

Abstract
A semidefined medium based on Casamino Acids allowed Lactococcus lactis ATCC 19435 to grow in the presence of oxygen at a slow rate (0.015 h−1). Accumulation of H2O2 in the culture prevented a higher growth rate. Addition of asparagine to the medium increased the growth rate, whereby H2O2 accumulated only temporarily during the lag phase. H2O2 is an inhibitor for several glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase being the most sensitive. Strain ATCC 19435 contained NADH oxidase (maximum specific rate under aerobic conditions, 426 nmol of NADH min−1 mg of protein−1), which reduced oxygen to water, whereby superoxide was formed as a by-product. H2O2 originated from the dismutation of superoxide by superoxide dismutase. Although H2O2 was rapidly destroyed under high metabolic fluxes, neither NADH peroxidase nor any other enzymatic H2O2-reducing activity was detected. However, pyruvate, the end product of glycolysis, reacted nonenzymatically and rapidly with H2O2 and hence was a potential alternative for scavenging of this oxygen metabolite intracellularly. Indeed, intracellular concentrations of up to 93 mM pyruvate were detected in aerobic cultures growing at high rates. It is hypothesized that self-generated pyruvate may serve to protect L. lactis strain ATCC 19435 from H2O2.
The effect of oxygen on the growth and metabolism of lactic acid bacteria (LAB) has been studied for several decades. Lactococci are capable of oxygen consumption (2, 17, 36) and contain flavin-type NADH oxidases (2, 11), NADH peroxidases, and superoxide dismutase (SOD) (2, 10, 19, 42). Because of a lack of catalase, hydrogen peroxide might accumulate and is therefore considered to be the primary cause of destruction of cell components under aerobic conditions. It has been reported that accumulation of H2O2 by lactococci occurred especially in poor growth media, e.g., milk, or under growth-limiting conditions and could be avoided by adding catalase, yeast extract, or reducing agents (2, 17, 33) to the medium.
The following reactions between NADH and O2 have been reported to occur in LAB (10, 21, 36, 37):
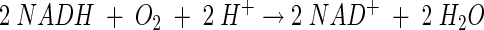 |
(1) |
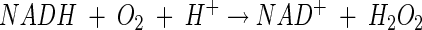 |
(2) |
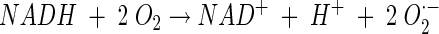 |
(3) |
At least two types of NADH oxidases are found, catalyzing either reaction 1 or reaction 2. Organisms possessing an NADH oxidase that carries out reaction 2 usually also possess an NADH peroxidase to produce water as the final product: NADH + H2O2 + H+ → NAD+ + H2O. Reaction 3 often occurs as a side reaction because the flavin group of NADH oxidase is known to carry out single-electron transfer besides the transfer of two or four electrons (3, 4, 13, 23). Subsequently, SOD catalyzes the dismutation of the superoxide radical as follows: O2·− + O2·− + 2 H+ → H2O2 + O2. Thus, hydrogen peroxide can be formed by NADH oxidase directly or via SOD. However, in general, the generation of H2O2 from O2·− is considered not to be of importance in lactococci (12). Instead, it is believed that H2O2 is directly formed by NADH oxidase and peroxide accumulation is a consequence of very low NADH peroxidase activity. This concluded, more or less, the discussion of the mechanism of oxygen metabolism of lactococci at the end of the 1980s.
During the development of a semidefined medium for our lactococcal strains, we observed that the extent of the influence of oxygen on growth depended on the medium composition. We examined the problem more closely for Lactococcus lactis subsp. lactis ATCC 19435 in a semidefined medium. It appeared that, besides the enzymes NADH oxidase and SOD, pyruvate is also involved in the oxygen metabolism of this strain.
It is well known that pyruvate scavenges H2O2 nonenzymatically according to the following reaction (20): pyruvate + H2O2 → acetate + CO2 + H2O. Fast-growing (aerobic) cultures of L. lactis ATCC 19435 contained significant concentrations of both extracellular and intracellular pyruvate. Some evidence is collected to show that L. lactis ATCC 19435 possesses an alternative mechanism of protection against H2O2 through self-generated intracellular pyruvate. The results signify that this mechanism is flawed at low growth rates, such as during lag phases and growth on poor media.
MATERIALS AND METHODS
Microorganism and media.
