

Abstract
Phosphatidylinositol 4, 5-bisphosphate (PIP2) at the inner leaflet of the plasma membrane has been proposed to locally regulate the actin cytoskeleton. Indeed, recent studies that use GFP-tagged pleckstrin homology domains (GFP-PH) as fluorescent PIP2 sensors suggest that this lipid is enriched in membrane microdomains. Here we report that this concept needs revision. Using three distinct fluorescent GFP-tagged pleckstrin homology domains, we show that highly mobile GFP-PH patches colocalize perfectly with various lipophilic membrane dyes and, hence, represent increased lipid content rather than PIP2-enriched microdomains. We show that bright patches are caused by submicroscopical folds and ruffles in the membrane that can be directly visualized at ∼15 nm axial resolution with a novel numerically enhanced imaging method. F-actin motility is inhibited significantly by agonist-induced PIP2 breakdown, and it resumes as soon as PIP2 levels are back to normal. Thus, our data support a role for PIP2 in the regulation of cortical actin, but they challenge a model in which spatial differences in PIP2 regulation of the cytoskeleton exist at a micrometer scale.
INTRODUCTION
Phosphatidylinositolbisphosphate (PIP2) controls many cell processes, ranging from channel gating (Hilgemann and Ball, 1996) to vesicle trafficking (De Camilli et al., 1996) and the actin cytoskeleton. In this latter case, PIP2 is thought to directly interact with actin binding proteins, influencing the equilibrium of monomeric (G-) to filamentous (F-) actin at the level of G-actin availability (e.g., plectin; Andra et al., 1998, profilin; Lassing and Lindberg, 1985, 1988) and at the level of polymerization into actin fibers. Examples are members of the F-actin–severing and –capping protein family including gelsolin and CapG (Sun et al., 1997), and protein β2 (DiNubile and Huang, 1997). Cytoskeletal interactions with integral membrane proteins can also be regulated by PIP2, as is the case with ezrin binding to ICAM and CD44 (Heiska et al., 1998). Many of these actin binding proteins interact with PIP2 via pleckstrin homology domains (Kavran et al., 1998; Wang et al., 1999).
The majority of data on the interaction of PIP2 with the cytoskeleton stem from in vitro binding studies and lipid biochemistry. If membrane lipids are to exert a local signaling function, local differences in content or availability must exist. However, although the phosphoinositide levels at the membrane appear to be tightly regulated by a multitude of specific reactions (Sakisaka et al., 1997; for review see Takenawa et al., 1999), little is known about their distribution along the membrane. Biochemical studies have reported the existence of separate PIP2 pools within cells (Koreh and Monaco, 1986; Varnai and Balla, 1998), and PIP2 was reportedly enriched in detergent-insoluble membrane fractions (rafts, caveolae; Hope and Pike, 1996; Laux et al., 2000), although a very recent electron microscopy study challenges these results (Watt et al., 2002). In addition to these biochemical reports, a limited number of in vivo studies have implicated PIP2 levels in the regulation of the cytoskeleton. For example, increasing [PIP2] by overexpression of the type I PIP-kinase α lead to an increase of stress fibers in CV1 cells (Yamamoto et al., 2001), and sequestering of this lipid using a membrane-permeable PIP2-binding peptide blocked motility (Cunningham et al., 2001).
A detailed understanding of a role for PIP2 as a local signal requires techniques to study cellular processes with spatial and temporal resolution in single living cells. Recently, an approach to image PIP2 in living cells was pioneered in the labs of Meyer and Balla (Stauffer et al., 1998; Varnai and Balla, 1998). Both groups used the PIP2-binding pleckstrin homology domain of PLCδ1, fused to GFP (GFP-PH), to study PIP2 in the membrane in vivo. In resting cells, GFP-PH is bound to the membrane, and it translocates to the cytosol after agonist-induced PIP2 hydrolysis. Translocation can be detected at very high sample rate using fluorescence resonance energy transfer (van der Wal et al., 2001). Imaging of GFP-PH bound to the membrane also provides spatial resolution. Interestingly, initial studies reported a rather uniform distribution of GFP-PH along the membrane in unstimulated cells (Stauffer et al., 1998), whereas a recent study (Tall et al., 2000) reported that GFP-PH displays distinct bright patches on a uniformly labeled background. These bright patches are highly dynamic and rich in F-actin content, and they often colocalize with membrane ruffles and microvilli-like structures. Development of new membrane ruffles was reported to start with local concentration of GFP-PH. Based on these observations, and in line with the hypothesized function of PIP2 as a local regulatory factor in cytoskeletal dynamics, GFP-PH patches were interpreted as local PIP2 enrichments (Tall et al., 2000).
