

Abstract
The fadD1 and macs1 genes of Streptomyces coelicolor are part of a two-gene operon. Both genes encode putative acyl coenzyme A synthetases (ACSs). The amino acid sequence of FadD1 has high homology with those of several ACSs, while MACS1 has the closest homology with medium-chain ACSs, broadly known as SA proteins. Like FadD of Escherichia coli, FadD1 also has a broad substrate specificity, although saturated long-chain fatty acids appears to be the preferred substrate. fadD1 is a growth-phase-regulated gene, and its mRNA is detected only during the stationary phase of growth. Interestingly, a mutation in fadD1 alters the levels of another ACS or ACSs, both at the stationary phase and at the exponential phase of growth, at least when glucose is used as a main carbon source. The mutant also shows a severe deficiency in antibiotic production, and at least for Act biosynthesis, this deficiency seems to be related to delayed expression of the Act biosynthetic genes. Antibiotic production is restored by the introduction of a wt fadD1 allele into the cell, demonstrating a strict link between ACS activity and the biosynthesis of secondary metabolites. The results of this study indicate that the ACSs may be useful targets for the design of rational approaches to improving antibiotic production.
Exogenous fatty acids represent an important class of hydrophobic compounds that can serve as a sole carbon and energy source to support growth in bacteria. In Streptomyces coelicolor, fatty acids with different chain lengths (C4 to C18) are efficiently degraded through the β-oxidation cycle, whose enzymes are constitutively expressed (1). In sharp contrast, the β-oxidation cycle enzymes of Escherichia coli are induced by long-chain fatty acids (29).
The first step in any further utilization of the fatty acids is the biosynthesis of their coenzyme A (CoA) derivatives. This enzymatic step is carried out by an acyl-CoA synthetase (ACS) that catalyzes the formation of fatty acyl-CoA from fatty acid, ATP, and CoA (39). Fatty acyl-CoAs represent bioactive compounds that are involved in protein transport (13, 30), enzyme activation (6, 24), protein acylation (14, 26), cell signaling (22), and transcriptional control (10); in addition, they serve as substrates for β oxidation as well as triacylglyceride (TAG) and phospholipid biosynthesis. Given the multiple roles of fatty acyl-CoAs, it is clear that fatty ACSs (FACSs) occupy a pivotal role in cellular homeostasis, particularly in lipid metabolism.
E. coli contains only one ACS with a broad substrate specificity (18). In the other extreme, the actinomycete Mycobacterium tuberculosis contains 36 genes annotated as putative ACS genes (8). In the S. coelicolor genome database (http://www.sanger.ac.uk/Projects/S_coelicolor/), at least 15 open reading frames (ORFs) have been annotated as encoding putative ACSs, but so far none of the gene products has been characterized.
Streptomycetes produce a wide variety of secondary metabolites, many of them having important applications as antibiotics or other useful compounds in human medicine and agriculture (27). While most of the published data related to antibiotic production refer to the elucidation of physiological signals and underlying regulatory mechanisms, very little is known about the carbon source for antibiotic formation. Interestingly, in several Streptomyces species, including S. coelicolor, drops of TAGs have been found to accumulate in the cytoplasm of the cells as storage compounds (28). The degradation of these neutral lipids by endogenous lipases is an internal source of fatty acids. It has been hypothesized that the β oxidation of the free fatty acids to acetyl-CoA is the source of the carbon units for the biosynthesis of many polyketide compounds (28, 34). If this is so, then the ACS activity or activities involved in the activation of the fatty acids to their CoA thioesters would play a key role in antibiotic production.
In an attempt to initiate the characterization of the ACSs of S. coelicolor and in order to understand the physiological role of this enzyme activity in this microorganism, we started our studies with the genetic and biochemical characterization of a putative ACS with high homology to the E. coli protein FadD. The choice of the latter enzyme for the homology search was based on the fact that FadD is probably one of the few well-characterized ACSs in bacteria (9).
MATERIALS AND METHODS
Strains, media, and growth conditions.
E. coli DH5α (33) and BL21(DE3) (36) were grown at 37°C in Luria-Bertani medium supplemented with 100 μg of ampicillin/ml, 50 μg of kanamycin/ml, or 100 μg of apramycin/ml when necessary.
E. coli K27 (fadD88 tyrT58 mel-1) (21, 29) was grown in M9 minimal medium (33) supplemented with glucose (1% [wt/vol]) or oleate (0.1% [wt/vol]) as a sole carbon source.
Cultures of S. coelicolor M145 were grown at 30°C on MS agar (20) or in SMM medium (38) supplemented with oleate (0.1% [wt/vol]) (SMM-oleate) or glucose (1% [wt/vol]) (SMM-glucose) as a carbon source and with thiostrepton or apramycin when necessary. Strain ET12567/pUZ8002 was used for E. coli-S. coelicolor conjugation experiments (3). For selection of Streptomyces exconjugants, media were overlaid with thiostrepton (300 μg/plate) or apramycin (1 mg/plate).
DNA manipulations.
Isolation of chromosomal and plasmid DNAs, restriction enzyme digestion, and agarose gel electrophoresis were carried out by conventional methods (17, 33). Southern blot analyses were performed by using the complete fadD1 ORF 32P labeled by random oligonucleotide priming (Prime-a-gene kit; Promega).
The nucleotide sequence of the fadD1 gene was determined by dideoxy sequencing (34) with a Promega Taq Track sequencing kit and double-stranded DNA templates.