L. lactis subsp. lactis ATCC 19435, L. lactis strain MG 5267, and L. lactis strain NZ2007 were stored in litmus milk at −80°C. Strain MG 5267 and its lactate dehydrogenase (LDH)-deficient derivative, strain NZ2007 (26), were obtained from NIZO, Ede, The Netherlands. For cultivations in shake flasks and a fermentor, two semidefined media, SD1 and SD3, were used (Table 1). The vitamin solution consisted of the following (milligrams per liter): biotin, 10; pyridoxal-HCl, 206; folic acid, 100; riboflavin, 100; niacinamide, 100; thiamine-HCl, 100; pantothenate, 95; p-aminobenzoic acid, 10. The trace element solution contained the following (grams per liter): EDTA (disodium), 15; ZnSO4 · 7H2O, 4.5; MnCl2 · 2H2O, 1; CoCl2 · 6H2O, 0.3; CuSO4 · 5H2O, 0.3; Na2MoO4 · H2O, 0.4; CaCl2 · 2H2O, 4.5; FeSO4 · 7H2O, 3; H3BO3, 1; KI, 0.1. Prior to sterilization, the pH of this solution was adjusted to 4.0. The nucleic acid bases were added as a solution in 0.1 N NaOH containing the following (grams per liter): uracil, 2; adenine, 1; guanine, 1. Because the plasmid construct contained resistance to tetracycline, strain NZ2007 required the antibiotic in the medium (1 mg/liter) (26). The solutions of vitamins, trace elements, nucleic acid bases, asparagine, glutamine, yeast nitrogen base (250 g/liter; Difco), glutathione (1 g/liter), and tetracycline (1 g/liter) were filter sterilized (0.2-μm pore size) and added to the media aseptically. Aerobic cultures of 100 ml were grown in 250-ml shake flasks plugged with cotton wool. Anaerobic cultures of 50 ml were grown in 100-ml crimp seal flasks with rubber stoppers. The medium of anaerobic cultures was heated and sparged with N2 gas for 15 min prior to closing of the flasks. After inoculation, the cultures were continuously shaken at 200 rpm in a orbital incubator at 30°C and growth was monitored without pH control. Cultures of 1 liter were grown in a jacketed 3-liter fermentor (Applikon) at a temperature of 30°C. The pH was kept at 6.5 by automatic titration with 250 g of NaOH/liter. The agitation speeds for anaerobic, oxygen-limited, and aerobic cultures were 200, 200, and 600 rpm, respectively. For anaerobic conditions, the headspace was continuously sparged with N2 at a rate of 0.1 liter/min. For oxygen-limited and aerobic conditions, air was sparged over and through the cultures, respectively. The oxygen concentration was measured with a polarographic oxygen electrode. The electrode was calibrated by sparging the medium with air (100% saturation) and with N2 (0% saturation). Under oxygen-limited conditions, no oxygen could be detected, and under aerobic conditions, the concentration was kept between 80 and 90% air saturation. Each growth condition was studied at least in duplicate.
TABLE 1.
Composition of media SD1 and SD3a
| Constituent | Concn (g/liter) or presence in SD1b |
|---|---|
| Glucose | 10 |
| KH2PO4 | 2.5 |
| K2HPO4 | 2.5 |
| MgSO4 · 7H2O | 0.5 |
| NaCl | 0.525 |
| Casamino Acids | 5 |
| Yeast nitrogen basec | 5 |
| Uracil | 0.06 |
| Adenine | 0.03 |
| Guanine | 0.03 |
| Glutathione | 0.01 |
| Vitaminsd | + |
| Trace elementsd | + |
For preparations, see Materials and Methods. The presence of a constituent is indicated by a plus sign.
SD3 is as SD1 but with the addition of 0.4 g of asparagine/liter.
Yeast nitrogen base without amino acids (Difco).
For ingredients, see Materials and Methods.
CE preparation.
At the end of the exponential growth phase, the cultures were harvested and centrifuged at 5,000 × g and 4°C for 10 min. The pellets were washed once and resuspended in 6 ml of 50 mM triethanolamine (TEA) buffer containing 5 mM MgCl2 · 6H2O (pH 7.2). The cell suspensions were mixed with glass beads (1:1, vol/vol) and stored at −20°C. Cell extracts (CE) were prepared by disintegrating the cells by the glass bead method (6 × 30 s of vortexing with 30-s intervals of cooling on ice). Cell debris was removed by centrifugation (10,000 × g, 10°C) for 5 min. The supernatants were kept on ice during analyses.
Enzyme assays. (i) NADH oxidase
. The rate of NADH oxidation in the presence of oxygen was measured spectrophotometrically at 340 nm (ɛ = 6.22 mM/cm). The activity was also determined with an oxygen electrode in a biological oxygen monitor (Strathkelvin Instruments, Glasgow, Scotland). The oxygen concentration was monitored on a paper recorder. The oxygen uptake rates were calculated from the slope of the tangents of the oxygen concentration versus time at various intervals. At the end of the reaction, catalase was added to the enzyme assay mixtures to determine the accumulated H2O2 that was formed during the oxidation of NADH.
(ii) NADH peroxidase.
The rate of NADH peroxidation was measured under anaerobic conditions at 340 nm in anaerobic cuvettes. For this assay, the reaction was started by the addition of H2O2 by injection with a syringe through the rubber stopper (final concentration, 50 to 100 μM). The enzyme activity detection limit of this assay was calculated to be 1 nmol of NADH/min/mg of protein.
(iii) SOD.
SOD activity was assayed as described by McCord and Fridovich (27) but with partially acetylated cytochrome c (0.6 mg/ml assay) and with a cuvette volume of 1 ml.
Detection of O2·− production by NADH oxidase.
