

Abstract
蛋白质结构决定功能,结构信息对蛋白质耐热性预测至关重要。本文提出一种融合图嵌入特征和网络拓扑特征的蛋白质耐热性预测模型。通过构建残基相互作用网络(RIN)表示蛋白质结构,计算网络拓扑特征,并利用深度神经网络(DNN)挖掘其隐含特征。通过DeepWalk和Node2vec算法获取节点嵌入,结合TopN策略与双向长短期记忆网络(BiLSTM)提取图嵌入特征;同时引入Doc2vec算法替代图嵌入算法中的Word2vec模块,得到图嵌入特征向量编码。基于注意力机制融合图嵌入特征与网络拓扑特征,构建高精度预测模型,在细菌来源的蛋白质数据集上实现了87.85%的预测准确率。此外,本文分析了网络拓扑特征在模型中的贡献差异及不同图嵌入方法的差异,发现结合Doc2vec的DeepWalk特征与全部拓扑特征是耐热蛋白质识别的关键。本研究为蛋白质耐热性预测提供了切实有效的新方法,这为探索蛋白质多样性、发现新的耐热蛋白质及常温蛋白质智能改造提供了理论指导。
Keywords: 残基相互作用网络, 图嵌入, 特征融合, 深度学习, 蛋白质耐热性
Abstract
Protein structure determines function, and structural information is critical for predicting protein thermostability. This study proposes a novel method for protein thermostability prediction by integrating graph embedding features and network topological features. By constructing residue interaction networks (RINs) to characterize protein structures, we calculated network topological features and utilize deep neural networks (DNN) to mine inherent characteristics. Using DeepWalk and Node2vec algorithms, we obtained node embeddings and extracted graph embedding features through a TopN strategy combined with bidirectional long short-term memory (BiLSTM) networks. Additionally, we introduced the Doc2vec algorithm to replace the Word2vec module in graph embedding algorithms, generating graph embedding feature vector encodings. By employing an attention mechanism to fuse graph embedding features with network topological features, we constructed a high-precision prediction model, achieving 87.85% prediction accuracy on a bacterial protein dataset. Furthermore, we analyzed the differences in the contributions of network topological features in the model and the differences among various graph embedding methods, and found that the combination of DeepWalk features with Doc2vec and all topological features was crucial for the identification of thermostable proteins. This study provides a practical and effective new method for protein thermostability prediction, and at the same time offers theoretical guidance for exploring protein diversity, discovering new thermostable proteins, and the intelligent modification of mesophilic proteins.
Keywords: Residue interaction networks, Graph embedding, Feature fusion, Deep learning, Protein heat tolerance
0. 引言
蛋白质的热稳定性是衡量蛋白质在高温条件下保持结构和功能特性的能力。当环境温度超出蛋白质能够承受的温度时,蛋白质的三维结构将遭到破坏,进而导致蛋白质发生变性和功能失活[1-2]。由于耐热蛋白质具有在高温下长时间保持活性的特点,在食品、制药、纺织、生物精炼、饲料等行业有着重要的应用。目前耐热蛋白质的获取主要通过实验方法[3-5]。随着计算机技术的飞速发展,用计算方法预测蛋白质的耐热性以及分析热稳定机制,为耐热蛋白质的开发与应用开辟了新路径。