Cloning of fadD1.
The synthetic oligonucleotides FadDup (5′-CGCATATGCGGCGAGCTGCCCAAG-3′) and FadDdown (5′-GAAGCTTATACCGGTGCCGAAAGCG-3′) were used to amplify the S. coelicolor fadD1 gene; the forward primer and the reverse primer were designed to contain an NdeI site (underlined) and an HindIII site (underlined), respectively. The reaction mixture contained 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 2 mM MgCl2, 25 mM each of the four deoxynucleoside triphosphates (dNTPs), 10% dimethyl sulfoxide, 2 U of Taq DNA polymerase, 20 pmol of each primer, and 50 ng of template DNA in a final volume of 100 μl. Samples were subjected to 30 cycles of denaturation (95°C, 1 min), annealing (65°C, 1 min), and extension (72°C, 1 min). PCR products were analyzed by agarose (0.9%) gel electrophoresis. The PCR-amplified DNA fragment was cloned into pGEM-T Easy (Promega) to yield pCB01. An NdeI-HindIII fragment containing fadD1 was isolated from pCB01 and cloned into NdeI-HindIII-digested pET22b(+) (Novagen) to generate pCB1.
Construction and complementation of a fadD1 in-frame deletion mutant.
We constructed a fadD1 mutant allele from which most of the fadD1 coding region (encoding amino acids 99 to 347) was deleted. For this, a 1.7-kb EcoRI-HindIII fragment containing the complete ORF was obtained from plasmid pCB01 and subcloned into pSET151 (3), yielding pCB13. In order to construct the in-frame deletion of fadD1, the 1-kb SmaI-SmaI fragment within this gene (Fig. 1) was released by digestion, and the remaining vector was religated. The final construction, named pCB14, was transformed into E. coli strain ET12567/pUZ8002 and then transferred into S. coelicolor by E. coli-S. coelicolor conjugation. Single-crossover exconjugants were selected on soya-flour-mannitol containing thiostrepton. Three such colonies underwent three rounds of nonselective growth on soya-flour-mannitol, and spores were plated for single colonies that were scored for thiostrepton sensitivity. The deletion within fadD1 was confirmed by Southern hybridization. The mutant was named MCB40.
FIG. 1.

Genetic organization of the macs1-fadD1 operon and the transcriptional regulator acsR. Arrows show oligonucleotides used for RT-PCR assays. The bar represents the fadD1 DNA fragment used as a probe in Northern and Southern analyses. S, SmaI sites within fadD1 that were used for the construction of the deletion mutant.
Integrative plasmid pSET152 (3) was used to complement the fadD1 mutation. For this, the 1.7-kb insert of pCB1 was released by digestion with XbaI and HindIII and cloned into XbaI-HindIII-cleaved pWHM3e (41), yielding pCB32, to leave the fadD1 gene under the transcriptional control of the ermE* promoter. This plasmid was digested with EcoRI and HindIII, and the insert was cloned into EcoRI-HindIII-cleaved pIJ2925, yielding pCB321. A 2.1-kb insert containing fadD1 was released from this vector by digestion with BglII and cloned into BamHI-cleaved pSET152 to yield pCB322. This plasmid was transformed into E. coli strain ET12567/pUZ8002 and introduced into S. coelicolor MCB40 by conjugation, with selection for Amr exconjugants.
RNA extraction.
A volume of 30 to 50 ml of S. coelicolor M145 (wild type [wt]) or S. coelicolor MCB40 culture was pelleted. The cells were washed and resuspended in 0.3 ml of cold Tris-EDTA buffer, and the mixture was added to 0.5 g of glass beads (212 to 300 μm; Sigma)- 0.1 ml of 2% Macaloid-0.05 ml of 10% sodium dodecyl sulfate (SDS)-0.25 ml of Tris-EDTA-saturated phenol-CHCl3. The cells were disrupted by sonication with an ultrasonic processor (Vibrocell VCX600 sonicator; Sonic & Materials, Inc.) for 10 s at 4°C. After centrifugation for 15 min at 4°C and 15,000 × g, supernatants were collected and treated with phenol-chloroform (1:1 [vol/vol]). RNA was precipitated at −20°C with a 1/10 volume of 3 M sodium acetate (pH 7) and 3 volumes of absolute ethanol, rinsed with cold 70% ethanol, and resuspended in water. The RNA concentration was determined by measuring the absorbance at 260 nm.
Reverse transcription (RT)-PCR.
For each reaction, 15 μg of RNA was hybridized for 5 min at 65°C with 1 pmol of RT-down primer (5′-GGTGTCGCCGAGCAGCGGGGTGGT-3′) (Fig. 1) and 0.66 μl of RNase inhibitor (Promega) in enzyme buffer. Then, 20 U of reverse transcriptase, 10 μM each dNTP, and 10 μM [α-32P]ATP were added, and the mixture was incubated for 1 h at 42°C. A 10-μl quantity of the mixture was used as a template for the PCR. The reaction mixture contained 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 2 mM MgCl2, 1.25 mM each of the four dNTPs, 10% dimethyl sulfoxide, 2 U of Taq DNA polymerase, and 20 pmol of each primer (RT-up [5′-AGCCCGGACCGGACACCGCGAAGGTGCT-3′] and RT-down) in a final volume of 50 μl. Samples were subjected to 30 cycles of denaturation (95°C, 1 min), annealing (65°C, 1 min), and extension (72°C, 1 min). PCR products were analyzed by agarose (0.9%) gel electrophoresis. A total of 50 ng of DNA and 15 μg of RNA were used as positive and negative controls, respectively.