O2·− formation by NADH oxidase in CE was studied with an assay based on cytochrome c reduction. For this, the assay for NADH oxidase activity was supplemented with partially acetylated cytochrome c (0.6 mg/ml of assay) and its reduction was monitored spectrophotometrically at 550 nm (ɛ = 19 mM/cm). The reaction was started by addition of 250 μM NADH. A possible reaction between pyruvate and O2·− was checked spectrophotometrically at 550 nm in an assay containing 0.03 U of xanthine oxidase per ml and 48 μM cytochrome c with and without 10 to 20 mM pyruvate. The reaction was started with 0.5 mM xanthine. It was shown that pyruvate did not reoxidize reduced cytochrome c.
Glucokinase (GK).
The initial reaction rate was measured spectrophotometrically at 340 nm by monitoring the reduction of NADP+ (5). One milliliter of reaction mixture consisted of the following: TEA (pH 7.6), 50 mM; MgCl2, 8 mM; glucose, 100 mM; ATP, 0.6 mM; glucose-6-phosphate dehydrogenase, 0.57 U; NADP+, 0.25 mM; CE, 160 mg of protein/liter (to start the reaction). To study the influence of H2O2 on the activity of the enzyme, the CE was incubated with H2O2 (0 to 44 mM) for 10 min prior to addition to the reaction mixture.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
The initial reaction rate was measured spectrophotometrically at 340 nm by monitoring the oxidation of NADH (6). One milliliter of reaction mixture consisted of the following: TEA (pH 7.2), 50 mM; glycerate-3-phosphate, 0.6 mM; ATP, 1 mM; phosphoglycerate kinase, 12 U (1 U = 1 μmol/min); CE, 16 mg of protein/liter; NADH, 0.25 mM (to start the reaction). To study the inactivation of the enzyme by H2O2, the CE was incubated with H2O2 (0 to 4.4 mM) for 10 min prior to addition to the reaction mixture.
Fructose-1,6-diphosphate aldolase.
The initial reaction rate was measured spectrophotometrically at 340 nm by monitoring the oxidation of NADH (7). One milliliter of reaction mixture consisted of the following: TEA (pH 7.2), 50 mM; iodoacetate, 0.3 mM; fructose-1,6-diphosphate, 3.6 mM; LDH, 1 U; glycerophosphate dehydrogenase, 0.28 U; triosephosphate isomerase, 3.6 U; NADH, 0.25 mM; CE, 160 mg of protein/liter (to start the reaction). To study the influence of H2O2 on the enzyme activity, CE was incubated with H2O2 (0 to 440 mM) for 10 min prior to addition to the reaction mixture.
Intracellular pyruvate.
Pyruvate in CE was measured spectrophotometrically (340 nm) by determining the conversion to lactate with NADH in anaerobic cuvettes and using anaerobically prepared solutions of TEA buffer (pH 7.2; 50 mM), NADH (50 mM), and LDH. The last traces of oxygen in the assays were removed by addition of NADH prior to the addition of LDH. All assays were carried out at least in duplicate.
Kinetics.
The kinetics of the reaction of pyruvate with H2O2 has been studied before under conditions physicochemically different from those used here (16). Therefore, the rate constant was measured aerobically at 30°C by determining the conversion of H2O2 at discrete time intervals in 50 ml of water, in TEA buffer (50 mM, pH 7.2), and in 10 ml of TEA buffer plus CE. The rate equation for the disappearance of H2O2 is −rH = kCPCH, where rH is the rate of H2O2 conversion, k is the second-order rate constant, and CP and CH are the concentrations of pyruvate and H2O2, respectively. Integration of this equation gives a relationship between concentration and time. The concentration of pyruvate in our system was much higher than that of H2O2; hence, CP,0 − CP ≅ CP,0 (with CP,0 being the start concentration of pyruvate), and the equation can be simplified to CH = CH,0 · e−kCp,ot, with CH,0 being the starting concentration of H2O2.
Analyses.
Cell growth was monitored by measuring the optical density (OD) at 620 nm spectrophotometrically. Cell mass concentration as dry weight (DW) was analyzed in selected samples as described by Åkerberg et al. (1). The relationship between DW (grams per liter) and OD was determined to be DW = 0.32 · OD − 0.1 g liter−1 for ODs of >0.5. Samples were centrifuged (10,000 × g, 3 min), and the supernatants were analyzed for glucose, lactate, formate, acetate, ethanol, and pyruvate by high-performance liquid chromatography. The compounds were detected with a refractive-index and UV detector after separation at 65°C on an Aminex HPX 87-H column (Bio-Rad Laboratories, Richmond, Calif.). As the mobile phase, 5 mM H2SO4 was used at a flow rate of 0.6 ml/min. All samples were injected twice. Standards were injected separately before the samples. A computer program (Unipoint; Gilson, Middletown, Wis.) was used for quantification by integrating the area under each chromatographic peak. Hydrogen peroxide was determined with 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid) (ABTS; Sigma) at 433 nm (ɛ = 17.1 mM/cm). To 1 ml of sample, 5 μl of ABTS solution (140 g/liter) and 5 μl of horseradish peroxidase (500 U/ml; Boehringer) were added and absorption was read immediately. No reaction with ABTS occurred with any of the components in the medium or CE other than H2O2. The protein concentration of CE was measured as described by Bradford (9) with bovine serum albumin as the standard.
