Q10, an antioxidant involved in energy production; Ca2+: calcium ion; Curcumin: a compound found in turmeric with anti-inflammatory properties; C15H12N4: a chemical formula, possibly referring to a specific compound; Capsaicin: a compound found in chili peppers with analgesic properties; CBD: Cannabidiol, a non-psychoactive compound found in cannabis; Creatine: a naturally occurring compound that provides energy to muscle cells; Deferoxamine: an iron chelator used in the treatment of iron overload; Ebselen: a synthetic antioxidant and anti-inflammatory agent; EGCG: Epigallocatechin gallate, a major catechin in green tea; F2.6BP: Fis1, a mitochondrial fission protein; Flavonoids: a class of plant compounds with antioxidant and anti-inflammatory properties; Fosgonimeton: a progestin drug; Glycyrrhizin: a compound found in licorice with anti-inflammatory properties; Green tea extract: an extract from green tea with potential health benefits; IONIS-HTTRx: Ionis Pharmaceuticals’ antisense oligonucleotide targeting huntingtin gene expression; Laquinimod: an immunomodulatory drug used in the treatment of multiple sclerosis; Lycopene: a carotenoid with antioxidant properties; Melatonin: a hormone that regulates sleep-wake cycles; Morin: a flavonol with potential health benefits; Monensin: an ionophore antibiotic used as a growth promoter in livestock; MTAxs: Methionine aminopeptidase inhibitors; Masitinib: a tyrosine kinase inhibitor used in the treatment of certain cancers; N-acetylcysteine (NAC): an antioxidant and mucolytic agent; Nrf-2: nuclear factor erythroid 2-related factor 2, a transcription factor that regulates antioxidant response; Omega-3 fatty acids: a class of polyunsaturated fatty acids with health benefits; Pepinemab: a monoclonal antibody targeting huntingtin protein; Polyscias guilfoylei: a plant species, possibly used in traditional medicine; PBT2: a small molecule that promotes the clearance of toxic proteins; PCG1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha, a transcriptional coactivator; Pridopidine: a drug used in the treatment of Huntington’s disease; Quercetin: a flavonoid with potential anti-inflammatory properties; Rhodiolarosea: Rhodiola rosea, a plant used in traditional medicine for its adaptogenic properties; Resveratrol: a polyphenol found in red wine with potential health benefits; Rosiglitazone: a thiazolidinedione antidiabetic drug; Reserpine: an antihypertensive drug; Riluzole: a drug used in the treatment of amyotrophic lateral sclerosis; Selisistat: a cathepsin inhibitor; Sodium phenylbutyrate: a drug used in the treatment of urea cycle disorders; Thioredoxin: an antioxidant enzyme; Troriluzole: an anthelmintic drug; Trehalose: a disaccharide with potential protective effects on cells; Vitamin E: a fat-soluble vitamin with antioxidant properties.
Among the 33 drugs targeting mitochondria for ALS intervention, their primary mechanistic strategy is to improve oxidative stress, accounting for 42.4% of such drugs. This approach is consistent with the focus of intervention strategies for AD, HD, and PD (14 drugs), indicating that modulating oxidative stress responses is a viable intervention method for patients with CNDs (Figure 15).
Figure 15.

Schematic representation of all drugs targeting mitochondrial dysfunction for ALS.
The figure shows the status of all drugs targeting mitochondrial dysfunction in ALS as of December 14, 2024. The outermost circle represents the stages of drug development, including the research phase, preclinical stage, and clinical Phases I, II, III, and IV. Different colored blocks represent ten aspects of mitochondrial improvement. Different fonts indicate the classification of drugs, such as drugs developed by our research group (indicated in bold red), traditional Chinese medicine and its extracts (italicized and underlined), natural compounds extracted from plants and gut microbiota (italicized), small-molecule compounds (indicated in black), substances produced by the human body, antibiotics and antibodies (underlined), and organic compounds independently developed or already marketed (without special formatting). Created with BioRender.com. Arctigenin derivative A-1: A derivative of arctigenin, a compound with potential anti-inflammatory properties; ALCAR: acetyl-L-carnitine, a compound involved in energy metabolism; Baricitinib: a Janus kinase inhibitor used in the treatment of rheumatoid arthritis; Ca2+: calcium ion; curcumin: a compound found in turmeric with anti-inflammatory properties; CoQ10: coenzyme Q10, an antioxidant involved in energy production; Diallyl trisulfide: a compound found in garlic with potential health benefits; Edaravone: a free radical scavenger used in the treatment of ALS; Flavonoids: a class of plant compounds with antioxidant and anti-inflammatory properties; Guanabenz: an alpha-2 adrenergic agonist used in the treatment of hypertension; Glutathione monoethyl ester: a derivative of glutathione, an antioxidant; Honokiol: a compound found in magnolia bark with potential health benefits; Melatonin: a hormone that regulates sleep-wake cycles; Methylene blue: a dye with potential therapeutic properties; MitoQ: a mitochondria-targeted antioxidant; Nilotinib: a tyrosine kinase inhibitor used in the treatment of certain cancers; N-terminal VDAC1-derived peptide: a peptide derived from the N-terminus of the voltage-dependent anion channel 1; NAC: N-acetylcysteine, an antioxidant and mucolytic agent; Oleuropein: a compound found in olives with potential health benefits; Phenazine: a class of compounds with potential antimicrobial properties; PB-TUDCA: a derivative of tauroursodeoxycholic acid, a compound with potential cytoprotective properties; RTA: Reelin signaling pathway activator; Riluzole: a drug used in the treatment of ALS; RTA-408: a specific compound, details not widely recognized; Resveratrol: a polyphenol found in red wine with potential health benefits; Salubrinal: a specific compound; TMZ: Temozolomide, a chemotherapy drug; Trolox: a synthetic analog of vitamin E with antioxidant properties; Tofersen: an antisense oligonucleotide targeting the SOD1 gene; Triheptanoin: a triglyceride used in the treatment of fatty acid oxidation disorders; Urate: a compound formed from the breakdown of purines, with potential neuroprotective properties.
In summary, self-developed drugs play a major role in mitochondrial interventions for various diseases, particularly AD and PD. However, in terms of specific mechanisms of action, drugs that improve oxidative stress significantly outnumber those that act via other mechanisms, indicating that this is the main current research direction for mitochondrial dysfunction interventions and has great potential. Notably, among all the drugs targeting mitochondrial dysfunction, those focused on “oxidative stress” are prolific, accounting for 45.8% (as shown in the green section of Figure 15). Although many drugs are still in the research phase, there is conclusive evidence that reducing oxidative stress can significantly improve the symptoms of CNDs. Additionally, it is encouraging to note that traditional Chinese medicines and their extracts, despite comprising only 7.7% of the total, have demonstrated unique efficacy. These formulations are attracting the attention of a growing number of scientists interested in exploring the potential roles of individual herbs and their combinations in preventing and managing the progression of CNDs.
Our research group has long been dedicated to studying neurodegenerative diseases, a field of immense importance not only to the medical community but also to society as a whole. In the post-pandemic era, global population aging has intensified, leading to a significant increase in the number of patients with dementia, which places a heavy burden on families and society (MacDonald and Penney, 2025). Concurrently, neurological inflammation caused by viral infections is becoming increasingly severe, further complicating neurodegenerative diseases (Pang et al., 2024). These challenges have compelled researchers to accelerate efforts to find new solutions to this growing health crisis. Our research group has, for the first time, isolated and purified a novel halogenated type-II polyketide compound from the fermentation products of Streptomyces sp. FJS31-2, which we named the zunyimycins series (Lü et al., 2016). This discovery is important because zunyimycins not only exhibit antibacterial and antitumor properties but also show potential for intervention in AD. Building on this finding, our group has continuously optimized zunyimycin extraction methods and further investigated its mechanisms of action as an AD interventions. Through a series of experiments, we found that zunyimycins can significantly ameliorate mitochondrial dysfunction (Lü et al., 2017; Wang et al., 2022). Specifically, our research indicates that zunyimycin C may be used to regulate mitophagy through the NF-κB and PI3K/Akt pathways, reduce the injury and death of microglia, and further reduce neuroinflammation, offering new insights and directions for AD intervention research (Wang et al., 2022).