Gene expression studies.
actIII and redD were expressed in slot blot assays. Before blotting, 5 μg of each RNA sample was treated with 50% formamide, 6.5% formaldehyde, and 1× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) for 15 min at 68°C. After 2 h at 80°C, the membrane was hybridized with [α-32P]ATP-labeled actIII or redD probe in 0.5% SDS- 5× Denhardt solution- 6× SSC at 60°C. The 32P-labeled actIII probe was prepared by random oligonucleotide priming of the BamHI fragment cloned in pIJ2329 (16). The redD probe was prepared as described previously (37).
Analysis of fadD1 gene expression was carried out by Northern blotting under the conditions described for the slot blot assays. The probe used was the same as that used for the Southern blot experiments and is described above. The accB probe was synthesized as described previously (32).
Growth conditions, protein expression, and preparation of cell extracts.
For measuring ACS activity, the S. coelicolor wt strain and MCB40 were grown at 30°C and 250 rpm in SMM-oleate supplemented with Brij 58 (1% [wt/vol]) or SMM-glucose. After different growth times, the mycelia were harvested by centrifugation and washed twice with SMM medium-Brij 58. Mycelia resuspended in 10 mM phosphate buffer (pH 7.6) containing 1 mM phenylmethylsulfonyl fluoride were disrupted by sonication with an ultrasonic processor (Vibrocell VCX600 sonicator). The lysates were centrifuged (30,000 × g at 4°C for 20 min), and the supernatants were used for ACS assays.
For the expression of fadD1, BL21(DE3) containing pCB1 was grown at 37°C in shake flasks containing Luria-Bertani medium with 100 μg of ampicillin ml−1 for plasmid maintenance. Overnight cultures were diluted 1:10 into fresh medium and grown to an A600 of 0.4 to 0.5 before the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. Induction was allowed to proceed for 4 to 8 h. The cells were then harvested, washed in 100 mM phosphate buffer (pH 8) containing 1 mM phenylmethylsulfonyl fluoride, and resuspended in 1 ml of the same buffer. The cells were disrupted by sonic treatment (four or five bursts) with an ultrasonic processor (Vibrocell VCX600 sonicator). Cell debris was removed by centrifugation, and the supernatants were used as cell extracts.
Protein methods and enzyme assays.
Cell extracts were analyzed by denaturing SDS-polyacrylamide gel electrophoresis (23) with a Bio-Rad minigel apparatus. The final acrylamide monomer concentrations were 12% (wt/vol) for the separating gel and 5% (wt/vol) for the stacking gel. Coomassie brilliant blue was used to stain protein bands.
Protein contents were determined by the method of Bradford (5) with bovine serum albumin as a standard. ACS activity was assayed as described by Kameda et al. (19). Briefly, the reaction mixture contained 200 mM K2PO4 (pH 7.5), 2.5 mM ATP, 0.5 mM CoASH, 5 mM MgCl2, 0.1% Triton X-100, and 20 μM 14C-palmitic acid (8,000 cpm/nmol). The reaction was initiated by the addition of a crude extract (200 mg of protein). At different times, the reaction was stopped by the addition of 0.5 ml of 2-propanol- hexane- 1 M H2SO4 (40:10:1). After the addition of 1 ml of hexane, the organic phase was separated and removed. After three extractions, the radioactivity present in the aqueous phase was measured with a liquid scintillation counter.
Spectrophotometric analysis of actinorhodin.
One milliliter of whole broth was added to KOH to give a final concentration of 1 N; the solution was mixed vigorously and centrifuged at 4,000 × g for 5 min. The A640 of the supernatants was determined, and the Act concentrations were calculated by using a molar extinction coefficient at 640 nm of 25,320 (7).
RESULTS
Sequence analysis of two putative ACSs.
A BLAST search carried out with the E. coli FadD amino acid sequence against the S. coelicolor database revealed several sequences with high similarity to the ACS sequence of E. coli. The most closely related enzyme was a putative long-chain ACS located in cosmid SC2G5 of the S. coelicolor cosmid library (31), with an overall identity of 35%. The amino acid sequence of this putative ACS contained an internal sequence(449GRWMHTGDLAVMREDGYVEIVGRIK472) with 15 amino acid residues that were either identical to or highly conserved in relation to the FACS signature motif common to all FACSs (4). This putative ACS sequence also contained a conserved AMP-binding motif (229YTSGTTGLPK238) commonly found in FACSs and more broadly in the family of adenylate-forming enzymes (40). We called the corresponding ORF of S. coelicolor fadD1. Immediately upstream of the S. coelicolor fadD1 ORF is an ORF that appears to be part of a two-gene operon (Fig. 1), judging by the fact that the stop codon of this ORF overlaps the most likely ATG initiation codon of the fadD1 ORF (data not shown). Database searching of the deduced amino acid sequence of the first gene of the putative operon revealed high similarity with the sequences of a variety of medium-chain ACSs, broadly known as SA proteins (12): 37, 36, and 35% identities overall with putative SA proteins from Thermoplasma acidophilum (Genbank accession no. AL445067), Archaeoglobus fulgidus (Genbank accession no. AE001015), and mammals (Genbank accession no. AF068246, BC015248, and AB022340), respectively. It also showed identities of <32% with putative acetyl-CoA synthetases from both prokaryotes and archaea. We called this gene macs1.