蛋白质三维结构转换为残基相互作用网络(residue interaction network,RIN),通过机器学习预测蛋白质的耐热性,为蛋白质耐热性研究提供了新视角[6-10]。RIN中,氨基酸以节点表示,氨基酸间的相互作用关系则由节点间的边表示。基于此,研究者们用网络拓扑特征提取RIN信息,挖掘影响蛋白质耐热性的关键因素。Hu等[11]计算了大量的特征用于分析影响蛋白质耐热性的因素,包括特征路径长度、聚类系数和直径在内的网络度量,还研究网络的全局拓扑参数,如小世界性质和堆积密度,计算中心性度量,包括介数、接近中心性等。Verkhivker[12]通过中心性度量以及社团检测寻找关键残基。Jiao等[13-14]以不同相互作用的能量为边,构建加权RIN用于识别关键残基和活性位点。但这类基于网络拓扑特征的分析与预测方法存在局限性,不能全面提取结构特征。
随着深度学习与复杂网络研究的融合,一系列能够捕捉并表征完整的图结构信息的图嵌入算法不断涌现,并在生物信息学领域展现出巨大的应用潜力[15]。图嵌入的核心思想是将图信息转化为序列信息,借助成熟的序列表征算法获取节点嵌入。其中DeepWalk算法通过随机游走策略将图转化成序列,并结合Word2vec算法获取节点嵌入[16]。随后改进算法Node2vec进一步优化随机游走机制,同时保留节点的邻域特征和网络结构[17]。这些算法的发展显著提升了复杂网络的表征能力,为多学科交叉研究提供了有力工具。
本文聚焦于细菌来源且已解析三维结构的蛋白质,以蛋白质原子的三维笛卡尔坐标构建加权RIN,计算五种网络拓扑特征并通过深度神经网络(deep neural network,DNN)学习隐藏特征。通过DeepWalk算法和Node2vec算法获取节点嵌入,结合TopN和双向长短期记忆网络(bidirectional long short-term memory,BiLSTM)提取图嵌入特征;同时探索Doc2vec算法替代传统图嵌入中Word2vec模块以实现图嵌入特征向量编码。最终基于注意力机制将图嵌入特征与网络拓扑特征融合,构建蛋白质耐热性预测模型,为蛋白质耐热性的精准预测提供新方法。
1. 数据与方法
1.1. 数据集的构建
本文从蛋白质数据库(Protein Data Bank,PDB)筛选含有结构信息的蛋白质数据[18],通过细菌元数据库Bacdive[19]检索其微生物来源的最适生长温度,将最适生长温度超过40 ℃的蛋白质标记为耐热蛋白质,其余标记为常温蛋白质。为保证数据集质量,采用以下步骤进行数据筛选:① 只保留蛋白质结构测量方法为X射线晶体衍射法的蛋白质。② 对于相似性高于40%的蛋白质,保留分辨率更小的蛋白质。③ 删除序列长度小于50的蛋白质。④ 只保留来自细菌的蛋白质。最终得到2 950个耐热蛋白质,并从符合条件的常温蛋白质中随机选出等数量的蛋白质作为负样本。所有的样本按正负样本等比分为无交集的训练集、验证集和测试集,分别占总数据集的81%、9%和10%。
1.2. 加权残基相互作用网络的构建
本文基于PDB数据库蛋白质原子的三维笛卡尔坐标,依据盐桥、二硫键、范德华力、氢键、π-π相互作用、π-阳离子相互作用的距离阈值(4.0 Å、3.0 Å、0.8 Å、3.5 Å、7.0 Å和7.0 Å),计算原子间相互作用,构建加权RIN[20]。以氨基酸为节点,相互作用关系为边,边权为两个氨基酸间所有原子相互作用的个数。对于图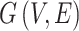 ,其中
,其中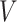 表示节点集,
表示节点集,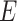 表示边集,邻接矩阵
表示边集,邻接矩阵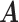 定义为:当节点
定义为:当节点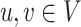 存在权值为
存在权值为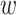 的边时,
的边时,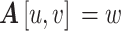 ,否则
,否则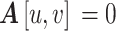 。原子间每类化学键的形成条件参见附件1。
。原子间每类化学键的形成条件参见附件1。
1.3. 网络拓扑特征的计算
1.3.1. 度中心性
表示节点与其他节点的连接紧密程度。若N为网络总节点数,度中心性表示为:
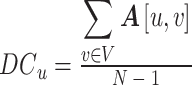 |
1 |
1.3.2. 接近中心性
评估节点的重要性。节点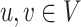 的最短路径用
的最短路径用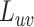 表示,则节点
表示,则节点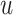 的接近中心性为:
的接近中心性为:
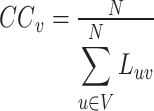 |
2 |
1.3.3. 介数中心性
可用于衡量节点重要性。对于网络中的节点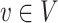 ,其介数中心性可表示为:
,其介数中心性可表示为:
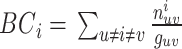 |
3 |
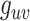 表示节点u、v间最短路径的数目,