RESULTS
Medium composition.
A semidefined medium (SD1) based on Casamino Acids was developed (Table 1). The quality of L. lactis ATCC 19435 growth was tested by measuring the growth rate of aerobic cultures in shake flasks. Growth on this medium was relatively slow, suggesting that it lacked one or more growth-stimulating compounds. By trial and error, it was found that asparagine (medium SD3; Table 1) shortened the lag phase by a factor of 2 and enhanced the growth rate by a factor of 3 compared to SD1.
Effect of oxygen on growth and product formation.
Further experiments were done with pH-controlled batch cultures. Growth, glucose consumption, product formation, and accumulation of H2O2 were monitored in an aerobic culture in medium SD1 (Fig. 1A and B). The maximum growth rate (μmax) was very low (Table 2), and it took about 100 h to reach the stationary phase (Fig. 1A). Throughout the experiment, no sparging with air was necessary to keep the culture air saturated. After inoculation, H2O2 immediately accumulated in the culture. A maximum of 140 μM H2O2 was found during the mid-exponential phase, after which it declined gradually. Under these conditions, growth was not possible without glutathione, indicating that it protected the cells to a certain extent. In contrast, H2O2 accumulated only temporarily during the lag phase of aerobic cultures on medium SD3 (Fig. 1C).
FIG. 1.

Growth of L. lactis ATCC 19435 and H2O2 accumulation in aerobic pH-controlled batch cultures on glucose in SD1 medium (A) and SD3 medium (C). Glucose consumption and product formation in SD1 (B) and SD3 (D) media. Symbols: ▪, OD of the culture; ○, H2O2; □, glucose; ♦, lactate; ▴, acetate.
TABLE 2.
Effects of additions to medium SD1 on the growth rate during the main exponential growth phase and biomass yield in pH-controlled batch cultures of L. lactis ATCC 19435a
| Medium | Condition | Growth rate (h−1) | DW (g/liter) | Pyruvate concn (mM) |
|---|---|---|---|---|
| SD1 | Aerobic | 0.015 | 0.09 | 0.0 |
| SD1 + catalase | Aerobic | 0.31 | 0.29 | 3.3 |
| SD1 + 5 mM pyruvate | Aerobic | 0.62 | 0.92 | 4.9 |
| SD1 | Anaerobic | 0.66 | 1.02 | 0.0 |
| SD3 | Aerobic | 0.58 | 0.89 | 8.6 |
| SD3 | O2 limitation | 0.75 | 1.02 | 0.0 |
| SD3 | Anaerobic | 0.86 | 1.21 | 0.0 |
For comparison, the results of growth on SD3 are included. The pyruvate concentration in the culture fluid was measured at the end of the exponential growth phase.
Addition of catalase or pyruvate to medium SD1 at the moment of inoculation prevented H2O2 accumulation, and the growth rate increased at least 20-fold (Table 2).
The presence of oxygen also affected the yield of biomass (Table 2) and lactate. On medium SD1, glucose was completely converted to lactate and acetate (Fig. 1B), whereas on medium SD3, less acetate was formed and pyruvate was excreted (Fig. 1D; Table 2). The latter also happened in an aerobic culture on medium SD1 when catalase was present.
Effect of H2O2 on glycolytic enzymes.
Several of the potentially glycolytic enzymes that are generally known to be inhibited by H2O2 were checked, i.e., GK, fructose-1,6-diphosphate aldolase, and GAPDH. The fructose-1,6-diphosphate aldolase of strain ATCC 19435 was not sensitive to H2O2 at concentrations of up to 440 mM, but both GK and GAPDH were inhibited by H2O2. GAPDH was the most sensitive, about 10 times more so than GK, being completely inactivated at a concentration of 2.2 mM H2O2 (Fig. 2).
FIG. 2.

Inactivation of GAPDH (□) and GK (▪) as a function of the concentration of H2O2. CE were incubated for 10 min with H2O2 prior to addition to the assay.
Reduction of oxygen.
The activities of NADH oxidase and NADH peroxidase were examined in CE by using NADH at a concentration of 250 μM. NADH oxidase was present under both aerobic and anaerobic conditions. The activity of NADH oxidase was relatively low in cells grown aerobically on medium SD1 (14.5 nmol of NADH/min/mg of protein). On medium SD3, the activity of this enzyme during the mid-exponential growth phase was three times as high under fully aerobic conditions (426 nmol of NADH/min/mg of protein) as under anaerobic or oxygen-limiting conditions. NADH peroxidase was not detected, implying either that it was not present or that its activities were below the detection limit (<1 nmol of NADH/min/mg of protein).