Upstream of macs1 and in the opposite orientation is a gene for a putative regulator with homology to proteins belonging to the LuxR-UhpA family of transcriptional regulators. Due to the physical proximity of this gene to the putative macs1-fadD1 operon and its probable role in the regulation of these genes, we called this ORF acsR.
fadD1 encodes a long-chain ACS of S. coelicolor.
In order to confirm that fadD1 encodes a functional ACS, we complemented the E. coli fadD mutant K27 (21, 29). This strain does not produce ACS and is therefore unable to utilize fatty acids as a sole carbon source. To carry out this experiment, we constructed plasmid pCB12 by subcloning the XbaI-HindIII fragment of pCB1 into XbaI-HindIII-digested pSKBluescript, leaving fadD1 under the control of the lacUV5 promoter. K27/pCB12 was able to grow in M9 minimal medium containing oleate or palmitate as a sole carbon source only when IPTG was present in the medium (data not shown). However, growth complementation appeared to be partial, and the colonies reached only small sizes; these results probably reflected a mislocation of the enzyme or a toxic effect exerted by the expression of the heterologous protein. In accordance with these observations, we also found that the overexpression of fadD1 from the strong φ10 promoter in BL21(DE3)/pCB1 was probably toxic to the cell, since no enzyme accumulation was observed in Coomassie brilliant blue-stained SDS-polyacrylamide gels after IPTG induction.
In order to gain more information on the substrate specificity of FadD1, we determined the apparent Km of the ACS for different substrates in crude extracts of K27/pCB12. The apparent Km and Vmax for palmitate were 28.6 ± 6.7 μM and 1.66 ± 0.11 pmol/μg/min, respectively, and those for octanoic acid were 229.5 ± 34.3 μM and 1.17 ± 0.13 pmol/μg/min, respectively. These results and those of the complementation studies carried out with the E. coli fadD mutant showed that FadD1 is an ACS that can recognize a wide range of acyl chain lengths, although the affinity is clearly higher for the C16:0 substrate. The apparent Km and Vmax for oleic acid (18:1) were 1.16 ± 0.24 mM and 0.14 ± 0.02 pmol/μg/min, respectively. This last result highlights a clear difference between the S. coelicolor and E. coli ACSs, since the affinities of E. coli FadD for oleate and palmitate are on the same order of magnitude, while FadD1 has a 100-fold higher Km for oleate.
Analysis of ACS activity during growth for the wt and FadD1 mutant strains.
To understand the role of FadD1 in the physiology of S. coelicolor, we generated an in-frame deletion mutant as described in Materials and Methods. Since several genes encoding putative long-chain ACSs are present in the S. coelicolor genome, we first studied the levels of this enzyme activity during growth for both M145 and the isogenic fadD1 mutant MCB40 in cultures grown in SMM-oleate and SMM-glucose. Since all these studies were carried out with palmitic acid as a substrate for the enzyme assay, we biased our determinations toward the pool of ACSs with long-chain fatty acids as preferred substrates. As shown in Fig. 2, the specific activity of the enzyme(s) changed with the growth phase and also with the main carbon source present in the medium. In crude extracts prepared from glucose-grown cultures of M145, the pool of ACS activity was high during the exponential phase and decreased sharply after the transition phase. The profile of ACS activity in oleate-grown cultures was opposite that in cultures grown with glucose. The ACS levels increased during the transition phase of growth and reached a maximum during the stationary phase, suggesting an important role of this enzyme activity during this specific growth phase, where most of the secondary metabolites are produced (15). These experiments revealed an interesting regulation of the ACSs present in S. coelicolor, not only by different carbon sources but also by growth phase.
FIG. 2.

ACS levels throughout growth. ACS activities in crude protein extracts prepared from cells of S. coelicolor M145 (filled symbols) and MCB40 (open symbols) grown in SMM-glucose (circles) or SMM-oleate (diamonds) were determined. Palmitate was used as a substrate in the enzyme assays. The values are the averages of three independent determinations. EP, TP, and SP, exponential, transition, and stationary phases of growth, respectively.
Determination of the ACS levels during growth for deletion mutant MCB40 revealed some unexpected complexity in the regulation of this enzyme activity in S. coelicolor. In SMM-glucose, for example, the levels of ACS during the exponential phase were much lower in MCB40 than in M145 (Fig. 2). This decrease in enzyme activity could not be explained by the simple absence of FadD1 since, as is shown below, fadD1 was not expressed during the exponential phase. Surprisingly, in the stationary phase, the levels of ACS were much higher in the mutant than in M145. These results revealed the existence of an intricate regulation net for these enzyme activities, since the absence of FadD1 provoked a significant imbalance in the other ACSs during growth. In SMM-oleate, the enzyme levels in MCB40 remained low and constant during the different growth phases and reached only 30% the levels seen with the wt strain during the stationary phase (Fig. 2). This last result suggests that FadD1 is probably the most abundant ACS in the stationary phase of oleate-grown cultures. However, considering the effect of the fadD1 mutation on other ACS activities, we cannot rule out the possibility that the levels of other ACSs in the stationary phase are also affected in the mutant.
fadD1 is regulated at the transcriptional level.