表示节点u、v间最短路径的数目,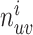 表示节点
表示节点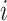 位于u、v间最短路径上的次数。
位于u、v间最短路径上的次数。
1.3.4. 特征向量中心性
对于网络中的节点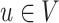 ,
,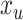 表示节点的重要性,特征向量中心性可表示为:
表示节点的重要性,特征向量中心性可表示为:
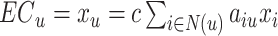 |
4 |
其中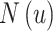 表示节点u的邻居节点,
表示节点u的邻居节点,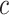 为常数,将网络中所有节点的特征向量中心性表示为一个列向量
为常数,将网络中所有节点的特征向量中心性表示为一个列向量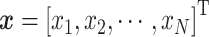 ,经过多次迭代达到稳态时,可写矩阵形式:
,经过多次迭代达到稳态时,可写矩阵形式:
 |
5 |
1.3.5. PageRank值
PageRank核心思想是网页被指向次数多则重要,且重要网页链接的网页重要性也高[21]。无向图的边可视为双向的有向边。第l次迭代,节点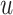 将前一次的PageRank值平分给其指向的节点,若节点
将前一次的PageRank值平分给其指向的节点,若节点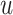 出度为
出度为 ,其邻接节点分得的PageRank值为
,其邻接节点分得的PageRank值为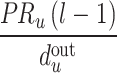 。更新得到:
。更新得到:
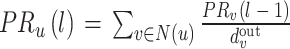 |
6 |
基于邻接矩阵定义一个新的矩阵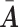 :
:
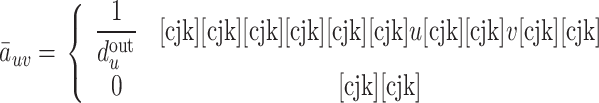 |
7 |
迭代规则可写成矩阵形式:
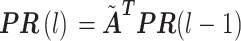 |
8 |
其中
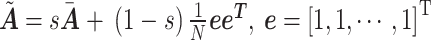 |
9 |
式中s表示阻尼系数,值为0.85。
1.4. 经典图嵌入方法
1.4.1. DeepWalk算法
DeepWalk思想类似Word2vec算法[22],使用图中节点与节点的共现关系学习节点的向量表示。使用随机游走算法从图中随机获取序列:以图中任意节点为起点,从当前节点邻域中随机选取未访问过的节点加入序列,重复此过程至序列达到目标长度或邻域无新节点。在每个节点随机游走多次后,获得大量的随机游走序列,将每个随机游走序列视为Word2vec中的句子,单个节点或一段连续的节点视为单词,构建语料库,使用Skip-Gram模型学习包含了节点与节点的共现关系的节点嵌入。
1.4.2. Node2vec算法
Node2vec算法优化了随机游走策略,使邻近节点更容易被选取。采取一种有偏的随机游走策略:对于任意的当前节点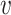 ,访问下一个节点
,访问下一个节点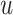 的概率为:
的概率为:
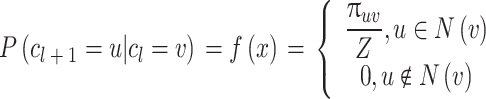 |
10 |
其中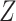 为归一化常数,
为归一化常数,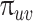 是节点v、u之间未归一化的转移概率,表示为:
是节点v、u之间未归一化的转移概率,表示为:
 |
11 |
 |
12 |
式中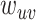 表示节点
表示节点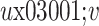 间边的权值,
间边的权值,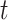 为随机游走序列中节点
为随机游走序列中节点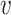 的前一个节点。
的前一个节点。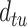 表示节点
表示节点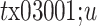 间的最短距离,示意图见图1。超参数
间的最短距离,示意图见图1。超参数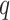 和
和 分别控制随机游走在局部游走的趋向和全局游走的趋向。若
分别控制随机游走在局部游走的趋向和全局游走的趋向。若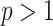 ,趋向于全局游走;若
,趋向于全局游走;若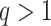 ,则更趋向于局部游走。随机游走后将所有提取的序列作为语料库,获取节点嵌入。
,则更趋向于局部游走。随机游走后将所有提取的序列作为语料库,获取节点嵌入。
图 1.