This study was undertaken to determine to what extent local differences in membrane PIP2 content influence the cytoskeleton in vivo. To this goal, we investigated 1) whether changes in PIP2 level induced by physiological PLC-activating agonists modulate cortical actin dynamics and 2) whether physiologically relevant differences in membrane [PIP2] (which, be it relative enrichments or depletions, will here be collectively termed PIP2 patches) exist locally at the plasma membrane of cultured cells.
MATERIALS AND METHODS
Materials
1-Oleoyl-LPA, endothelin, bradykinin, histamine, phenylarsine oxide, and quercetin were from Sigma Chemical Co. (St. Louis, MO), and ionomycin and neurokinin A were from Calbiochem-Novabiochem Corp. (La Jolla, CA). H2O2 was from Riedel-deHaën (Germany), and thrombin receptor activating peptide (SFLLRN) was synthesized in house. Membrane dyes were from Molecular Probes Inc. (Eugene, OR); listed are the dye name, with the structures in parentheses: diphenyl DiI (1,1′-dioctadecyl-5,5′-diphenyl-3,3,3′,3′-tetramethylindocarbocyanine chloride); SP-DiIC18(3) (1,1′-dioctadecyl-6,6′-di(4-sulfophenyl)-3,3,3′,3′-tetramethylindocarbocyanine); SP-DiOC18(3) (3,3′-dioctadecyl-5,5′-di(4- sulfophenyl) oxacarbocyanine, sodium salt); DiIC18(5) oil (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate); TRITC DHPE (N-(6-tetramethylrhodaminethiocarbamoyl) -1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt); bis-BODIPY FL C11-PC (1,2-bis-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-undecanoyl)-sn-glycero-3-phosphocholine); BODIPY 564/570 C11 (4,4-difluoro-5-styryl-4-bora-3a,4a-diaza-s-indacene-3-undecanoic acid); NBD-PA (7-nitrobenz-2-oxa-1,3-diazol-PA).
Constructs and Transfection
The pcDNA3 expression vectors with inserts eGFP-PH(PLCδ1), eCFP-PH(PLCδ1), eYFP-PH(PLCδ1), and eGFP-CAAX were described elsewhere (van der Wal et al., 2001). pEYFP-Mem was from Clontech (Palo Alto, CA), pcDNA3 with insert GFP-Actin (from the N-terminus: GFP, flexible linker [GGGLDPRVR] and actin) was obtained from Dr. J. Neefjes, Division of Tumor Biology, and vectors containing the endothelin B receptor, and a human NK2 receptor with C-terminal truncation at position 328 (Alblas et al., 1995) were obtained from Dr. W. Moolenaar, Division of Cellular Biochemistry at our institute. Constructs were transfected using calcium phosphate precipitate, at ∼0.8 μg DNA/well. After transfection for 12 h, cells were washed with fresh medium and incubated for 4–24 h until usage.
Cell Culture and Stimulation
N1E-115 neuroblastoma cells and NIH-3T3 fibroblast cells were seeded in six-well plates at ∼25.000 cells per well on 25-mm glass coverslips and cultured in 3 ml DMEM supplemented with 10% serum and antibiotics. Agonists and inhibitors were added from concentrated stock solutions. It was verified that the PIP kinase inhibitors PAO, quercetin and H2O2 did not noticeably affect cell viability over the time course of the experiments.
Determination of PLC-mediated PIP2 Breakdown by Fluorescence Resonance Energy Transfer
Monitoring of dynamics of PLC activation with FRET was described in detail elsewhere (van der Wal et al., 2001). In brief, cells were transiently transfected with YFP-PH and CFP-PH, at 1:1 ratio. When bound to PIP2 at the membrane, these constructs are in close proximity and show FRET; upon PIP2 hydrolysis, CFP-PH and YFP-PH dilute out into the cytosol and FRET ceases. Excitation of CFP-PH was at 425 ± 5 nm, and emission was collected simultaneously at 475 ± 15 (CFP) and 540 ± 20 nm (YFP). FRET was expressed as ratio of CFP to YFP signals, and changes were expressed as percent deviation from the initial value.