In the future, our group will continue to explore the potential of zunyimycins in the intervention of neurodegenerative diseases and investigate their applicability in clinical trials. Simultaneously, we have adopted network pharmacology methods to conduct in-depth modifications and adjustments to Qi Fu Yin, as recorded in “Jing Yue Quan Shu,” and Jing Fang Baidu San from “She Sheng Zhong Miao Fang,” Volume Eight. In this process, we combined modern medical insights into disease mechanisms with the essence of ancient Chinese medicine theories, aiming to optimize the therapeutic efficacy of these formulations. After repeated screening and validation, we obtained several promising traditional Chinese medicine formulations, including the YA3D series (Cheng et al., 2024).
Notably, YA3D4 and YA3D5 successfully completed preliminary clinical trial verification. The trial data indicated that these formulations demonstrated significant effects in regulating human immune function, reducing inflammation, and providing antioxidant benefits, all of which effectively combat neurodegenerative diseases by modulating mitochondrial function (Figure 13). YA3D4 and YA3D5 not only enhance mitochondrial energy metabolism but also mitigate oxidative stress-induced neuronal damage, thereby delaying disease progression. This provides important directions and hope for future drug development (Cheng et al., 2024). Furthermore, ongoing research and clinical trials suggest that more traditional Chinese medicine formulations will play an increasingly significant role in the intervention of neurodegenerative diseases and related fields.
Limitations
Unfortunately, to date, no drug has been able to cure CNDs, which may be attributed to their complex pathogenesis. Although many drugs are currently in Phase IV clinical trials, as well as in Phases II and III, the efficacy of these medications is often diminished due to the lack of obvious clinical symptoms in the early stages of CNDs. Even after a diagnosis is made, the therapeutic effects frequently fall short of expectations.
Therefore, there is an urgent need for new targeted therapies. Given the long asymptomatic period during the early stages of CNDs and the low cure rate after diagnosis, current research is focusing on early interventions for CNDs. Mitochondrial dysfunction is closely associated with CNDs and has emerged as a significant research topic in recent years, particularly in the context of improving oxidative stress. This suggests that mitochondria could serve as a promising entry point for exploring the pathogenesis of CNDs. To improve patient quality of life, the development of new drugs may help alleviate patient concerns. Additionally, early detection of CND indicators, such as Aβ protein testing, should be integrated into clinical practice to facilitate timely interventions. This approach aims to achieve “early detection, early intervention, and early treatment,” ultimately reducing mortality rates and improving the prognosis and quality of life for patients with CNDs.
Summary and Outlook
Summary
This paper also elucidates the role of mitochondrial dysfunction—characterized by reduced mitochondrial biogenesis, impaired dynamics, abnormal autophagy, Ca2+ overload, and ROS-mediated oxidative stress—in the pathogenesis of CNDs. These findings not only enhance our understanding of the pathological mechanisms underlying CNDs but also provide important theoretical support for the development of new intervention strategies. Although there is no single treatment that can completely cure CNDs (Zong et al., 2024), interventions targeting mitochondria have shown significant potential. By restoring mitochondrial biogenesis, improving mitochondrial dynamics, inhibiting abnormal autophagy, and reducing oxidative stress, these interventions may not only help delay disease progression but also offer new ideas and methods for future treatments.
Outlook
Looking to the future, with continuous advancements in science and technology and deeper research, our research group aims to gain a more comprehensive understanding of the relationship between mitochondria and CNDs. Several key areas should be the focus of future research: First, we strive to elucidate the exact mechanisms of mitochondrial dysfunction in the pathogenesis of CNDs. Second, we aim to develop more precise and effective mitochondrial intervention strategies. Third, we seek to explore the role of the interconnections between mitochondria and other organelles or signaling pathways in CNDs. Finally, we will conduct large-scale clinical trials to verify the effectiveness and safety of interventions targeting mitochondria.
Based on the previous analysis, oxidative stress is a current research hotspot and a focal point for future studies, providing a new direction for subsequent investigations. Intervention strategies targeting mitochondrial oxidative stress, which emphasize “multiple pathways, targets, and drugs,” show great potential for the intervention of CNDs. This comprehensive research approach not only deepens our understanding of the pathogenesis of CNDs but also supports the development of new drugs and intervention methods. The relationship between mitochondria and neurodegenerative diseases presents both challenges and opportunities. Through continuous efforts and innovations, we believe that more groundbreaking discoveries and intervention strategies will emerge in the future, ultimately improving patient prognosis and enhancing the quality of life for those affected by CNDs.
Funding Statement
Funding: This work was partly supported by the Yan’an University Qin Chuanyuan “Scientist + Engineer” Team Special Fund, No. 2023KXJ-012 (to YL); Yan’an University Transformation of Scientific and Technological Achievements Fund, No. 2023CGZH-001 (to YL); College Students Innovation and Entrepreneurship Training Program, Nos. D2023158, 202410719056 (to XS, JM); Yan’an University Production and Cultivation Project, No. CXY202001 (to YL); Kweichow Moutai Hospital Research and Talent Development Fund Project, No. MTyk2022-25 (to XO).
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
C-Editor: Zhao M; S-Editors: Wang J, Li CH; L-Editor: Song LP; T-Editor: Jia Y
Data availability statement:
Not applicable.
References
- Abramov AY, Bachurin SO. Neurodegenerative disorders-Searching for targets and new ways of diseases treatment. Med Res Rev. 2021;41:2603–2605. doi: 10.1002/med.21794. [DOI] [PubMed] [Google Scholar]
- Adam D, Langerscheidt F, Zempel H. Amyloid-β-induced disruption of axon-initial-segment mitochondria localization: consequences for TAU missorting in Alzheimer’s disease pathology. Neural Regen Res. 2025;20:1407–1408. doi: 10.4103/NRR.NRR-D-24-00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agirman G, Yu KB, Hsiao EY. Signaling inflammation across the gut-brain axis. Science. 2021;374:1087–1092. doi: 10.1126/science.abi6087. [DOI] [PubMed] [Google Scholar]
- Amorim JA, Coppotelli G, Rolo AP, Palmeira CM, Ross JM, Sinclair DA. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat Rev Endocrinol. 2022;18:243–258. doi: 10.1038/s41574-021-00626-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antico O, Thompson PW, Hertz NT, Muqit MMK, Parton LE. Targeting mitophagy in neurodegenerative diseases. Nat Rev Drug Discov. 2025 doi: 10.1038/s41573-024-01105-0. doi: 10.1038/s41573-024-01105-0. [DOI] [PubMed] [Google Scholar]