The diagram of different shortest distance relationships between node t and node u in the Node2vec algorithm
Node2vec算法中节点t与节点u不同最短距离关系示意图
1.5. 基于节点嵌入的图嵌入方法
1.5.1. TopN的图嵌入
TopN选取高频的节点间共现关系来表示图的特征。在DeepWalk算法框架下,首先利用随机游走获取长序列,用于训练Word2vec模型。训练Word2vec时,对长序列进行k-mer分词,以获得更丰富的词嵌入向量。随机游走时起始点为孤立点或游走到度为1的节点,则保留长度超过k的长序列来丰富Word2vec的语料库。随后再次运用随机游走提取长度为k的短序列。起始点为孤立点或游走到度为1的节点时,保留长度为k的短序列。为降低随机因素导致的偏差,每个节点被多次遍历。统计每种k-mer在整个网络中出现的频次。将出现频率中前N个k-mer作为图的特征,结合Word2vec算法学习的k-mer嵌入,实现整个网络的表示。
与DeepWalk算法类似,在Node2vec算法框架下,TopN同样通过对随机游走提取的序列进行处理,实现图特征表示。DeepWalk算法在提取长序列和短序列时均采用无偏随机游走策略,而在Node2vec算法中,提取长序列和短序列则同时采用有偏的随机游走策略。
1.5.2. Doc2vec的图嵌入
Doc2vec算法能够将不同的RIN表示成固定长度的特征向量。Doc2vec在Word2vec的基础上进行扩展[23],将多个句子整合成段落,并为每段赋予一个段向量,在训练词嵌入的同时,段向量作为每个词所处的上下文环境一同被学习。在DeepWalk和Node2vec算法中,将每个RIN视为一段,基于随机游走提取的长序列视为句子,分词后的k-mer视为词,使用PV-DM模型将每个网络编码成固定长度的向量。
1.6. 耐热性预测的深度学习模型构建
1.6.1. 基于网络拓扑的深度学习模型
为了探索利用网络拓扑特征预测蛋白质耐热性的能力,首先构建了基于网络拓扑特征的蛋白质耐热性预测深度学习模型。基于五种网络拓扑特征,使用DNN学习每个网络拓扑特征,并将隐藏特征嵌入到相同维度。对于任意的网络拓扑特征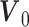 ,通过多个全连接的隐藏层后,嵌入后的特征表示为:
,通过多个全连接的隐藏层后,嵌入后的特征表示为:
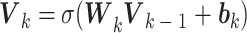 |
13 |
其中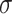 为激活函数,
为激活函数,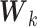 和
和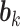 是可训练的权和偏置,
是可训练的权和偏置,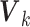 可用于后续的分类任务。
可用于后续的分类任务。
为了增强拓扑特征的融合,通过注意力机制计算各个特征间的注意力,基于注意力自动分配权重[24]。融合后的特征经过激活函数和神经元分类器,得到最终的预测。为系统对比深度学习模型的预测性能,将支持向量机(support vector machine,SVM)作为基线模型。
1.6.2. 图嵌入驱动的深度学习模型
为了探索两种节点嵌入算法和两种基于节点嵌入的图嵌入方法对蛋白质耐热性预测的影响,设计四种图嵌入方法,针对不同的图嵌入使用匹配的深度学习算法提取隐藏特征用于蛋白质耐热性分类:① DeepWalk + TopN模型利用DeepWalk算法获取节点嵌入,基于TopN获取图嵌入特征矩阵,随后借助BiLSTM提取图嵌入特征预测蛋白质的耐热性。② Node2vec + TopN模型使用Node2vec算法获取节点嵌入并基于TopN获取图嵌入,得到特征矩阵,随后用BiLSTM学习隐藏特征,用于分类耐热蛋白质和常温蛋白质。③ DeepWalk + Doc2vec模型将DeepWalk算法中Word2vec部分替换成Doc2vec,将每个图编码成特征向量,后用DNN学习隐藏特征,用于分类耐热蛋白质和常温蛋白质。④ Node2vec + Doc2vec模型将Node2vec算法中的Word2vec部分替换成Doc2vec,将每个图编码成特征向量,随后使用DNN学习隐藏特征,分类耐热蛋白质和常温蛋白质。
1.6.3. 融合网络拓扑和图嵌入的模型
为了更精准地预测蛋白质的耐热性,进一步将图嵌入信息与网络拓扑特征融合,构建基于融合特征的蛋白质耐热性预测模型。分别构建了使用TopN获取的图嵌入并结合网络拓扑特征的预测模型(见图2)以及使用Doc2vec获取的图嵌入与网络拓扑特征融合的预测模型(见图3)。
图 2.