Northern blot analysis performed with RNA samples obtained at different times from cultures grown with oleate or glucose as the main carbon source showed very similar expression profiles (Fig. 3A). In both instances, fadD1 mRNA was detected exclusively during the stationary phase of growth. However, the levels of expression of fadD1 were much lower in glucose-grown cultures than in oleate-grown cultures, a finding that may partially explain the lower levels of ACS activity observed during the stationary phase in cultures grown with glucose as the main carbon source (Fig. 2). As a control experiment, we monitored the expression of accB (Fig. 3B), the gene that encodes the β subunit of an essential acyl-CoA carboxylase. As shown before (32), this gene is actively transcribed during the exponential phase of growth. Curiously, for the two probes used, fadD1 and accB, two mRNA species of approximately 2.9 and 1.5 kb were detected (Fig. 3). These hybridizing bands coincided with the 23S and 16S rRNAs; however, since hybridization in both instances occurred only in a time-dependent fashion, we believe that the hybridization was probe specific. One explanation for this result could be that the specific mRNAs are trapped by the abundant rRNAs and are not separated properly in the agarose gel. Interestingly, for the RNA sample obtained during the stationary phase from oleate-grown cultures, a third band of approximately 3.5 kb was detected when fadD1 was used as a probe (Fig. 3A). This band probably represents the real size of the transcript and, if so, would be sufficiently long to include the MACS1- and FadD1-coding regions.
FIG. 3.

Expression of fadD1 throughout growth. Northern blot analyses of total RNAs isolated during the exponential phase (EP), the transition phase (TP), and the stationary phase (SP) from cultures of S. coelicolor grown in SMM-glucose or SMM-oleate. The probes used were specific for fadD1 (A) or accB (B). Size markers in kilobases are indicated to the left of the panels.
To confirm this hypothesis, we performed an RT-PCR experiment. For the reverse transcriptase reaction, we used the RT-down oligonucleotide corresponding to a sequence within fadD1. The PCR was carried out with the oligonucleotides RT-down for the 3′end and RT-up corresponding to a sequence within macs1. As shown in Fig. 4, PCR fragments of the correct size were detected in the RNA samples obtained from stationary-phase cultures grown in SMM-oleate and in SMM-glucose. No amplification was observed when RNA samples obtained from exponential-phase cultures were used as a template for the reverse transcriptase reaction. These results confirmed that macs1 and fadD1 are part of the same operon, whose expression is tightly regulated during the exponential phase of growth.
FIG. 4.

RT-PCR analysis for determination of the transcriptional linkage of macs1 and fadD1. The pair of primers used is indicated in Fig. 1. RNAs were isolated from stationary-phase cultures grown in SMM-oleate (lane 1) or SMM-glucose (lane 2) or from exponential-phase SMM-oleate-grown cultures (lane 3). No specific amplification occurred when reverse transcriptase was omitted from the reaction mixture of RNA from stationary-phase cultures (lane 4). DNA was used as a positive control for PCR (lane 5). Lane 6, size markers (SM).
Analysis of Act and undecylprodigiosin (Red) production in M145 and MCB40: involvement of FadD1 in the normal onset of antibiotic production.
M145 growing in SMM-glucose produces the colored antibiotics Act and Red after the cultures enter the stationary phase of growth. At this time, the specific activators actIIORF4 and redD reach the threshold levels that allow the expression of the specific biosynthetic pathways (2, 15, 37). Interestingly, when M145 was grown in SMM-oleate, only Act was synthesized (data not shown). To understand the level at which Red production was blocked, we first studied the expression of the pathway-specific activator redD at different times. We observed that redD was switched on at the same time in both oleate- and glucose-grown cultures, reaching similar levels of expression at 41 h (Fig. 5). This result indicates that in SMM-oleate, as in SMM-glucose, the Red biosynthetic pathway must have been switched on in the cells. Therefore, the failure to produce Red when oleate was used as a carbon source could be related to posttranslational regulation of the Red biosynthetic enzymes or to a deficiency of the specific substrates involved in the biosynthesis of this secondary metabolite.
FIG. 5.
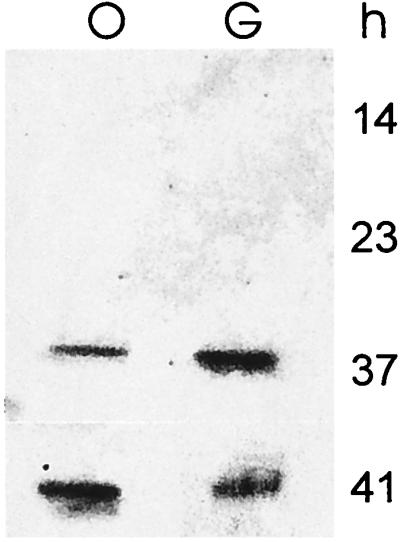
Effect of the growth medium on redD expression. RNAs (5 μg) were isolated at the times indicated from cultures grown in SMM-oleate (lane O) or SMM-glucose (lane G). The samples were blotted on a nitrocellulose filter and hybridized with 32P-labeled redD.