Confocal Microscopy
For imaging, coverslips with cells were transferred to a culture chamber and mounted on an inverted microscope. All experiments were performed in bicarbonate-buffered saline (containing in mM: 140 NaCl, 5 KCl, 1 MgCl2, 10 glucose, and 10 HEPES Ca2+), pH 7.2, kept under 5% CO2, at 37°C. Confocal imaging was with a DM-IRBE inverted microscope fitted with TCS-SP scanhead (Leica, Mannheim, Germany). Excitation of eGFP, eYFP, DiO, NBD-PA, and BODIPY-FL was with the 488-nm laserline, and emission was collected at 500–560 nm. For DiI, Bodipy 564/570, and TRITC, excitation was with 568 nm, and emission was collected at 590–650 nm. DiD was excited at 633 nm, whereas emission was collected at 645–700 nm. Cross-talk between channels was checked and where necessary corrected using Leica Confocal Software.
Image Analysis
Timelapse Analysis.
For time lapse studies, series of confocal images were taken at 5–30-s time intervals and stored on harddisc. Visualization and analysis was performed off-line using TCS and Qwin software (Leica) and a suite of analysis routines that were written by one of the authors using the APL+Win development platform (APL2000 Inc., Bethesda, MD), as detailed below.
Motility Assay.
To detect motility of actin and GFP-PH patches, pairs of images from a stored timelapse series (8-bit grayscale values) were analyzed essentially for correlation of pixel intensities, using the following steps: (i) Within a series of N images (1…N), pairewise comparison was carried out for images (1 with J + 1), (2 with J + 2),… ([N − J] with N), with J selected to obtain a lapse of 30–120 s between the image pair, which appeared optimal for the detection of changes. (ii) Analysis of movement is best understood by referring to Figure 1A, lower scatterplot. Pixels that are intensely fluorescent in the first but not in the second image appear in the scatterplot below the blue diagonal line. Slope and abscissa of this line can be set for optimal rejection of unaltered pixels; in addition, a threshold can be included for rejection of background noise. If If is the intensity of a given pixel in the first image, and Is that in the second image, then for these pixels,
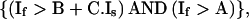 |
1 |
where A is the parameter that determines detection threshold (background; usually <30), B the X-abscissa (range, 5–40) and C the slope (range, 1–1.5). Similarly, for pixels above the red diagonal
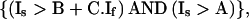 |
2 |
where B is the Y-abscissa. The degree of dissimilarity DD is then calculated as
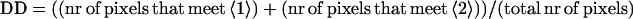 |
3 |
(iii) Finally, motility traces are constructed by plotting corresponding DD versus image index for (1…[N − J]). The reproducibility and sensitivity of this algorithm were checked using simulated and real data. Further details are available upon request from K.J.
Figure 1.

Influence of agonist-induced PLC activation on cortical actin dynamics. (A) Detection of actin motility using intensity scatter plot. Time-lapse series of confocal images of the basal membrane of GFP-actin expressing N1E-115 cells were collected. For each pair of subsequent images X and Y, intensities of each pixel were plotted as dots in a (X, Y) scatterplot (see MATERIALS AND METHODS for further details). Multiple occurrence of identical coordinates is color-coded. For a pair of images collected 2 s apart, the scatterplot shows a distribution along the diagonal (left scatterplot), with some divergence due to the inherent photon noise. For pairs of images taken at longer time intervals (top panel, t = 0 and 2 min), bright actin structures that have moved in between collection of the images will be apparent as off-diagonal clusters of dots (right scatterplot). Dots within the blue-dashed area in this plot represent pixels of which the intensity has significantly decreased in 2 min; the red-dashed area contains all pixels whose intensity has increased after 2 min. Taking a fixed time interval (usually 30–120 s), actin motility as a function of time can be quantified extremely sensitively by calculating the fraction of pixels that are off-diagonal for subsequent images in a time-lapse series. In the composite (rightmost) photomicrograph, pixels from the dashed areas are superimposed on the image. Scale bar, 0.5 μm. (B–F) Comparison of agonist-induced PIP2 hydrolysis (left panels) and actin motility (right panels) in separate N1E-115 cells. Shown are responses from single cells (B) expressing the endothelin B receptor and stimulated with 20 nM endothelin (ET); (C) stimulated with 10 μM histamine (HIS); (D) stimulated with 1 μM bradykinin (BK); and (E) expressing a desensitization-defective neurokinin A receptor and stimulated with 1 μM neurokinin A (NKA) and (F) pretreated with 1 μM phenylarsine oxide (PAO) for 5 min and stimulated with 1 μM bradykinin (BK). Red marks indicate the moment of agonist addition.