Prediction model based on graph embedding obtained using TopN and fused with network topology features
使用TopN获取的图嵌入和网络拓扑特征融合的预测模型
图 3.

Prediction model based on graph embedding obtained using Doc2vec and fused with network topology features
使用Doc2vec获取的图嵌入与网络拓扑特征融合后的预测模型
1.7. 超参数设置
本文使用的DNN模型共两层,每种图特征嵌入到100维。DeepWalk和Node2vec算法中随机序列长度为15,Word2vec算法窗口大小为9,节点嵌入维度为100。Node2vec算法中p = 0.25,q = 4。BiLSTM的隐藏层维度为64。
1.8. 评价指标
使用十折交叉验证评估模型的分类能力,模型性能通过准确性(accuracy,ACC)、精度(precision,PR)、召回率(recall,REC)、F1分数(F1-score,F1)、马修斯相关系数(Matthews Correlation Coefficient,MCC)、接收者操作特征曲线下面积(area under the receiver operating characteristic curve,AUROC)衡量。十次重复实验计算指标的平均值,以确保评估结果的可靠性。各个指标的计算公式如下:
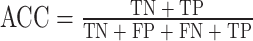 |
14 |
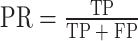 |
15 |
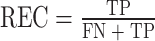 |
16 |
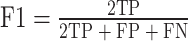 |
17 |
 |
18 |
其中TP代表耐热蛋白质中正确分类的个数,FP代表耐热蛋白质中错误分类的个数,TN代表常温蛋白质中正确分类的个数,FN代表常温蛋白质中错误分类的个数。
2. 结果与分析
2.1. 基于网络拓扑特征模型性能分析
五种网络拓扑特征和全部拓扑特征在预测耐热蛋白质时的分类结果见表1。五种网络拓扑特征在DNN模型和SVM模型上呈现出相同的规律,介数中心性在两种模型中均表现最佳,接近中心性表现最差,其他三种网络拓扑特征也展现出一定的预测能力。比较单一特征在两种模型中的性能,除特征向量中心性外,其他网络特征在SVM模型上的表现优于DNN模型,因此对于单一的网络拓扑特征,SVM模型表现出更优秀的分类性能。基于全部网络拓扑特征的模型优于单一特征模型,表明不同的网络拓扑特征涵盖了蛋白质不同方面信息,多种特征的融合能更全面地描述蛋白质。基于全部网络拓扑特征的DNN模型的性能优于SVM模型,证实了深度学习模型在多特征融合时的优势,即自动为每个特征分配权值,实现与特征筛选相似的功能。
表 1. Performance of different network topology features on the test set (%).