- Arnold PK, Jackson BT, Paras KI, Brunner JS, Hart ML, Newsom OJ, Alibeckoff SP, Endress J, Drill E, Sullivan LB, Finley LWS. A non-canonical tricarboxylic acid cycle underlies cellular identity. Nature. 2022;603:477–481. doi: 10.1038/s41586-022-04475-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai A, Bharathi V, Patel BK. Therapeutic targeting of the oxidative stress generated by pathological molecular pathways in the neurodegenerative diseases, ALS and Huntington’s. Eur J Pharmacol. 2024;987:177187. doi: 10.1016/j.ejphar.2024.177187. [DOI] [PubMed] [Google Scholar]
- Behl T, Makkar R, Anwer MK, Hassani R, Khuwaja G, Khalid A, Mohan S, Alhazmi HA, Sachdeva M, Rachamalla M. Mitochondrial dysfunction: a cellular and molecular hub in pathology of metabolic diseases and infection. J Clin Med. 2023;12:2882. doi: 10.3390/jcm12082882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatti JS, Kaur S, Mishra J, Dibbanti H, Singh A, Reddy AP, Bhatti GK, Reddy PH. Targeting dynamin-related protein-1 as a potential therapeutic approach for mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. 2023;1869:166798. doi: 10.1016/j.bbadis.2023.166798. [DOI] [PubMed] [Google Scholar]
- Blagov A, Postnov A, Sukhorukov V, Popov M, Uzokov J, Orekhov A. Significance of mitochondrial dysfunction in the pathogenesis of parkinson’s disease. Front Biosci. 2024;29:36. doi: 10.31083/j.fbl2901036. [DOI] [PubMed] [Google Scholar]
- Borković-Mitić S, Stojsavljević A, Vujotić L, Matić S, Mitić B, Manojlović D, Pavlović S. Differences between antioxidant defense parameters and specific trace element concentrations in healthy, benign, and malignant brain tissues. Sci Rep. 2021;11:14766. doi: 10.1038/s41598-021-94302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsche M, Märtens A, Hörmann P, Brückmann T, Lohmann K, Tunc S, Klein C, Hiller K, Balck A. In vivo investigation of glucose metabolism in idiopathic and prkn-related Parkinson’s disease. Mov Disord. 2023;38:697–702. doi: 10.1002/mds.29333. [DOI] [PubMed] [Google Scholar]
- Boulos A, Maroun D, Ciechanover A, Ziv NE. Peripheral sequestration of huntingtin delays neuronal death and depends on N-terminal ubiquitination. Commun Biol. 2024;7:1014. doi: 10.1038/s42003-024-06733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Heneka MT. The endotoxin hypothesis of Alzheimer’s disease. Mol Neurodegener. 2024;19:30. doi: 10.1186/s13024-024-00722-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burté F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- Burtscher J, Strasser B, Pepe G, Burtscher M, Kopp M, Di Pardo A, Maglione V, Khamoui AV. Brain-periphery interactions in huntington’s disease: mediators and lifestyle interventions. Int J Mol Sci. 2024;25:4696. doi: 10.3390/ijms25094696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante-Barrientos FA, Luque-Campos N, Araya MJ, Lara-Barba E, de Solminihac J, Pradenas C, Molina L, Herrera-Luna Y, Utreras-Mendoza Y, Elizondo-Vega R, Vega-Letter AM, Luz-Crawford P. Mitochondrial dysfunction in neurodegenerative disorders: potential therapeutic application of mitochondrial transfer to central nervous system-residing cells. J Transl Med. 2023;21:613. doi: 10.1186/s12967-023-04493-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai P, Li W, Xu Y, Wang H. Drp1 and neuroinflammation: deciphering the interplay between mitochondrial dynamics imbalance and inflammation in neurodegenerative diseases. Neurobiol Dis. 2024;198:106561. doi: 10.1016/j.nbd.2024.106561. [DOI] [PubMed] [Google Scholar]
- Cai R, Zhang Y, Simmering JE, Schultz JL, Li Y, Fernandez-Carasa I, Consiglio A, Raya A, Polgreen PM, Narayanan NS, Yuan Y, Chen Z, Su W, Han Y, Zhao C, Gao L, Ji X, Welsh MJ, Liu L. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J Clin Invest. 2019;129:4539–4549. doi: 10.1172/JCI129987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo-Rodriguez M, Bacskai BJ. Mitochondria and calcium in Alzheimer’s disease: from cell signaling to neuronal cell death. Trends Neurosci. 2021;44:136–151. doi: 10.1016/j.tins.2020.10.004. [DOI] [PubMed] [Google Scholar]
- Cao M, Zou J, Shi M, Zhao D, Liu C, Liu Y, Li L, Jiang H. A promising therapeutic: Exosome-mediated mitochondrial transplantation. Int Immunopharmacol. 2024;142:113104. doi: 10.1016/j.intimp.2024.113104. [DOI] [PubMed] [Google Scholar]
- Cao Y, Zheng J, Wan H, Sun Y, Fu S, Liu S, He B, Cai G, Cao Y, Huang H, Li Q, Ma Y, Chen S, Wang F, Jiang H. A mitochondrial SCF-FBXL4 ubiquitin E3 ligase complex degrades BNIP3 and NIX to restrain mitophagy and prevent mitochondrial disease. EMBO J. 2023;42:e113033. doi: 10.15252/embj.2022113033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso SM, Empadinhas N. The microbiome-mitochondria dance in prodromal Parkinson’s disease. Front Physiol. 2018;9:471. doi: 10.3389/fphys.2018.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castora FJ, Kerns KA, Pflanzer HK, Shelton MG, Coleman RA. Differential expression of mitochondrial energy producing genes in AD brains compromise ATP Synthesis and neuronal viability and stimulate inflammatory pathways and ROS synthesis. Alzheimers Dement. 2023 doi:10.1002/alz.077104. [Google Scholar]
- Cenini G, Lloret A, Cascella R. Oxidative stress in neurodegenerative diseases: from a mitochondrial point of view. Oxid Med Cell Longev. 2019;2019:2105607. doi: 10.1155/2019/2105607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Mandal SM, Mankevich M, Sreenivasmurthy SG, Hegde ML, Krishnan B, Ghosh G, Hazra T. F2,6BP restores mitochondrial genome integrity in Huntington’s disease. bioRxiv [Preprint] 2024 doi: 10.1101/2024.11.04.621834. [Google Scholar]
- Chanaday NL, Nosyreva E, Shin OH, Zhang H, Aklan I, Atasoy D, Bezprozvanny I, Kavalali ET. Presynaptic store-operated Ca (2+) entry drives excitatory spontaneous neurotransmission and augments endoplasmic reticulum stress. Neuron. 2021;109:1314–1332.e15. doi: 10.1016/j.neuron.2021.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Chao YC, Chang HS, Wu YL, Chang HJ, Lin YS, Cheng WL, Lin TT, Liu CS. Intranasal delivery of mitochondria for treatment of Parkinson’s disease model rats lesioned with 6-hydroxydopamine. Sci Rep. 2021;11:10597. doi: 10.1038/s41598-021-90094-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Cao J, Zha L, Wang P, Liu Z, Guo B, Zhang G, Sun Y, Zhang Z, Wang Y. Neuroprotective and neurogenic effects of novel tetramethylpyrazine derivative T-006 in Parkinson’s disease models through activating the MEF2-PGC1α and BDNF/CREB pathways. Aging. 2020;12:14897–14917. doi: 10.18632/aging.103551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Cross AC, Thakkar A, Xu H, Li A, Paull D, Noggle SA, Kruger L, Denton TT, Gibson GE. Selective linkage of mitochondrial enzymes to intracellular calcium stores differs between human-induced pluripotent stem cells, neural stem cells, and neurons. J Neurochem. 2021;156:867–879. doi: 10.1111/jnc.15160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Majerníková N, Marmolejo-Garza A, Trombetta-Lima M, Sabogal-Guáqueta AM, Zhang Y, Ten Kate R, Zuidema M, Mulder P, den Dunnen W, Gosens R, Verpoorte E, Culmsee C, Eisel ULM, Dolga AM. Mitochondrial transplantation rescues neuronal cells from ferroptosis. Free Radic Biol Med. 2023;208:62–72. doi: 10.1016/j.freeradbiomed.2023.07.034. [DOI] [PubMed] [Google Scholar]
- Chen YH, Lin S, Jin SY, Gao TM. Extracellular ATP is a homeostatic messenger that mediates cell-cell communication in physiological processes and psychiatric diseases. Biol Psychiatry. 2025;97:41–53. doi: 10.1016/j.biopsych.2024.04.013. [DOI] [PubMed] [Google Scholar]