The initial rates of NADH oxidation and oxygen reduction in air-saturated CE of aerobic (Fig. 3) and anaerobic cultures (results not shown because of similar kinetics) were measured as a function of NADH concentration. The stoichiometry of the reaction was calculated from the ratio of the initial specific rates of NADH oxidation and O2 reduction. The NADH/O2 ratios appeared to be 1.99 (r2 = 0.93) and 2.10 (r2 = 0.99) for aerobic and anaerobic cultures, respectively, indicating that water was the end product. The reaction followed Michaelis-Menten-type kinetics between 25 and 200 μM NADH. The Km values for NADH and O2 were 39 and 5 μM, respectively. H2O2 formation in air-saturated CE increased with the supplied NADH concentration (Fig. 3). NADH oxidase was inhibited neither by NAD+ concentrations of up to 500 μM nor by hydrogen peroxide concentrations of up to 4 mM (results not shown).
FIG. 3.

NADH oxidase activity in air-saturated CE of L. lactis ATCC 19435 cultures grown aerobically in medium SD3 as a function of the NADH concentration. Symbols: ⋄, specific rate of NADH oxidation (nanomoles of NADH per minute per milligram of protein); ▪, specific rate of O2 reduction (nanomoles of O2 per minute per milligram of protein); ○, concentration of H2O2 formed (micromolar).
Sources of oxygen metabolites.
One of the possible sources of H2O2 is the dismutation of O2· − catalyzed by SOD. The culture grown aerobically on SD3 had a SOD activity of 31 μmol of O2· −/min/mg of protein. Nevertheless, it was still possible to detect O2· − through the reduction of acetylated cytochrome c by NADH oxidase-mediated oxidation of NADH in CE. The origin of H2O2 was determined in a second manner with air-saturated CE in the biological oxygen monitor. To scavenge any O2· −, cytochrome c was added to the assay mixture prior to the addition of NADH (250 μM). The oxidation of NADH was followed to completion, and catalase was subsequently added to determine the amount of H2O2 being produced. In the absence of cytochrome c, 20 μM H2O2 was formed (Fig. 4A), but when cytochrome c was present, no H2O2 was detected (Fig. 4B). This indicated that in L. lactis ATCC 19435, H2O2 was produced solely by SOD.
FIG. 4.

Effect of cytochrome c (Cyt. c) on the formation of H2O2 during oxidation of NADH (250 μM) by NADH oxidase in CE of anaerobically grown cells as measured with an oxygen electrode. Panels: A, assay without initial addition of cytochrome c; B, assay with initial addition of cytochrome c. After complete oxidation of NADH, catalase was added to the assay to detect H2O2.
Elimination of H2O2.
Since neither (pseudo)catalase nor NADH peroxidase activity was present in cells of strain ATCC 19435, another mechanism had to be responsible for the disappearance of H2O2 during growth on SD3 (Fig. 1C). Therefore, the rate of disappearance of H2O2 in CE of anaerobically grown cells was studied. Plain addition of 100 μM H2O2 to CE resulted only in a very slow decline of H2O2. Subsequent addition of NADH only increased the H2O2 concentration, confirming the absence of NADH peroxidase activity. The following catabolic metabolites were tested for a reaction with H2O2: lactate, phosphoenolpyruvate, 3-phosphoglycerate, and pyruvate. Only the latter reacted, and its kinetics was subsequently investigated at 30°C by using 100 μM H2O2 and various concentrations of pyruvate (1 to 20 mM). The second-order rate constant of the reaction was determined in TEA buffer (50 mM, pH 7.2). The reaction was about 7.5 times faster in the buffered solution (k = 5.9 · 10−3 min/mM) than in water (k = 7.9 · 10−3 min/mM). The presence of CE (552 mg of protein/liter) had no significant effect, ruling out the involvement of any enzyme. Overall, it implied that the reaction with pyruvate was not mediated by an enzyme.
Intracellular pyruvate concentration.
At high growth rates in the presence of oxygen, L. lactis strain ATCC 19435 excreted relatively high concentrations of pyruvate (Table 2). Under those conditions, the intracellular concentration of pyruvate was as high as 93 mM. When the bacteria were grown on medium SD1, no intracellular pyruvate could be found and thus its concentration remained below the detection limit (<1.2 mM).
Aerobic growth of an LDH− strain.
In an LDH− strain such as L. lactis strain NZ2007, it is expected that intracellular levels of pyruvate are elevated under all growth conditions. Therefore, growth of such mutants will be less restricted by oxygen. Like its parental type strain MG 5267 and strain ATCC 19435, strain NZ2007 contains an H2O-producing NADH oxidase and no NADH peroxidase activity. In the assays with CE of strain NZ2007, low concentrations of H2O2 were detectable (ca. 10 μM). Both the mutant and parental type strains were compared for growth on SD1 and SD3 media under aerobic conditions. Indeed, strain NZ2007 grew well on SD1 in the presence of oxygen and no accumulation of H2O2 in the culture was observed at any time. The μmax (0.24 h−1) was similar to that obtained under anaerobic conditions. Pyruvate was excreted in the early exponential phase up to 4.5 mM. With growth on medium SD3 under aerobic conditions, a μmax of 0.45 h−1 was obtained and no H2O2 accumulated. Pyruvate was excreted up to 4 mM. On the other hand, the parental type strain grew slowly on SD1 (μmax = 0.14 h−1). Growth ceased prematurely, possibly because of accumulation of H2O2 (10 to 13 μM). On medium SD3, growth was fast under air saturation (μmax = 0.88 h−1) and no H2O2 accumulation was observed throughout the fermentation.