不同网络拓扑特征在测试集上的性能(%)
| 特征 | 模型 | ACC↑ | PR↑ | REC↑ | F1↑ | MCC↑ |
| 注:加粗数字表示最优结果;↑表示该指标越大越好 | ||||||
| 度中心性 | SVM | 63.24 | 67.19 | 52.17 | 56.18 | 28.02 |
| DNN | 63.06 | 61.08 | 72.19 | 66.14 | 26.59 | |
| 特征向量中心性 | SVM | 55.98 | 55.70 | 55.56 | 55.52 | 12.04 |
| DNN | 56.11 | 55.40 | 62.94 | 58.89 | 12.35 | |
| 接近中心性 | SVM | 54.84 | 60.33 | 28.00 | 38.18 | 11.39 |
| DNN | 52.73 | 52.30 | 61.60 | 56.56 | 5.56 | |
| 介数中心性 | SVM | 75.30 | 73.87 | 78.90 | 76.25 | 50.79 |
| DNN | 73.02 | 72.82 | 73.69 | 73.17 | 46.16 | |
| PageRank | SVM | 58.07 | 61.38 | 53.94 | 57.38 | 16.24 |
| DNN | 55.82 | 54.09 | 76.99 | 63.52 | 12.90 | |
| 全部拓扑特征 | SVM | 78.92 | 80.19 | 78.82 | 79.47 | 57.86 |
| DNN | 79.59 | 81.65 | 76.52 | 78.87 | 59.39 | |
2.2. 基于图嵌入的模型的性能分析
四种图嵌入模型在验证集和测试集的结果见表2和表3,其中Node2vec + Doc2vec模型预测性能最优。
表 2. Performance of different graph embedding methods on the validation set (%).
不同图嵌入方法在验证集上的性能(%)
| 模型 | ACC↑ | PR↑ | REC↑ | F1↑ | MCC↑ |
| 注:加粗数字表示最优结果;↑表示该指标越大越好 | |||||
| DeepWalk + TopN | 80.48 | 84.19 | 75.24 | 79.36 | 61.42 |
| Node2vec + TopN | 80.18 | 81.12 | 79.12 | 79.95 | 60.60 |
| DeepWalk + Doc2vec | 80.22 | 79.86 | 80.87 | 80.34 | 60.48 |
| Node2vec + Doc2vec | 81.99 | 81.08 | 83.48 | 82.25 | 64.03 |
表 3. Performance of different graph embedding methods on the test set (%).
不同图嵌入方法在测试集上的性能(%)
| 模型 | ACC↑ | PR↑ | REC↑ | F1↑ | MCC↑ | AUROC↑ |
| 注:加粗数字表示最优结果;↑表示该指标越大越好 | ||||||
| DeepWalk + TopN | 79.44 | 79.12 | 80.00 | 79.55 | 58.89 | 86.26 |
| Node2vec + TopN | 79.22 | 80.77 | 77.24 | 78.76 | 58.78 | 86.38 |
| DeepWalk + Doc2vec | 80.99 | 81.18 | 80.79 | 80.96 | 62.03 | 88.42 |
| Node2vec + Doc2vec | 83.51 | 82.83 | 84.57 | 83.68 | 67.05 | 90.84 |
比较两种节点嵌入方法,Node2vec的整体表现优于DeepWalk,说明Node2vec的有偏游走策略有助于获取更充分的图信息。而DeepWalk + TopN与Node2vec + TopN模型的性能差异不显著,可能是前N个明显的特征容易捕获,不同的游走策略对该类特征的获取能力相近,因此在分类时未体现出明显差异。
在提取图嵌入特征方法的对比上,Doc2vec明显优于TopN。Doc2vec基于整个语料库进行训练,不仅能捕捉到高频图结构,还提取了低频的图结构信息,从而获取更全面的图特征表示。从蛋白质分子结构角度分析,蛋白质结构决定其功能,耐热蛋白质通常具备在高温环境下更稳定的分子结构模式,TopN和Doc2vec能从这类高频的特征中提取隐藏信息用于预测。然而,非高频的蛋白质结构特征上也存在关键信息,Doc2vec凭借从全局获取信息的能力,能够更充分地挖掘这些潜在信息,表现优于只保留高频蛋白质结构特征TopN的方法。
2.3. 融合图嵌入和网络拓扑的模型分析
将图嵌入特征与网络拓扑特征融合,验证集和测试集的结果见表4和表5。融合拓扑特征后模型性能提升,说明网络拓扑特征作为耐热蛋白质分类的有效特征,既能单独作为特征分类耐热蛋白质,又能与图嵌入特征协同作用,辅助提升分类精度。本文的蛋白质拓扑特征更关注网络的全局信息,与p > 1时的Node2vec所提取的特征存在较高相似性,这种特征冗余性可能是Node2vec+Doc2vec模型在融合拓扑特征后性能提升幅度有限的关键原因。因此在多特征融合过程中,减少特征冗余是优化模型性能的重要方向。
表 4. Performance of models fused with different features on the validation set (%).