- Cheng J, Li W, Wang L, Gao Y, Ma Y, Zhou M, Yang T, Yue C, Yan L, Lyu Y. Jiawei Qifuyin enhances immunity and improves cognitive impairment in app/ps1 mice through modulation of neuroinflammatory pathways. J Inflamm Res. 2024;17:9021–9040. doi: 10.2147/JIR.S479899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini M, Lopez-Molina L, Gines S. Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca (2+) efflux and Reactive Oxygen Species (ROS) homeostasis. Neurobiol Dis. 2020;136:104741. doi: 10.1016/j.nbd.2020.104741. [DOI] [PubMed] [Google Scholar]
- Choi H, Mook-Jung I. Functional effects of gut microbiota-derived metabolites in Alzheimer’s disease. Curr Opin Neurobiol. 2023;81:102730. doi: 10.1016/j.conb.2023.102730. [DOI] [PubMed] [Google Scholar]
- Cossu D, Yokoyama K, Sato S, Noda S, Sechi LA, Hattori N. PARKIN modifies peripheral immune response and increases neuroinflammation in active experimental autoimmune encephalomyelitis (EAE) J Neuroimmunol. 2021;359:577694. doi: 10.1016/j.jneuroim.2021.577694. [DOI] [PubMed] [Google Scholar]
- Cummings J, Zhou Y, Lee G, Zhong K, Fonseca J, Cheng F. Alzheimer’s disease drug development pipeline: 2024. Alzheimers Dement (N Y) 2024;10:e12465. doi: 10.1002/trc2.12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cunha Germano BC, de Morais LCC, Idalina Neta F, Fernandes ACL, Pinheiro FI, do Rego ACM, Araújo Filho I, de Azevedo EP, de Paiva Cavalcanti JRL, Guzen FP, Cobucci RN. Vitamin E and its molecular effects in experimental models of neurodegenerative diseases. Int J Mol Sci. 2023;24:11191. doi: 10.3390/ijms241311191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Wang H, Lian A, Li J, Zhao G, Hu S, Li B. A comprehensive perspective of Huntington’s disease and mitochondrial dysfunction. Mitochondrion. 2023;70:8–19. doi: 10.1016/j.mito.2023.03.001. [DOI] [PubMed] [Google Scholar]
- Dash UC, Bhol NK, Swain SK, Samal RR, Nayak PK, Raina V, Panda SK, Kerry RG, Duttaroy AK, Jena AB. Oxidative stress and inflammation in the pathogenesis of neurological disorders: mechanisms and implications. Acta Pharm Sin B. 2025;15:15–34. doi: 10.1016/j.apsb.2024.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martino M, Rathmell JC, Galluzzi L, Vanpouille-Box C. Cancer cell metabolism and antitumour immunity. Nat Rev Immunol. 2024;24:654–669. doi: 10.1038/s41577-024-01026-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewanjee S, Chakraborty P, Bhattacharya H, Chacko L, Singh B, Chaudhary A, Javvaji K, Pradhan SR, Vallamkondu J, Dey A, Kalra RS, Jha NK, Jha SK, Reddy PH, Kandimalla R. Altered glucose metabolism in Alzheimer’s disease: Role of mitochondrial dysfunction and oxidative stress. Free Radic Biol Med. 2022;193:134–157. doi: 10.1016/j.freeradbiomed.2022.09.032. [DOI] [PubMed] [Google Scholar]
- Dhapola R, Sarma P, Medhi B, Prakash A, Reddy DH. Recent advances in molecular pathways and therapeutic implications targeting mitochondrial dysfunction for Alzheimer’s disease. Mol Neurobiol. 2022;59:535–555. doi: 10.1007/s12035-021-02612-6. [DOI] [PubMed] [Google Scholar]
- Douiev L, Sheffer R, Horvath G, Saada A. Bezafibrate improves mitochondrial fission and function in DNM1L-deficient patient cells. Cells. 2020;9:301. doi: 10.3390/cells9020301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebadpour N, Mahmoudi M, Kamal Kheder R, Abavisani M, Baridjavadi Z, Abdollahi N, Esmaeili SA. From mitochondrial dysfunction to neuroinflammation in Parkinson’s disease: pathogenesis and mitochondrial therapeutic approaches. Int Immunopharmacol. 2024;142:113015. doi: 10.1016/j.intimp.2024.113015. [DOI] [PubMed] [Google Scholar]
- Eghbalsaied S, Lawler C, Petersen B, Hajiyev RA, Bischoff SR, Frankenberg S. CRISPR/Cas9-mediated base editors and their prospects for mitochondrial genome engineering. Gene Ther. 2024;31:209–223. doi: 10.1038/s41434-023-00434-w. [DOI] [PubMed] [Google Scholar]
- Eo H, Yu SH, Choi Y, Kim Y, Kang YC, Lee H, Kim JH, Han K, Lee HK, Chang MY, Oh MS, Kim CH. Mitochondrial transplantation exhibits neuroprotective effects and improves behavioral deficits in an animal model of Parkinson’s disease. Neurotherapeutics. 2024;21:e00355. doi: 10.1016/j.neurot.2024.e00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktäschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22:401–412. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Revilla J, Ruiz R, Espinosa-Oliva AM, Santiago M, García-Domínguez I, Camprubí-Ferrer L, Bachiller S, Deierborg T, Joseph B, de Pablos RM, Rodríguez-Gómez JA, Venero JL. Dopaminergic neurons lacking Caspase-3 avoid apoptosis but undergo necrosis after MPTP treatment inducing a Galectin-3-dependent selective microglial phagocytic response. Cell Death Dis. 2024;15:625. doi: 10.1038/s41419-024-07014-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Kumar A. Harnessing mitophagy for therapeutic advances in aging and chronic neurodegenerative diseases. Neuroglia. 2024;5:391–409. [Google Scholar]
- Giorgi C, Marchi S, Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018;19:713–730. doi: 10.1038/s41580-018-0052-8. [DOI] [PubMed] [Google Scholar]
- Gómez-Deza J, Nebiyou M, Alkaslasi MR, Nadal-Nicolás FM, Somasundaram P, Slavutsky AL, Li W, Ward ME, Watkins TA, Le Pichon CE. DLK-dependent axonal mitochondrial fission drives degeneration after axotomy. Nat Commun. 2024;15:10806. doi: 10.1038/s41467-024-54982-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granatiero V, Manfredi G. Mitochondrial transport and turnover in the pathogenesis of amyotrophic lateral sclerosis. Biology. 2019;8:36. doi: 10.3390/biology8020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grel H, Woznica D, Ratajczak K, Kalwarczyk E, Anchimowicz J, Switlik W, Olejnik P, Zielonka P, Stobiecka M, Jakiela S. Mitochondrial dynamics in neurodegenerative diseases: unraveling the role of fusion and fission processes. Int J Mol Sci. 2023;24:13033. doi: 10.3390/ijms241713033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffioen G. Calcium dyshomeostasis drives pathophysiology and neuronal demise in age-related neurodegenerative diseases. Int J Mol Sci. 2023;24:13243. doi: 10.3390/ijms241713243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu YY, Zhao XR, Zhang N, Yang Y, Yi Y, Shao QH, Liu MX, Zhang XL. Mitochondrial dysfunction as a therapeutic strategy for neurodegenerative diseases: current insights and future directions. Ageing Res Rev. 2024;102:102577. doi: 10.1016/j.arr.2024.102577. [DOI] [PubMed] [Google Scholar]
- Guan S, Zhao L, Peng R. Mitochondrial respiratory chain supercomplexes: from structure to function. Int J Mol Sci. 2022;23:13880. doi: 10.3390/ijms232213880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardia-Laguarta C, Liu Y, Lauritzen KH, Erdjument-Bromage H, Martin B, Swayne TC, Jiang X, Przedborski S. PINK1 content in mitochondria is regulated by ER-associated degradation. J Neurosci. 2019;39:7074–7085. doi: 10.1523/JNEUROSCI.1691-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang Z, Gu J, Ke P, Liu J, Meng Y, Zheng W, Que W, Fan R, Luo J, Xiao F. FUDNC1-dependent mitophagy ameliorate motor neuron death in an amyotrophic lateral sclerosis mouse model. Neurobiol Dis. 2024;197:106534. doi: 10.1016/j.nbd.2024.106534. [DOI] [PubMed] [Google Scholar]