DISCUSSION
It is generally thought that H2O2 is the main inhibiting product from oxygen metabolism in LAB (12). H2O2 affected the growth rate strongly since addition of catalase improved growth in the presence of oxygen many-fold (2, 17, 34) (Table 2). The growth rate could also be increased with asparagine, which is a strong growth stimulator for various lactococcal strains (28, 30). The glycolytic enzyme GAPDH of L. lactis subsp. lactis ATCC 19435, like that of mitochondria (22), was very sensitive to H2O2 (Fig. 2). It has been calculated that GAPDH of lactococci possesses a high metabolic control coefficient; the value for growing cells may even reach 0.9 (29). It is therefore dominant in determining glycolytic flux. It can be hypothesized that small quantities of H2O2 might have a strong effect on metabolic flux and, hence, the growth rate (32). However, this should be further supported by investigating the effect of H2O2 on the enzymes in intact cells.
Accumulation of H2O2 in LAB is thought to be due to greater overall activity of oxidases that produce it than peroxidases (2, 12, 36, 37). It has been found frequently that NADH peroxidase activity is about 10 to 30 times lower than that of NADH oxidase. However, L. lactis strain ATCC 19435 did not contain any detectable NADH peroxidase activity under any of the growth conditions studied. This is consistent with the finding that strain ATCC 19435 possesses no gene for that enzyme (42).
According to the stoichiometry, NADH oxidase of strain ATCC 19435 produces water, and assay results showed that O2· − is also formed. It is known that flavin-type NADH oxidases of various organisms, LAB among them, can produce several oxygen metabolites (3, 13, 37). Results of assays that successfully scavenged O2· − (Fig. 3) suggested that H2O2 is formed by SOD in cultures of strain ATCC 19435.
Since strain ATCC 19435 did not contain NADH peroxidase or (pseudo)catalase, another mechanism should be present that is able to destroy H2O2. The differences in growth on the two media suggested that this mechanism is linked to high metabolic fluxes. A similar link was suggested by Smart and Thomas (36), who hypothesized that NADH oxidase should be outcompeted by LDH for NADH when metabolic fluxes are high. However, this is only part of the explanation. It has been found that at high metabolic fluxes, concentrations of various glycolysis intermediates increase to detectable levels (38). Of these compounds, only pyruvate reacted with H2O2 but in a nonenzymatic manner (20).
Under the various growth conditions applied to strain ATCC 19435, extracellular pyruvate accumulated at relatively high concentrations in the presence of oxygen and high metabolic fluxes (Table 2). Similar results have been obtained with other lactococcal strains (36). Under those conditions, the intracellular pyruvate concentration was more than adequate to destroy any H2O2 formed. Sjöberg et al. (35) showed that for strain ATCC 19435 in glucose-limited continuous cultures, the internal concentration of pyruvate increased with the growth rate. Other anaerobically grown lactococcal strains also contained relatively high intracellular concentrations of pyruvate (15, 24). For LDH− strain NZ2007, the metabolic flux does not need to be high for pyruvate to be present at elevated concentrations under all growth conditions. This was exemplified by the excretion of pyruvate even under the worst growth conditions: on SD1 and in the presence of oxygen.
The use of pyruvate as a protective agent has been studied for a long time, especially by chemists and medical biologists (14, 16, 39, 40). Although it has been suggested by Grufferty and Condon (18), nobody has made the link between metabolically generated pyruvate and H2O2 destruction for lactococci. We therefore propose the following scheme of reactions as it might happen in L. lactis ATCC 19435 at high glycolytic fluxes. NADH oxidase reduces oxygen mainly to water, and superoxide is formed as a by-product. Subsequently, superoxide is dismutated to hydrogen peroxide by SOD and metabolically produced pyruvate reacts with the peroxide to form water and acetate. Similar reactions of keto acids with hydrogen peroxide may be present in other organisms (e.g., diacetyl [E. W. J. van Niel and B. Hahn-Hägerdal, unpublished data], a by-product formed in aerobic cultures of several L. lactis strains, especially at high growth rates [25] and NADH oxidase activities [26]). The protection mechanism described here might play a role in the event of the viable-but-nonculturable state of the cell. It is hypothesized that a process of cellular self-destruction caused by oxygen metabolites in an excess of oxidation brings about nonculturability (8), e.g., during inoculum preparation. Indeed, protection by pyruvate has been observed with nonculturable cells (41) and against apoptosis (31).