不同特征融合后的模型在验证集上的性能(%)
| 模型 | ACC↑ | PR↑ | REC↑ | F1↑ | MCC↑ |
| 注:加粗数字表示最优结果;↑表示该指标越大越好 | |||||
| 全部拓扑特征 + DeepWalk + TopN | 84.47 | 85.02 | 83.80 | 84.37 | 69.02 |
| 全部拓扑特征 + Node2vec + TopN | 84.21 | 84.38 | 84.03 | 84.19 | 68.44 |
| 全部拓扑特征 + DeepWalk + Doc2vec | 86.54 | 86.11 | 87.25 | 86.63 | 73.17 |
| 全部拓扑特征 + Node2vec + Doc2vec | 86.81 | 85.68 | 88.41 | 87.02 | 73.67 |
表 5. Performance of models fused with different features on the test set (%).
不同特征融合后的模型在测试集上的性能(%)
| 模型 | ACC↑ | PR↑ | REC↑ | F1↑ | MCC↑ | AUROC↑ |
| 注:加粗数字表示最优结果;↑表示该指标越大越好 | ||||||
| 全部拓扑特征 + DeepWalk + TopN | 82.40 | 81.96 | 84.00 | 82.70 | 65.32 | 91.09 |
| 全部拓扑特征 + Node2vec + TopN | 84.04 | 83.55 | 85.03 | 84.21 | 68.22 | 91.75 |
| 全部拓扑特征 + DeepWalk + Doc2vec | 87.85 | 87.31 | 88.71 | 87.95 | 75.80 | 93.84 |
| 全部拓扑特征 + Node2vec + Doc2vec | 86.82 | 85.92 | 88.08 | 86.97 | 73.71 | 93.06 |
3. 结论
本文将蛋白质三维结构信息转化为RIN,提出基于蛋白质结构特征预测蛋白质耐热性的方法。通过计算五种网络拓扑特征,基于DNN获取隐藏特征,并基于注意力机制融合,构建的深度学习分类模型的预测能力强于传统的机器学习方法,且介数中心性起着最重要的作用。进一步的,为了全面捕获蛋白质结构信息,使用DeepWalk算法和Node2vec算法获取节点嵌入并提出获取图嵌入的方法。基于网络融合特征的蛋白质耐热性预测模型同时使用了网络拓扑特征和图嵌入特征,融合后的预测性能明显提升。
重要声明
利益冲突声明:本文全体作者均声明不存在利益冲突。
作者贡献声明:潘书宜、向孝洋、颜群芳进行实验设计;向孝洋进行数据收集;丁彦蕊、潘书宜、向孝洋共同进行数据分析和论文写作。
本文附件见本刊网站的电子版本(biomedeng.cn)。
Funding Statement
国家自然科学基金(62161050)
References
- 1.Finch A J, Kim J R Thermophilic proteins as versatile scaffolds for protein engineering. Microorganisms. 2018;6(4):97. doi: 10.3390/microorganisms6040097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silva N H, Vilela C, Marrucho I M, et al Protein-based materials: from sources to innovative sustainable materials for biomedical applications. Mater Chem B. 2014;2(24):3715–3740. doi: 10.1039/c4tb00168k. [DOI] [PubMed] [Google Scholar]
- 3.Guta M, Abebe G, Bacha K, et al Screening and characterization of thermostable enzyme-producing bacteria from selected hot springs of Ethiopia. Microbiol Spectr. 2024;12(3):e0371023. doi: 10.1128/spectrum.03710-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venev S V, Zeldovich K B Thermophilic adaptation in prokaryotes is constrained by metabolic costs of proteostasis. Mol Biol Evol. 2018;35(1):211–224. doi: 10.1093/molbev/msx282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rousseau M, Oulavallickal T, Williamson A, et al Characterisation and engineering of a thermophilic RNA ligase from Palaeococcus pacificus. Nucleic Acids Res. 2024;52(7):3924–3937. doi: 10.1093/nar/gkae149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barabasi A L, Oltvai Z N Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5(2):101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 7.常珊, 焦雄, 王美华, 等 蛋白质氨基酸网络研究进展. 现代生物医学进展. 2011;11(001):190–193. [Google Scholar]
- 8.Grewal R K, Roy S Modeling proteins as residue interaction networks. Protein Pept Lett. 2015;22(10):923–933. doi: 10.2174/0929866522666150728115552. [DOI] [PubMed] [Google Scholar]
- 9.Guan Ruining, Liu Wencheng, Li Ningqi, et al Machine learning models based on residue interaction network for ABCG2 transportable compounds recognition. Environ Pollut. 2023;337:122620. doi: 10.1016/j.envpol.2023.122620. [DOI] [PubMed] [Google Scholar]