- Haque ME, Akther M, Azam S, Kim IS, Lin Y, Lee YH, Choi DK. Targeting α-synuclein aggregation and its role in mitochondrial dysfunction in Parkinson’s disease. Br J Pharmacol. 2022;179:23–45. doi: 10.1111/bph.15684. [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer B. The effects of nuclear DNA mutations on mitochondrial function. J Am Assoc Nurse Pract. 2023;35:2–4. doi: 10.1097/JXX.0000000000000827. [DOI] [PubMed] [Google Scholar]
- Hilton BJ, Griffin JM, Fawcett JW, Bradke F. Neuronal maturation and axon regeneration: unfixing circuitry to enable repair. Nat Rev Neurosci. 2024;25:649–667. doi: 10.1038/s41583-024-00849-3. [DOI] [PubMed] [Google Scholar]
- Houldsworth A. Role of oxidative stress in neurodegenerative disorders: a review of reactive oxygen species and prevention by antioxidants. Brain Commun. 2024;6:fcad356. doi: 10.1093/braincomms/fcad356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, et al. A novel protein CYTB-187AA encoded by the mitochondrial gene CYTB modulates mammalian early development. Cell Metab. 2024;36:1586–1597.e1587. doi: 10.1016/j.cmet.2024.04.012. [DOI] [PubMed] [Google Scholar]
- Hui L, Just Ja. Brain tissue energy metabolism disorders and their mechanisms in Alzheimer’s disease. Advances in physiological science. 2022;53:276–280. [Google Scholar]
- Idrizaj E, Traini C, Vannucchi MG, Baccari MC. Nitric oxide: from gastric motility to gastric dysmotility. Int J Mol Sci. 2021;22:9990. doi: 10.3390/ijms22189990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang A, Handley RR, Lehnert K, Snell RG. From pathogenesis to therapeutics: a review of 150 years of huntington’s disease research. Int J Mol Sci. 2023;24:13021. doi: 10.3390/ijms241613021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Li K, Li X, Xu L, Yang Z. Sodium butyrate ameliorates the impairment of synaptic plasticity by inhibiting the neuroinflammation in 5XFAD mice. Chem Biol Interact. 2021;341:109452. doi: 10.1016/j.cbi.2021.109452. [DOI] [PubMed] [Google Scholar]
- Jurcau A, Jurcau CM. Mitochondria in Huntington’s disease: implications in pathogenesis and mitochondrial-targeted therapeutic strategies. Neural Regen Res. 2023;18:1472–1477. doi: 10.4103/1673-5374.360289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Isaacs-Trepanier C, Elmi N, Rapoport SI, Andreazza AC. Mitochondrial dysfunction and lipid peroxidation in rat frontal cortex by chronic NMDA administration can be partially prevented by lithium treatment. J Psychiatr Res. 2016;76:59–65. doi: 10.1016/j.jpsychires.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Lee S, Kim WK, Han BS. Mitochondrial transplantation: an overview of a promising therapeutic approach. BMB Rep. 2023;56:488–495. doi: 10.5483/BMBRep.2023-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemmensen MM, Borrowman SH, Pearce C, Pyles B, Chandra B. Mitochondrial dysfunction in neurodegenerative disorders. Neurotherapeutics. 2024;21:e00292. doi: 10.1016/j.neurot.2023.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D, Li C, Ma L, Du L, Jiang N, Zhao X, Zhang S, Zhao Z, Fang L, Du G. Identifying genetic targets in clinical subtypes of Parkinson’s disease for optimizing pharmacological treatment strategies. Signal Transduct Target Ther. 2024;9:320. doi: 10.1038/s41392-024-02020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotrys AV, Durham TJ, Guo XA, Vantaku VR, Parangi S, Mootha VK. Single-cell analysis reveals context-dependent, cell-level selection of mtDNA. Nature. 2024;629:458–466. doi: 10.1038/s41586-024-07332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryńska K, Kuliś K, Mazurek W, Gudowska-Sawczuk M, Zajkowska M, Mroczko B. The influence of SARS-CoV-2 infection on the development of selected neurological diseases. Int J Mol Sci. 2024;25:8715. doi: 10.3390/ijms25168715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubat GB, Picone P. Skeletal muscle dysfunction in amyotrophic lateral sclerosis: a mitochondrial perspective and therapeutic approaches. Neurol Sci. 2024;45:4121–4131. doi: 10.1007/s10072-024-07508-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuźniar J, Kozubek P, Czaja M, Leszek J. Correlation between Alzheimer’s disease and gastrointestinal tract disorders. Nutrients. 2024;16:2366. doi: 10.3390/nu16142366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Zhang Y, Zhou S, Xu J, Du Z, Feng Z, Yu L, Zhao Z, Wang W, Tang Y, Yang X, Guddat LW, Liu F, Gao Y, Rao Z, Gong H. Structure of the human ATP synthase. Mol Cell. 2023;83:2137–2147.e4. doi: 10.1016/j.molcel.2023.04.029. [DOI] [PubMed] [Google Scholar]
- Li X, Straub J, Medeiros TC, Mehra C, den Brave F, Peker E, Atanassov I, Stillger K, Michaelis JB, Burbridge E, Adrain C, Münch C, Riemer J, Becker T, Pernas LF. Mitochondria shed their outer membrane in response to infection-induced stress. Science. 2022;375:eabi4343. doi: 10.1126/science.abi4343. [DOI] [PubMed] [Google Scholar]
- Li Y, Xia X, Wang Y, Zheng JC. Mitochondrial dysfunction in microglia: a novel perspective for pathogenesis of Alzheimer’s disease. J Neuroinflammation. 2022;19:248. doi: 10.1186/s12974-022-02613-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Cui L, Gao J, Zhu M, Zhang Y, Zhang HL. Gut microbial metabolites in parkinson’s disease: implications of mitochondrial dysfunction in the pathogenesis and treatment. Mol Neurobiol. 2021;58:3745–3758. doi: 10.1007/s12035-021-02375-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MW, Lin CC, Chen YH, Yang HB, Hung SY. Celastrol inhibits dopaminergic neuronal death of Parkinson’s disease through activating mitophagy. Antioxidants (Basel, Switzerland) 2019;9:37. doi: 10.3390/antiox9010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Li C, Chen Y, Gao J, Li J, Huang C, Liu Z, Wang W, Zheng X, Song X, Wu J, Wu J, Luo OJ, Tu Z, Li S, Li XJ, Lai L, Yan S. RNA-targeting CRISPR/CasRx system relieves disease symptoms in Huntington’s disease models. Mol Neurodegener. 2025;20:4. doi: 10.1186/s13024-024-00794-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liu W, Li R, Yang H. Mitophagy in Parkinson’s disease: from pathogenesis to treatment. Cells. 2019;8:712. doi: 10.3390/cells8070712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhuang W, Cai M, Lv E, Wang Y, Wu Z, Wang H, Fu W. Kaemperfol protects dopaminergic neurons by promoting mtor-mediated autophagy in Parkinson’s disease models. Neurochem Res. 2023;48:1395–1411. doi: 10.1007/s11064-022-03819-2. [DOI] [PubMed] [Google Scholar]
- Long S, Zheng Y, Deng X, Guo J, Xu Z, Scharffetter-Kochanek K, Dou Y, Jiang M. Maintaining mitochondrial DNA copy number mitigates ROS-induced oocyte decline and female reproductive aging. Commun Biol. 2024;7:1229. doi: 10.1038/s42003-024-06888-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186:243–278. doi: 10.1016/j.cell.2022.11.001. [DOI] [PubMed] [Google Scholar]