Acknowledgments
We acknowledge the excellent technical assistance of E. Behtoye and C. Larsson.
We acknowledge the Swedish National Board for Industrial and Technical Development for economic support.
REFERENCES
- 1.Åkerberg, C., K. Hofvendahl, G. Zacchi, and B. Hahn-Hägerdal. 1998. Modeling the influence of pH, temperature, glucose and lactic acid concentrations on the kinetics of lactic acid production by Lactococcus lactis ssp. lactis ATCC 19435 in whole-wheat flour. Appl. Microbiol. Biotechnol. 49:682-690. [Google Scholar]
- 2.Anders, R. F., D. M. Hogg, and G. R. Jago. 1970. Formation of hydrogen peroxide by group N streptococci and its effect on their growth and metabolism. Appl. Microbiol. 19:608-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Badwey, J. A., and M. L. Karnovsky. 1979. Production of superoxide and hydrogen peroxide by an NADH-oxidase in guinea pig polymorphonuclear leukocytes. J. Biol. Chem. 254:11530-11537. [PubMed] [Google Scholar]
- 4.Balou, D., G. Palmer, and V. Massey. 1969. Direct demonstration of superoxide anion production during the oxidation of reduced flavin and of its catalytic decomposition by erythrocuprein. Biochem. Biophys. Res. Commun. 36:898-904. [DOI] [PubMed] [Google Scholar]
- 5.Bergmeyer, H. U. (ed.). 1974. Methods of enzymatic analysis, 2nd ed., vol. 1, p. 473. Verlag Chemie GmbH, Weinheim, Germany.
- 6.Bergmeyer, H. U. (ed.). 1974. Methods of enzymatic analysis, 2nd ed., vol. 1, p. 466-467. Verlag Chemie GmbH, Weinheim, Germany.
- 7.Bergmeyer, H. U., and E. Bernt. 1974. Fructose-1,6 diphosphate, p. 1100-1105. In H. U. Bergmeyer (ed.), Methods of enzymatic analysis, 2nd ed., vol. 2. Verlag Chemie GmbH, Weinheim, Germany.
- 8.Bloomfield, S. F., G. S. A. B. Stewart, C. E. R. Dodd, I. R. Booth, and E. G. M. Power. 1998. The viable but non-culturable phenomenon explained? Microbiology 144:1-3. [DOI] [PubMed] [Google Scholar]
- 9.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 10.Britton, L., D. D. Malinowski, and I. Fridovich. 1978. Superoxide dismutase and oxygen metabolism in Streptococcus faecalis and comparisons with other organisms. J. Bacteriol. 134:229-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruhn, J. C., and E. B. Collins. 1970. Reduced nicotinamide adenine dinucleotide oxidase of Streptococcus diacetilactis. J. Dairy Sci. 53:857-860. [DOI] [PubMed] [Google Scholar]
- 12.Condon, S. 1987. Responses of lactic acid bacteria to oxygen. FEMS Microbiol. Rev. 46:269-280. [Google Scholar]
- 13.Fridovich, I. 1970. Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J. Biol. Chem. 245:4053-4057. [PubMed] [Google Scholar]
- 14.Fujita, A., and T. Kodama. 1935. Untersuchungen über Atmung und Gärung pathogener Bakterien. Biochem. Z. 277:17-31. [Google Scholar]
- 15.Garrigues, C., P. Loubiere, N. D. Lindley, and M. Cocaign-Bousquet. 1997. Control of the shift from homolactic acid to mixed-acid fermentation in Lactococcus lactis: predominant role of the NADH/NAD+ ratio. J. Bacteriol. 179:5282-5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giancomenico, A. R., G. E. Cerniglia, J. E. Biaglow, C. W. Stevens, and C. J. Koch. 1997. The importance of sodium pyruvate in assessing damage produced by hydrogen peroxide. Free Radic. Biol. Med. 23:426-434. [DOI] [PubMed] [Google Scholar]
- 17.Gilliland, S. E., and M. L. Speck. 1969. Biological response of lactic streptococci and lactobacilli to catalase. Appl. Microbiol. 17:797-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grufferty, R. C., and S. Condon. 1983. Effect of fermentation sugar on hydrogen peroxide accumulation by Streptococcus lactis C10. J. Dairy Res. 50:481-489. [Google Scholar]
- 19.Hansson, L., and M. H. Häggström. 1984. Effects of growth conditions on the activities of superoxide dismutase and NADH-oxidase/NADH-peroxidase in Streptococcus lactis. Curr. Microbiol. 10:345-352. [Google Scholar]
- 20.Holleman, M. A. F. 1904. Note on the action of oxygenated water on α-ketoacids and 1,2-diketones. Recl. Trav. Chim. Pays-Bas Belg. 23:169-172. [Google Scholar]
- 21.Hoskins, D. D., H. R. Whiteley, and B. Mackler. 1962. The reduced diphosphopyridine nucleotide oxidase of Streptococcus faecalis: purification and properties. J. Biol. Chem. 237:2647-2651. [PubMed] [Google Scholar]
- 22.Hyslop, P. A., D. B. Hinshaw, W. A. Halsey, Jr., I. U. Schraufstätter, R. D. Saurheber, R. G. Spragg, J. H. Jackson, and C. G. Cochrane. 1988. Mechanisms of oxidant-mediated cell injury. J. Biol. Chem. 263:1665-1675. [PubMed] [Google Scholar]
- 23.Imlay, J. A., and I. Fridovich. 1991. Assay of metabolic superoxide production in Escherichia coli. J. Biol. Chem. 266:6957-6965. [PubMed] [Google Scholar]
- 24.Lohmeier-Vogel, E., M. Häggström, H.-B. Wittgren, and B. Hahn-Hägerdal. 1983. Levels of metabolic intermediates in Streptococcus lactis grown on different carbon sources and the effect on product formation. Acta Chem. Scand. B37:751-753.