Interestingly, Act production in SMM-oleate was remarkably delayed in mutant strain MCB40 compared to the wt strain, and only very low levels of the antibiotic were detected after 50 h (Fig. 6A). To learn whether the delay in antibiotic production in the mutant strain was due to an absence of transcription of the Act biosynthetic pathway or whether it was caused by a deficiency of substrate availability associated with the absence of FadD1 in the stationary phase, we examined the levels of actIII mRNA in both the wt and the mutant MCB40 at different times. In M145, actIII mRNA was clearly detected at 36 h, in coincidence with the presence of Act in the medium. On the other hand, in the mutant strain, transcripts of the actIII gene were not detected until the late stationary phase (60 h) (Fig. 6B). The delay in the expression of the act cluster in MCB40 did not cause a difference in growth rate, since the growth curve of the mutant strain was very similar to that of M145 (Fig. 6A). Complementation of mutant MCB40 with plasmid pCB32, which expresses fadD1 constitutively from the ermE* promoter, restored the production of Act. This result confirms that the delay in antibiotic production is directly related to the absence of FadD1. Surprisingly, Act biosynthesis started earlier in the complemented mutant strain than in the wt strain, and the final yield was 40% higher (Fig. 6A). The relationship between fadD1 and antibiotic production was not restricted to the biosynthesis of Act in a particular medium, since no antibiotic production was observed in cultures of MCB40 grown for 140 h either in YEME or in liquid R5 (20 and data not shown).
FIG. 6.

Effect of the fadD1 mutation on Act production and on the expression of act biosynthetic genes. (A) Open and filled circles represent the growth curves for M145 and MCB40, respectively. Open, filled, and grey squares represent the levels of Act production in MCB40, M145, and the complemented mutant MCB40/pCB32, respectively. (B) RNAs were isolated at the times indicated, and the samples were dot blotted on nitrocellulose membranes. The probe used was 32P-labeled actIII.
DISCUSSION
We determined, by genetic and biochemical studies, that fadD1 encodes a long-chain ACS in S. coelicolor. The amino acid sequence analysis of this protein revealed the presence of a 25-amino-acid consensus sequence common to all FACSs. A more detailed analysis of this consensus motif revealed some important differences from some of the characteristics proposed to be general features of the FACS signature motif. For example, of the two invariant glycine residues (at positions 2 and 7), only the one at position 7 is found in FadD1. Position 2 is occupied by an Arg, and at position 1, a glycine was found. The highly conserved Gly at position 16 is also present. Of the additional six residues that are proposed to be invariant in the family of ACSs, five are present in FadD1: Trp at position 3, Thr at position 6, Asp at position 8, Arg at position 23, and Lys at position 25. Asp at position 22 is replaced by Gly. Mutagenesis analysis carried out with the E. coli fadD gene determined that the FACS signature motif is essential for catalytic activity and functions in part to promote fatty acid chain length specificity (4). In this regard, six of the seven amino acids found to be involved in chain specificity in E. coli FadD are different in S. coelicolor FadD1. Although we have not performed an extensive analysis of the fatty acid preferences for FadD1, we have proved that this enzyme has a very low specific activity with oleic acid, while this unsaturated fatty acid is a very good substrate for FadD (18).
Gene expression studies clearly showed that the operon is tightly regulated in S. coelicolor and that its expression occurs only when the cultures reach the stationary phase, independent of the carbon source present in the media (Fig. 3A). This result revealed an important difference from the regulation of fadD in E. coli, where the negative regulation exerted by fadR is released by the presence of long-chain fatty acids in the growth medium. The regulation of the macs1-fadD1 operon may be controlled by the putative transcriptional regulator located 269 bp upstream of macs1.
Our results indicate that the inactivation of fadD1 in S. coelicolor leads to profound metabolic phenotypes, such as the inhibition of antibiotic production and the alteration of ACS activity levels. Considering the multiple regulatory roles assigned to acyl-CoA molecules at both the transcriptional and the enzymatic levels (11), we can hypothesize that in the wt strain, FadD1 synthesizes an acyl-CoA derivative with the ability to modulate the expression or the activity of other ACSs (Fig. 2) and the activation of the Act biosynthetic pathway (Fig. 6). Another possibility is that in the absence of FadD1, endogenous precursors (like free fatty acids) accumulate and somehow repress gene expression. We cannot rule out the possibility that FadD1 itself possesses regulatory properties; however, this possibility is less convincing, since there is no significant homology between FadD1 and any regulatory protein in the database.
Recent reports have also related FadD activity to the regulation of gene expression in other bacteria. For example, it has been shown that a mutation in the fadD gene of Sinorhizobium meliloti produces a defect in the expression of the nod and flaA genes (37). Also, FadD-mediated regulation of gene expression has been associated with the establishment of a pathogenic association. For instance, in Salmonella enterica serovar Typhimurium, the loss of ACS represses the expression of hisA, an activator of invasion genes (25).
Although we clearly showed that the absence of Act production in the FadD1 mutant was due to a lack of expression of the Act biosynthetic pathway, we also believe that the absence of this enzyme could affect the carbon flux that provides the carbon units needed for antibiotic biosynthesis. Evidence of the accumulation of TAGs in several Streptomyces species during the exponential phase and their further degradation during the stationary phase has led Olukoshi and Packter (28) to postulate that the catabolism of these compounds could provide the C2 units needed for the biosynthesis of acetate-derived antibiotics (28, 35). ACS activity should have an important role in the degradation of TAGs, since the free fatty acids released from TAGs by lipases first must be converted into their acyl-CoA derivatives to be further oxidized. In this regard, we found that TAG levels in oleate-grown cultures of mutant MCB40 during the stationary phase were two times higher than those of M145 (data not shown), suggesting a lower rate of TAG degradation and probably lower levels of the C2 units needed for the biosynthesis of Act.
Remarkably, the severe constraint in the production of both colored antibiotics exhibited by the FadD1 mutant was not medium dependent; no Act or Red production was observed in either of the rich media tested (YEME or liquid R5). This observation, together with the improved production of Act in the complemented mutant (Fig. 6A), highlights the important role that ACS activity may play in the production of secondary metabolites and suggests a new rationale for regulating antibiotic production in streptomycetes.