- Lü Y, Yue C, Shao M, Qian S, Liu N, Bao Y, Wang M, Liu M, Li X, Wang Y, Huang Y. Molecular genetic characterization of an anthrabenzoxocinones gene cluster in streptomyces Sp. FJS31-2 for the biosynthesis of BE-24566B and zunyimycin ale. Molecules. 2016;21:711. doi: 10.3390/molecules21060711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lü Y, Shao M, Wang Y, Qian S, Wang M, Wang Y, Li X, Bao Y, Deng C, Yue C, Liu D, Liu N, Liu M, Huang Y, Chen Z, Hu Y. Zunyimycins B and C, new chloroanthrabenzoxocinones antibiotics against methicillin-resistant staphylococcus aureus and enterococci from streptomyces sp. FJS31-2. Molecules. 2017;22:251. doi: 10.3390/molecules22020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald DS, Penney J. Microglial dysfunction and genetic risk for neurodegenerative disease. Neural Regen Res. 2025;20:1401–1402. doi: 10.4103/NRR.NRR-D-24-00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson A, Wu R, Demirel I. Porphyromonas gingivalis triggers microglia activation and neurodegenerative processes through NOX4. Front Cell Infect Microbiol. 2024;14:1451683. doi: 10.3389/fcimb.2024.1451683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesan E, Nardin A, Mauri S, Bernardo G, Chander V, Di Paola S, Chinellato M, von Stockum S, Chakraborty J, Herkenne S, Basso V, Schrepfer E, Marin O, Cendron L, Medina DL, Scorrano L, Ziviani E. Activation of Ca (2+) phosphatase Calcineurin regulates Parkin translocation to mitochondria and mitophagy in flies. Cell Death Differ. 2024;31:217–238. doi: 10.1038/s41418-023-01251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareckova K, Mendes-Silva AP, Jáni M, Pacinkova A, Piler P, Gonçalves VF, Nikolova YS. Mitochondrial DNA variants and their impact on epigenetic and biological aging in young adulthood. Transl Psychiatry. 2025;15:16. doi: 10.1038/s41398-025-03235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx N, Ritter N, Disse P, Seebohm G, Busch KB. Detailed analysis of Mdivi-1 effects on complex I and respiratory supercomplex assembly. Sci Rep. 2024;14:19673. doi: 10.1038/s41598-024-69748-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, Fazel A, Bergeron JJ, Trudeau LE, Burelle Y, Gagnon E, McBride HM, Desjardins M. Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell. 2016;166:314–327. doi: 10.1016/j.cell.2016.05.039. [DOI] [PubMed] [Google Scholar]
- McFarthing K, Buff S, Rafaloff G, Pitzer K, Fiske B, Navangul A, Beissert K, Pilcicka A, Fuest R, Wyse RK, Stott SRW. Parkinson’s disease drug therapies in the clinical trial pipeline: 2024 update. J Parkinsons Dis. 2024;14:899–912. doi: 10.3233/JPD-240272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei STR, Yanling M. Advances in the mechanism of mitochondrial dysfunction and its related diseases. Life Sci. 2018;30:87–93. [Google Scholar]
- Menacho C, Prigione A. Tackling mitochondrial diversity in brain function: from animal models to human brain organoids. Int J Biochem Cell Biol. 2020;123:105760. doi: 10.1016/j.biocel.2020.105760. [DOI] [PubMed] [Google Scholar]
- Miao R, et al. Gasdermin D permeabilization of mitochondrial inner and outer membranes accelerates and enhances pyroptosis. Immunity. 2023;56:2523–2541.e2528. doi: 10.1016/j.immuni.2023.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzel AS, Enríquez JA, Picard M. Multifaceted mitochondria: moving mitochondrial science beyond function and dysfunction. Nat Metab. 2023;5:546–562. doi: 10.1038/s42255-023-00783-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradi Vastegani S, Nasrolahi A, Ghaderi S, Belali R, Rashno M, Farzaneh M, Khoshnam SE. Mitochondrial dysfunction and Parkinson’s disease: pathogenesis and therapeutic strategies. Neurochem Res. 2023;48:2285–2308. doi: 10.1007/s11064-023-03904-0. [DOI] [PubMed] [Google Scholar]
- Moskal N, Riccio V, Bashkurov M, Taddese R, Datti A, Lewis PN, Angus McQuibban G. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat Commun. 2020;11:88. doi: 10.1038/s41467-019-13781-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Pinto MF, Candeias E, Melo-Marques I, Esteves AR, Maranha A, Magalhães JD, Carneiro DR, Sant’Anna M, Pereira-Santos AR, Abreu AE, Nunes-Costa D, Alarico S, Tiago I, Morgadinho A, Lemos J, Figueiredo PN, Januário C, Empadinhas N, Cardoso SM. Gut-first Parkinson’s disease is encoded by gut dysbiome. Mol Neurodegener. 2024;19:78. doi: 10.1186/s13024-024-00766-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naia L, Rosenstock TR, Oliveira AM, Oliveira-Sousa SI, Caldeira GL, Carmo C, Laço MN, Hayden MR, Oliveira CR, Rego AC. Comparative mitochondrial-based protective effects of resveratrol and nicotinamide in Huntington’s disease models. Mol Neurobiol. 2017;54:5385–5399. doi: 10.1007/s12035-016-0048-3. [DOI] [PubMed] [Google Scholar]
- Narendra DP, Youle RJ. The role of PINK1-Parkin in mitochondrial quality control. Nat Cell Biol. 2024;26:1639–1651. doi: 10.1038/s41556-024-01513-9. [DOI] [PubMed] [Google Scholar]
- Newton K, Strasser A, Kayagaki N, Dixit VM. Cell death. Cell. 2024;187:235–256. doi: 10.1016/j.cell.2023.11.044. [DOI] [PubMed] [Google Scholar]
- Nhu NT, Xiao SY, Liu Y, Kumar VB, Cui ZY, Lee SD. Neuroprotective effects of a small mitochondrially-targeted tetrapeptide elamipretide in neurodegeneration. Front Integr Neurosci. 2021;15:747901. doi: 10.3389/fnint.2021.747901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoliche T, Bartolomeo CS, Lemes RMR, Pereira GC, Nunes TA, Oliveira RB, Nicastro ALM, Soares É N, da Cunha Lima BF, Rodrigues BM, Maricato JT, Okuda LH, de Sairre MI, Prado CM, Ureshino RP, Stilhano RS. Antiviral, anti-inflammatory and antioxidant effects of curcumin and curcuminoids in SH-SY5Y cells infected by SARS-CoV-2. Sci Rep. 2024;14:10696. doi: 10.1038/s41598-024-61662-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntetsika T, Papathoma PE, Markaki I. Novel targeted therapies for Parkinson’s disease. Mol Med. 2021;27:17. doi: 10.1186/s10020-021-00279-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onisiforou A, Zanos P. From viral infections to Alzheimer’s disease: unveiling the mechanistic links through systems bioinformatics. J Infect Dis. 2024;230:S128–140. doi: 10.1093/infdis/jiae242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgen S, Krigman J, Zhang R, Sun N. Significance of mitochondrial activity in neurogenesis and neurodegenerative diseases. Neural Regen Res. 2022;17:741–747. doi: 10.4103/1673-5374.322429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma FR, He C, Danes JM, Paviani V, Coelho DR, Gantner BN, Bonini MG. Mitochondrial superoxide dismutase: what the established, the intriguing, and the novel reveal about a key cellular redox switch. Antioxid Redox Signal. 2020;32:701–714. doi: 10.1089/ars.2019.7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma FR, Gantner BN, Sakiyama MJ, Kayzuka C, Shukla S, Lacchini R, Cunniff B, Bonini MG. ROS production by mitochondria: function or dysfunction? Oncogene. 2024;43:295–303. doi: 10.1038/s41388-023-02907-z. [DOI] [PubMed] [Google Scholar]
- Parikh D, Shah M. Values of epigenetic markers in Parkinson’s disease as biomarkers and therapeutic targets: A narrative review. NeuroMarkers. 2025;2:100037. [Google Scholar]
- Pang Z, Tang A, He Y, Fan J, Yang Q, Tong Y, Fan H. Neurological complications caused by SARS-CoV-2. Clin Microbiol Rev. 2024;37:e0013124. doi: 10.1128/cmr.00131-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Lee CH. The impact of pulmonary disorders on neurological health (lung-brain axis) Immune Netw. 2024;24:e20. doi: 10.4110/in.2024.24.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzotti-Jametti L, et al. Mitochondrial complex I activity in microglia sustains neuroinflammation. Nature. 2024;628:195–203. doi: 10.1038/s41586-024-07167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Shirihai OS. Mitochondrial signal transduction. Cell Metab. 2022;34:1620–1653. doi: 10.1016/j.cmet.2022.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picca A, Calvani R, Coelho-Junior HJ, Landi F, Bernabei R, Marzetti E. Mitochondrial dysfunction, oxidative stress, and neuroinflammation: intertwined roads to neurodegeneration. Antioxidants (Basel) 2020;9:647. doi: 10.3390/antiox9080647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihán P, Carreras-Sureda A, Hetz C. BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017;24:1478–1487. doi: 10.1038/cdd.2017.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov LD. Mitochondria as intracellular signalling organelles. An update. Cell Signal. 2023;109:110794. doi: 10.1016/j.cellsig.2023.110794. [DOI] [PubMed] [Google Scholar]