- 10.Inan T, Yuce M, Mackerell JR A D, et al Exploring druggable binding sites on the Class A GPCRs using the residue interaction network and site identification by ligand competitive saturation. ACS Omega. 2024;9(38):40154–40171. doi: 10.1021/acsomega.4c06172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Guang, Yan Wenying, Zhou Jianhong, et al Residue interaction network analysis of Dronpa and a DNA clamp. J Theor Biol. 2014;348:55–64. doi: 10.1016/j.jtbi.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 12.Verkhivker G Molecular dynamics simulations and modelling of the residue interaction networks in the BRAF kinase complexes with small molecule inhibitors: probing the allosteric effects of ligand-induced kinase dimerization and paradoxical activation. Mol Biosyst. 2016;12(10):3146–3165. doi: 10.1039/C6MB00298F. [DOI] [PubMed] [Google Scholar]
- 13.Jiao Xiong, Chang Shan, Li Chunhua, et al. Construction and application of the weighted amino acid network based on residue fluctuations. Phys Rev E Stat Nonlin Soft Matter Phys, 2007, 75(5 Pt 1): 051903.
- 14.Jiao Xiong, Yang Lifeng, An Meiwen, et al A modified amino acid network model contains similar and dissimilar weight. Comput Math Methods Med. 2013;2013(1):197892. doi: 10.1155/2013/197892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Ziqi, Zhang Yuanyuan, Wang Shudong, et al SINE: second-order information network embedding. IEEE Access. 2020;8:139044–139051. doi: 10.1109/ACCESS.2020.3007886. [DOI] [Google Scholar]
- 16.Perozzi B, Al-Rfou R, Skiena S. Deepwalk: Online learning of social representations// Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. New York: ACM SIGKDD, 2014: 701-710.
- 17.Grover A, Leskovec J. node2vec: Scalable feature learning for networks// Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. San Francisco: ACM SIGKDD, 2016: 855-864.
- 18.Berman H M, Westbrook J, Feng Z, et al The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schober I, Koblitz J, Sardà C J, et al BacDive in 2025: the core database for prokaryotic strain data. Nucleic Acids Res. 2025;53(D1):D748–D756. doi: 10.1093/nar/gkae959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piovesan D, Minervini G, Tosatto S C The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res. 2016;44(W1):W367–W74. doi: 10.1093/nar/gkw315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Page L, Brin S, Motwani R, et al. The PageRank citation ranking: Bringing order to the web. stanford digital libraries working paper, 1999.
- 22.Mikolov T, Chen K, Corrado G, et al. Efficient estimation of word representations in vector space. arXiv preprint arXiv, 2013: 1301.3781.
- 23.Le Q, Mikolov T. Distributed representations of sentences and documents// Proceedings of the 31th International Conference on Machine Learning. Beijing: IMLS, 2014: 1188-1196.
- 24.Shaw P, Uszkoreit J, Vaswani A. Self-attention with relative position representations. arXiv preprint arXiv, 2018: 1803.02155.