Acknowledgments
We are grateful to D. Catalano for helpful assistance with the ACS assays and A. Uttaro and C. Risso for critical reading of the manuscript.
This work was supported by the National Research Council of Argentina (CONICET) (grant PIP 1005-98), ANPCyT grants 01-00078-01686 and 01-06622, and the Universidad Nacional de Rosario.
REFERENCES
- 1.Banchio, C., and H. Gramajo. 1997. Medium-and long chain fatty acid uptake and utilization by Streptomyces coelicolor A3(2): first characterization of a Gram-positive bacterial system. Microbiology 143:2439-2447. [DOI] [PubMed] [Google Scholar]
- 2.Bibb, M. J. 1996. The regulation of antibiotic production in Streptomyces coelicolor A3(2). Microbiology 142:837-845. [DOI] [PubMed] [Google Scholar]
- 3.Bierman, M., R. Logan, K. O'Brien, E. T. Seno, R. Nagaranja Rao, and B. E. Shoner. 1992. Plasmid Cloning vectors for the conjugal transfer of DNA from E. coli to Streptomyces spp. Gene 116:43-49. [DOI] [PubMed] [Google Scholar]
- 4.Black, P. N., Q. Zhang, J. D. Weimar, and C. DiRusso. 1997. Mutational analysis of a fatty acyl-coenzyme A synthetase signature motif identifies seven amino acid residues that modulate fatty acid substrate specificity. J. Biol. Chem. 272:4896-4903. [DOI] [PubMed] [Google Scholar]
- 5.Bradford, M. 1976. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 6.Brofman, M., A. Orenella, M. N. Morales, F. Bieri, F. Waechter, W. Staubli, and P. Bently. 1989. Potentiation of diacylglycerol-activated protein kinase C by acyl-coenzyme A thioesters of hypolipidaemic drugs. Biochem. Biophys. Res. Commun. 159:1026-1031. [DOI] [PubMed] [Google Scholar]
- 7.Bystrykh, L. V., M. A. Fernandez Moreno, J. K. Herrema, F. Malpartida, D. A. Hopwood, and I. Dijkhuizen. 1996. Production of actinorhodin-related blue pigments by Streptomyces coelicolor A3(2). J. Bacteriol. 178:2238-2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 9.DiRusso, C. C., P. N. Black, and J. D. Weimar. 1999. Molecular inroads into the regulation and metabolism of fatty acid: lessons from bacteria. Prog. Lipid Res. 38:129-197. [DOI] [PubMed] [Google Scholar]
- 10.DiRusso, C. C., T. L. Heimert, and A. K. Metzger. 1992. Characterization of FadR, a global transcriptional regulator of fatty acid metabolism in Escherichia coli. Interaction with the fadB promoter is prevented by long chain fatty acyl coenzyme A. J. Biol. Chem. 267:8685-8691. [PubMed] [Google Scholar]
- 11.Faergeman, N. J., and J. Knudsen. 1997. Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signaling. Biochem. J. 323:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujino, T., Y. Takei, H. Sone, R. Ioke, A. Kamataki, K. Magoori, S. Takahashi, J. Saka, and T. Yamamoto. 2001. Molecular identification and characterization of two medium-chain acyl-CoA synthetases, MACS1 and the Sa gene product. J. Biol. Chem. 276:35961-35966. [DOI] [PubMed] [Google Scholar]
- 13.Glick, B. S., and J. E. Rothman. 1987. Possible role for fatty acyl-coenzyme A in intracellular protein transport. Nature 326:309-312. [DOI] [PubMed] [Google Scholar]
- 14.Gordon, J. I., R. J. Duronio, D. A. Rudnick, S. P. Adams, and G. W. Gokel. 1991. Protein N-myristoylation. J. Biol. Chem. 266:8647-8650. [PubMed] [Google Scholar]
- 15.Gramajo, H. C., E. Takano, and M. J. Bibb. 1993. Studies of the growth-phase dependent expression of genes for actinorhodin production in Streptomyces coelicolor. Mol. Microbiol. 7:837-845. [DOI] [PubMed] [Google Scholar]
- 16.Hallan, S. E., F. Malpartida, and D. A. Hopwood. 1993. Nucleotide sequence, transcription and deduced function of gene involved in poliketide biosynthesis in Streptomyces coelicolor. Gene 74:305-320. [DOI] [PubMed] [Google Scholar]
- 17.Hopwood, D. A., M. J. Bibb, K. F. Chater, T. Kieser, C. J. Bruton, H. M. Keiser, D. J. Lydiate, C. P. Smith, J. M. Ward, and H. Schrempf. 1985. Genetic manipulation of Streptomyces: a laboratory manual. The John Innes Foundation, Norwich, United Kingdom.
- 18.Kameda, K., and W. D. Nunn. 1981. Purification and characterization of acyl-coenzyme A synthetase from Escherichia coli. J. Biol. Chem. 256:5702-5707. [PubMed] [Google Scholar]
- 19.Kameda, K., L. K. Suzuki, and Y. Imai. 1985. Further purification, characterization and salt activation of acyl-CoA-synthetase from Escherichia coli. Biochim. Biophys. Acta 840:29-36. [DOI] [PubMed] [Google Scholar]
- 20.Kieser, T., M. J. Bibb, M. J. Buttner, K. F. Chater, and D. A. Hopwood. 2000. Practical Streptomyces genetics. John Innes Centre, Norwich, United Kingdom.