- Princen K, et al. Pharmacological modulation of septins restores calcium homeostasis and is neuroprotective in models of Alzheimer’s disease. Science. 2024;384:eadd6260. doi: 10.1126/science.add6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pszczołowska M, Walczak K, Miśków W, Mroziak M, Chojdak-Łukasiewicz J, Leszek J. Mitochondrial disorders leading to Alzheimer’s disease-perspectives of diagnosis and treatment. Geroscience. 2024;46:2977–2988. doi: 10.1007/s11357-024-01118-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin P, Sun Y, Li L. Mitochondrial dysfunction in chronic neuroinflammatory diseases (Review) Int J Mol Med. 2024;53:47. doi: 10.3892/ijmm.2024.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raefsky SM, Mattson MP. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic Biol Med. 2017;102:203–216. doi: 10.1016/j.freeradbiomed.2016.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios L, Pokhrel S, Li SJ, Heo G, Haileselassie B, Mochly-Rosen D. Targeting an allosteric site in dynamin-related protein 1 to inhibit Fis1-mediated mitochondrial dysfunction. Nat Commun. 2023;14:4356. doi: 10.1038/s41467-023-40043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizea RE, Corlatescu AD, Costin HP, Dumitru A, Ciurea AV. Understanding amyotrophic lateral sclerosis: pathophysiology, diagnosis, and therapeutic advances. Int J Mol Sci. 2024;25:9966. doi: 10.3390/ijms25189966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues F, Reis M, Ferreira L, Grosso C, Ferraz R, Vieira M, Vasconcelos V, Martins R. The neuroprotective role of cyanobacteria with focus on the anti-inflammatory and antioxidant potential: current status and perspectives. Molecules. 2024;29:4799. doi: 10.3390/molecules29204799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo AI, Pajares M, Rada P, Nuñez A, Nevado-Holgado AJ, Killik R, Van Leuven F, Ribe E, Lovestone S, Yamamoto M, Cuadrado A. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017;13:444–451. doi: 10.1016/j.redox.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KW, Fung TS, Baker DC, Saoi M, Park J, Febres-Aldana CA, Aly RG, Cui R, Sharma A, Fu Y, Jones OL, Cai X, Pasolli HA, Cross JR, Rudin CM, Thompson CB. Cellular ATP demand creates metabolically distinct subpopulations of mitochondria. Nature. 2024;635:746–754. doi: 10.1038/s41586-024-08146-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta S, Chakraborty S, Bagchi D. Pathogenesis of neurodegenerative diseases and the protective role of natural bioactive components. J Am Nutr Assoc. 2024;43:20–32. doi: 10.1080/27697061.2023.2203235. [DOI] [PubMed] [Google Scholar]
- Shaheen N, Shaheen A, Osama M, Nashwan AJ, Bharmauria V, Flouty O. MicroRNAs regulation in Parkinson’s disease, and their potential role as diagnostic and therapeutic targets. NPJ Parkinsons Dis. 2024;10:186. doi: 10.1038/s41531-024-00791-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Zhao M, Fu C, Fu A. Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion. 2017;34:91–100. doi: 10.1016/j.mito.2017.02.005. [DOI] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D’Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S, Counts SE, Auwerx J. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature. 2017;552:187–193. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Shen R, Agnihotri SK, Chen Y, Huang Z, Büeler H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci Rep. 2018;8:383. doi: 10.1038/s41598-017-18786-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suski J, Lebiedzinska M, Bonora M, Pinton P, Duszynski J, Wieckowski MR. Relation between mitochondrial membrane potential and ROS formation. Methods Mol Biol. 2018;1782:357–381. doi: 10.1007/978-1-4939-7831-1_22. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis. 2018;62:1403–1416. doi: 10.3233/JAD-170585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tábara LC, Burr SP, Frison M, Chowdhury SR, Paupe V, Nie Y, Johnson M, Villar-Azpillaga J, Viegas F, Segawa M, Anand H, Petkevicius K, Chinnery PF, Prudent J. MTFP1 controls mitochondrial fusion to regulate inner membrane quality control and maintain mtDNA levels. Cell. 2024;187:3619–3637.e3627. doi: 10.1016/j.cell.2024.05.017. [DOI] [PubMed] [Google Scholar]
- Tan BG, Gustafsson CM, Falkenberg M. Mechanisms and regulation of human mitochondrial transcription. Nat Rev Mol Cell Biol. 2024;25:119–132. doi: 10.1038/s41580-023-00661-4. [DOI] [PubMed] [Google Scholar]
- Tan X, Su X, Wang Y, Liang W, Wang D, Huo D, Wang H, Qi Y, Zhang W, Han L, Zhang D, Wang M, Xu J, Feng H. BRD7 regulates cellular senescence and apoptosis in ALS by modulating p21 expression and p53 mitochondrial translocation respectively. Neuroscience. 2024;563:51–62. doi: 10.1016/j.neuroscience.2024.11.004. [DOI] [PubMed] [Google Scholar]
- Tang C, Dong Z. Chapter 2 - Ubiquitin-independent mitophagy: mechanisms and pathophysiological functions. In: Lemasters JJ, editor. Mitophagy in Health and Disease. Academic Press; 2025. pp. 25–41. [Google Scholar]
- Teleanu RI, Niculescu AG, Roza E, Vladâcenco O, Grumezescu AM, Teleanu DM. Neurotransmitters-key factors in neurological and neurodegenerative disorders of the central nervous system. Int J Mol Sci. 2022;23:5954. doi: 10.3390/ijms23115954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theparambil SM, Kopach O, Braga A, Nizari S, Hosford PS, Sagi-Kiss V, Hadjihambi A, Konstantinou C, Esteras N, Gutierrez Del Arroyo A, Ackland GL, Teschemacher AG, Dale N, Eckle T, Andrikopoulos P, Rusakov DA, Kasparov S, Gourine AV. Adenosine signalling to astrocytes coordinates brain metabolism and function. Nature. 2024;632:139–146. doi: 10.1038/s41586-024-07611-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong H, Yang T, Xu S, Li X, Liu L, Zhou G, Yang S, Yin S, Li XJ, Li S. Huntington’s disease: complex pathogenesis and therapeutic strategies. Int J Mol Sci. 2024;25:a024240. doi: 10.3390/ijms25073845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh J, et al. Mitochondrial DNA heteroplasmy distinguishes disease manifestation in PINK1/PRKN-linked Parkinson’s disease. Brain. 2023;146:2753–2765. doi: 10.1093/brain/awac464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbauer E, et al. Mitochondrial perturbation in the intestine causes microbiota-dependent injury and gene signatures discriminative of inflammatory disease. Cell Host Microbe. 2024;32:1347–1364.e1310. doi: 10.1016/j.chom.2024.06.013. [DOI] [PubMed] [Google Scholar]
- Vaglio-Garro A, Kozlov AV, Smirnova YD, Weidinger A. Pathological interplay between inflammation and mitochondria aggravates glutamate toxicity. Int J Mol Sci. 2024;25:2276. doi: 10.3390/ijms25042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venderova K, Park DS. Programmed cell death in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009365. doi: 10.1101/cshperspect.a009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercellino I, Sazanov LA. The assembly, regulation and function of the mitochondrial respiratory chain. Nat Rev Mol Cell Biol. 2022;23:141–161. doi: 10.1038/s41580-021-00415-0. [DOI] [PubMed] [Google Scholar]