- 25.Lopez de Felipe, F., M. J. C. Starrenburg, and J. Hugenholtz. 1997. The role of NADH-oxidation in acetoin and diacetyl production from glucose in Lactococcus lactis. FEMS Microbiol. Lett. 156:15-19. [Google Scholar]
- 26.Lopez de Felipe, F., M. Kleerebezem, W. M. de Vos, and J. Hugenholtz. 1998. Cofactor engineering: a novel approach to metabolic engineering in Lactococcus lactis by controlled expression of NADH oxidase. J. Bacteriol. 180:3804-3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCord, J. M., and I. Fridovich. 1969. Superoxide dismutase: an enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244:6049-6055. [PubMed] [Google Scholar]
- 28.Niven, C. F., Jr. 1944. Nutrition of Streptococcus lactis. J. Bacteriol. 47:343-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poolman, B., B. Bosman, J. Kiers, and W. N. Konings. 1987. Control of glycolysis by glyceraldehyde-3-phosphate dehydrogenase in Streptococcus cremoris and Streptococcus lactis. J. Bacteriol. 169:5887-5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poolman, B., and W. N. Konings. 1988. Relation of growth of Streptococcus lactis and Streptococcus cremoris to amino acid transport. J. Bacteriol. 170:700-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramakrishnan, N., R. Chen, D. E. McClain, R. Bunger. 1998. Pyruvate prevents hydrogen peroxide-induced apoptosis. Free Radic. Res. 29:283-295. [DOI] [PubMed] [Google Scholar]
- 32.Ramasarma, T. 1990. H2O2 has a role in cellular regulation. Ind. J. Biochem. Biophys. 27:269-274. [PubMed] [Google Scholar]
- 33.Selby Smith, J., A. J. Hillier, and G. J. Lees. 1975. The nature of the stimulation of the growth of Streptococcus lactis by yeast extract. J. Dairy Res. 42:123-138. [DOI] [PubMed] [Google Scholar]
- 34.Sevag, M. G., and L. Maiweg. 1934. The respiration mechanism of Pneumococcus III. J. Exp. Med. 60:95-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sjöberg, A., I. Persson, M. Quednau, and B. Hahn-Hägerdal. 1995. The influence of limiting and non-limiting growth conditions on glucose and maltose metabolism in Lactococcus lactis ssp. lactis strains. Appl. Microbiol. Biotechnol. 42:931-938. [Google Scholar]
- 36.Smart, J. B., and T. D. Thomas. 1987. Effect of oxygen on lactose metabolism in lactic streptococci. Appl. Environ. Microbiol. 53:533-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas, E. L., and K. A. Pera. 1983. Oxygen metabolism of Streptococcus mutans: uptake of oxygen and release of superoxide and hydrogen peroxide. J. Bacteriol. 154:1236-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thompson, J., and T. D. Thomas. 1977. Phosphoenolpyruvate and 2-phospoglycerate: endogenous energy source(s) for sugar accumulation by starved cells of Streptococcus lactis. J. Bacteriol. 130:583-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varma, S. D., P. S. Devamanoharan, and A. H. Ali. 1998. Prevention of intracellular oxidative stress to lens by pyruvate and its ester. Free Radic. Res. 28:131-135. [DOI] [PubMed] [Google Scholar]
- 40.Vásquez-Vivar, J., A. Denicola, R. Radi, and O. Augusto. 1997. Peroxynitrite-mediated decarboxylation of pyruvate to both carbon dioxide and carbon dioxide radical anion. Chem. Res. Toxicol. 10:786-794. [DOI] [PubMed] [Google Scholar]
- 41.Wai, S. N., Y. Mizunoe, A. Takade, S. Yoshida. 2000. A comparison of solid and liquid media for resuscitation of starvation- and low-temperature-induced nonculturable cells of Aeromonas hydrophila. Arch. Microbiol. 173:307-310. [DOI] [PubMed] [Google Scholar]
- 42.Zitzelsberger, W., F. Götz, and K. H. Schleifer. 1984. Distribution of superoxide dismutases, oxidases, and NADH peroxidase in various streptococci. FEMS Microbiol. Lett. 21:243-246. [Google Scholar]