- 21.Klein, K., B. Steinberg, B. Fiethen, and P. Overath. 1971. Fatty acid degradation in Escherichia coli. An inducible system for the uptake of fatty acids and further characterization of old mutants. Eur. J. Biochem. 19:442-450. [DOI] [PubMed] [Google Scholar]
- 22.Korchak, H. M., L. H. Kane, M. W. Rossi, and B. E. Corkey. 1994. Long chain acyl coenzyme A and signaling in neutrophils. An inhibitor of acyl coenzyme A synthetase, triacsin C, inhibits superoxide anion generation and degranulation by human neutrophils. J. Biol. Chem. 269:30281-30287. [PubMed] [Google Scholar]
- 23.Laemmli, U. K. 1970. Levage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 24.Lai, J. C. K., B. B. Liang, E. J. Jarri, A. J. L. Cooper, and D. R. Lu. 1993. Differential affectsof fatty acyl coenzyme A derivatives on citrate synthase and glutamate dehydrogenase. Res. Commun. Chem. Pathol. Pharmacol. 82:331-338. [PubMed] [Google Scholar]
- 25.Lucas, R. L., C. P. Lostroh, C. C. DiRusso, M. P. Spector, B. L. Wanner, and C. A. Lee. 2000. Multiple factors independently regulate hilA and invasion gene expression in Salmonella enterica serovar Typhimurium. J. Bacteriol. 182:1872-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLaughlin, S., and A. Aderem. 1995. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 20:272-276. [DOI] [PubMed] [Google Scholar]
- 27.Miyadoh, S. 1993. Research on antibiotic screening in Japan over the last decade: a producing microorganism approach. Actinomycetology 7:100-106. [Google Scholar]
- 28.Olukoshi, E. R., and N. M. Packter. 1994. Importance of stored triacylglycerols in Streptomyces: possible carbon source for antibiotics. Microbiology 140:931-943. [DOI] [PubMed] [Google Scholar]
- 29.Overath, P., G. Pauli, and U. Scharett. 1969. Fatty acid degradation in Escherichia coli. An inducible acyl-CoA synthetase, the mapping of old-mutations, and the isolation of regulatory mutants. Eur. J. Biochem. 7:559-574. [PubMed] [Google Scholar]
- 30.Pfanner, N., L. Orci, B. S. Glick, M. Amherdt, S. R. Arden, V. Malhotra, and J. E. Rothman. 1989. Fatty acyl-coenzyme A is required for budding of transport vesicles from Golgi cisternae. Cell 59:95-102. [DOI] [PubMed] [Google Scholar]
- 31.Redenbach, M., H. M. Kieser, A. Danapaite, A. Eichner, J. Cullum, H. Kinashi, and D. A. Hopwood. 1996. A set of ordered cosmids and a detailed genetic and physical map for the 8 Mb Streptomyces coelicolor A3(2) chromosome. Mol. Microbiol. 21:77-96. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez, E., C. Banchio, L. Diacovich, M. J. Bibb, and H. Gramajo. 2001. Role of an essential acyl coenzyme A carboxylase in the primary and secondary metabolism of Streptomyces coelicolor A3(2). Appl. Environ. Microbiol. 67:4166-4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 34.Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shim, M., W. Kim, and J. Kim. 1997. Neutral lipids and lipase activity for actinorhodin biosynthesis of Streptomyces coelicolor A3(2). Biotechnol. Lett. 19:221-223. [Google Scholar]
- 36.Soto, M. J., M. Fernánez-Pascual, J. San Juan, and J. Olivares. 2002. A fadD mutant of Sinorhizobium meliloti shows multicellular swarming migration and is impaired in nodulation efficiency on alfalfa roots. Mol. Microbiol. 43:371-382. [DOI] [PubMed] [Google Scholar]
- 37.Studier, F. W., and B. A. Moffatt. 1986. Use of the bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113-130. [DOI] [PubMed] [Google Scholar]
- 38.Takano, E., H. C. Gramajo, E. Strauch, N. Andres, J. White, and M. J. Bibb. 1992. Transcriptional regulation of the redD transcriptional activator gene accounts for growth-phase-dependent production of the antibiotic undecylprodigiosin in Streptomyces coelicolor A3(2). Mol. Microbiol. 6:2797-2804. [DOI] [PubMed] [Google Scholar]
- 39.Watkins, P. A. 1997. Fatty acid activation. Prog. Lipid Res. 36:55-83. [DOI] [PubMed] [Google Scholar]
- 40.Watkins, P. A., J. F. Lu, S. J. Steinberg, S. J. Gould, K. D. Smith, and L. T. Braiterman. 1998. Disruption of the Saccharomyces cerevisiae FAT1 gene decreases very long-chain fatty acyl-CoA synthetase activity and elevates intracellular very long-chain fatty acid concentrations. J. Biol. Chem. 273:18210-18219. [DOI] [PubMed] [Google Scholar]
- 41.Wezel, G. P., J. Meulen Kawamoto, S. R. Luiten, H. K. Koerten, and B. Kraal. 2000. ssgA is essential for sporulation of Streptomyces coelicolor A3(2) and affects hyphal development by stimulating septum formation. J. Bacteriol. 192:5653-5662. [DOI] [PMC free article] [PubMed] [Google Scholar]