- Verma M, Lizama BN, Chu CT. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl Neurodegener. 2022;11:3. doi: 10.1186/s40035-021-00278-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Li G, Cui G, Duan S, Chang S. Reprogramming of thyroid cancer metabolism: from mechanism to therapeutic strategy. Mol Cancer. 2025;24:74. doi: 10.1186/s12943-025-02263-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Z, Sun R, Li X, Guo R, Cui X, Liu B, Li W, Yang Y, Huang X, Qu H, Liu C, Wang Z, Lü Y, Yue C. Zunyimycin C enhances immunity and improves cognitive impairment and its mechanism. Front Cell Infect Microbiol. 2022;12:1081243. doi: 10.3389/fcimb.2022.1081243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xu E, Musich PR, Lin F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci Ther. 2019;25:816–824. doi: 10.1111/cns.13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YP, Sharda A, Xu SN, van Gastel N, Man CH, Choi U, Leong WZ, Li X, Scadden DT. Malic enzyme 2 connects the Krebs cycle intermediate fumarate to mitochondrial biogenesis. Cell Metab. 2021;33:1027–1041.e1028. doi: 10.1016/j.cmet.2021.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li P, Xu Y, Feng L, Fang Y, Song G, Xu L, Zhu Z, Wang W, Mei Q, Xie M. Lactate metabolism and histone lactylation in the central nervous system disorders: impacts and molecular mechanisms. J Neuroinflammation. 2024;21:308. doi: 10.1186/s12974-024-03303-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang R, Niu L, Zhou X, Han J, Li K. EPB41L4A-AS1 is required to maintain basal autophagy to modulates Aβ clearance. NPJ Aging. 2024;10:24. doi: 10.1038/s41514-024-00152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang C, Yuan B, Liu L, Zhang H, Zhu M, Chai H, Peng J, Huang Y, Zhou S, Liu J, Wu L, Wang W. Akkermansia muciniphila and its metabolite propionic acid maintains neuronal mitochondrial division and autophagy homeostasis during Alzheimer’s disease pathologic process via GPR41 and GPR43. Microbiome. 2025;13:16. doi: 10.1186/s40168-024-02001-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidinger A, Milivojev N, Hosmann A, Duvigneau JC, Szabo C, Törö G, Rauter L, Vaglio-Garro A, Mkrtchyan GV, Trofimova L, Sharipov RR, Surin AM, Krasilnikova IA, Pinelis VG, Tretter L, Moldzio R, Bayır H, Kagan VE, Bunik VI, Kozlov AV. Oxoglutarate dehydrogenase complex controls glutamate-mediated neuronal death. Redox Biol. 2023;62:102669. doi: 10.1016/j.redox.2023.102669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells RG, Neilson LE, McHill AW, Hiller AL. Dietary fasting and time-restricted eating in Huntington’s disease: therapeutic potential and underlying mechanisms. Transl Neurodegener. 2024;13:17. doi: 10.1186/s40035-024-00406-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Deng H, Li B, Chen J, Zhu J, Zhang X, Yoshida S, Zhou Y. Mitochondrial diseases: from molecular mechanisms to therapeutic advances. Signal Transduct Target Ther. 2025;10:9. doi: 10.1038/s41392-024-02044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen P, Sun Z, Gou F, Wang J, Fan Q, Zhao D, Yang L. Oxidative stress and mitochondrial impairment: key drivers in neurodegenerative disorders. Ageing Res Rev. 2025 doi: 10.1016/j.arr.2025.102667. doi: 10.1016/j.arr.2025.102667. [DOI] [PubMed] [Google Scholar]
- Wilson DM, 3rd, Cookson MR, Van Den Bosch L, Zetterberg H, Holtzman DM, Dewachter I. Hallmarks of neurodegenerative diseases. Cell. 2023;186:693–714. doi: 10.1016/j.cell.2022.12.032. [DOI] [PubMed] [Google Scholar]
- Wu J, Xu W, Su Y, Wang GH, Ma JJ. Targeting chaperone-mediated autophagy in neurodegenerative diseases: mechanisms and therapeutic potential. Acta Pharmacol Sin. 2024 doi: 10.1038/s41401-024-01416-3. doi: 10.1038/s41401-024-01416-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Liu Y, Ou L, Gan T, Zhangding Z, Yuan S, Liu X, Liu M, Li J, Yin J, Xin C, Tian Y, Hu J. Transfer of mitochondrial DNA into the nuclear genome during induced DNA breaks. Nat Commun. 2024;15:9438. doi: 10.1038/s41467-024-53806-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Liu X, Jiang R, Yan X, Ling Z. Roles and mechanisms of gut microbiota in patients with Alzheimer’s disease. Front Aging Neurosci. 2021;13:650047. doi: 10.3389/fnagi.2021.650047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Wang Y, Zheng J. COVID-19 and Alzheimer’s disease: how one crisis worsens the other. Transl Neurodegener. 2021;10:15. doi: 10.1186/s40035-021-00237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat Biomed Eng. 2022;6:76–93. doi: 10.1038/s41551-021-00819-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Jia J, Mao R, Cao X, Xu Y. Mitophagy in acute central nervous system injuries: regulatory mechanisms and therapeutic potentials. Neural Regen Res. 2025;20:2437–2453. doi: 10.4103/NRR.NRR-D-24-00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Baolu Z. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev. 2013;2013:316523. doi: 10.1155/2013/316523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Jia X, Yang X, Wang J, Fang Y, Ying X, Zhang M, Wei J, Pan Y. Targeting VDAC: a potential therapeutic approach for mitochondrial dysfunction in Alzheimer’s disease. Brain Res. 2024;1835:148920. doi: 10.1016/j.brainres.2024.148920. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zou Y, Wang L. Neurotransmitters in prevention and treatment of Alzheimer’s disease. Int J Mol Sci. 2023;24:3841. doi: 10.3390/ijms24043841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao MF, Dang T, Wang HJ, Zhu XZ, Qiao C. Mitochondrial homeostasis regulation: a promising therapeutic target for Parkinson’s disease. Behav Brain Res. 2024;459:114811. doi: 10.1016/j.bbr.2023.114811. [DOI] [PubMed] [Google Scholar]
- Ye J, Duan C, Han J, Chen J, Sun N, Li Y, Yuan T, Peng D. Peripheral mitochondrial DNA as a neuroinflammatory biomarker for major depressive disorder. Neural Regen Res. 2025;20:1541–1554. doi: 10.4103/NRR.NRR-D-23-01878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Zhao J, Yan L, Qi Y, Guo X, Lou Z, Hu J, Rao Z. Structural insights into G domain dimerization and pathogenic mutation of OPA1. J Cell Biol. 2020;219:e201907098. doi: 10.1083/jcb.201907098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CH, et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 2020;183:636–649.e618. doi: 10.1016/j.cell.2020.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Al-Nusaif M, Ding C, Zhao L, Dong C. The potential of the gut microbiome for identifying Alzheimer’s disease diagnostic biomarkers and future therapies. Front Neurosci. 2023;17:1130730. doi: 10.3389/fnins.2023.1130730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang X, Li W, Yang Y, Wu Z, Lyu Y, Yue C. Intestinal microbiota: a new perspective on delaying aging? Front Microbiol. 2023;14:1268142. doi: 10.3389/fmicb.2023.1268142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Liu H, Gao Y, Cao G, Wang Y, Li Z. Ameliorating mitochondrial dysfunction for the therapy of Parkinson’s disease. Small. 2024;20:e2311571. doi: 10.1002/smll.202311571. [DOI] [PubMed] [Google Scholar]
- Zhou LL, Kang Q, Peng Y, Qizhi Z. Progress in mitochondrial oxidative stress and targeted delivery system in Alzheimer’s disease. J Pharm. 2022;57:1630–1640. [Google Scholar]
- Zong Y, Li H, Liao P, Chen L, Pan Y, Zheng Y, Zhang C, Liu D, Zheng M, Gao J. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024;9:124. doi: 10.1038/s41392-024-01839-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.