Imaging of Reconstructed Axial PSF (RAP Imaging).
Stacks of X/Y images of the basal membrane were captured at 40- or 80-nm axial distance, using a 63×, 1.32 NA oil immersion Planapochromatic objective and a pinhole setting of 1 airy disk. Excitation was at 488 nm, and emission was collected at 525 nm. Because the axial resolution at these conditions is ∼1.05 μm, the objective point spread function (PSF; i.e., the gaussian intensity profile that is detected when a true point source is imaged with an objective) is thus oversampled up to 25 times (see Figure 5). Before processing, individual images were smoothed once. For each pixel (x,y), plotting its intensity in image 1 to n of the stack of n images (I1…In) versus axial position reconstructs the axial PSF, because the thickness of the membrane (∼5 nm) can be ignored. The “center of intensity” of the axial fluorescence intensity profiles was then determined by a calculation-efficient, simplified fitting algorithm as follows. First, image numbers were sorted and arranged in order of decreasing intensity. Then, these numbers were multiplied by an array of n weighting values (W1…Wn). Values for W were chosen to progressively reduce the influence of out-of-focus (dim) images (see below). The sum P of these products is proportional to the axial position of the center of intensity. For example, be the order of intensity for a given (x,y) pixel:
 |
then the expression for P is:
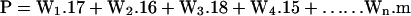 |
Three-dimensional surface profiles (see Figure 5) were then constructed by plotting P as a function of x and y, using the Surface Plotter plugin of the public domain software ImageJ, version 1.24t (Wayne Rasband, National Institutes of Health, Bethesda, MD).
Figure 5.

Direct visualization of subresolution membrane folds at the patches. (A) Stack of confocal XY-images, taken 80 nm apart in Z direction, showing the basal membrane of GFP-CAAX expressing N1E-115 cell. (B) Using image analysis software, an area was manually assigned within (blue) and just outside (red) a patch, and applied to the entire image stack. Scale bar, 2 μm. (C) Plotting the measured mean intensity within the blue and red area versus the image number in the stack reconstructs the axial pointspread function of the objective. The PSF (full-width at half-maximum) of the used Leica 63× oil 1.32 NA planapochromatic objective is 1.05 μm, at a confocal pinhole setting of 0.58 μm and at emission λ = 525 nm. By analysis of the offset of the normalized curves, the Z-position of the membrane can be estimated with high precision. (D) The three-dimensional surface profile of a patch is visualized with ∼15 nm resolution by applying this analysis on a point-by-point basis to the image stack.
The array of weighting values W was chosen so that
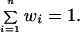 |
A trivial example is the case where W1 = 1, and W2…Wn = 0. In this case, P will simply be the image number of the highest pixel intensity. Because of the large Poisson-distributed photon noise in the confocal images, this results in extremely noisy surface plots. Good results were obtained with linear weighting arrays that emphasize the images of highest intensity, such as the array Wi = 0.14, 0.13, 0.12, 0.11, 0.10, 0.09, 0.08, 0.07, 0.06, 0.05, 0.03, 0.02, 0, 0, 0, …, 0. It should be noted that the described calculation-effective algorithm performed equally well as pixel-by-pixel curve-fitting algorithms that took dramatically longer computer processing. P was calibrated numerically as well as experimentally, using a slightly tilted mirror as the object. It is estimated that a Z-axis resolution up to ∼15 nm is obtained. Further details are available on request from K.J.
Membrane Staining
Membrane dye stocks were mixed by vigorous pipetting with bicarbonate-buffered saline to a final concentration of 1 μM. Cells were incubated with the mix for 5–15 min at 37°C. Before imaging, cells were washed three times with bicarbonate-buffered saline.
Fluorescence Recovery after Photobleaching
For fluorescence recovery after photobleaching (FRAP) experiments, cells were imaged using a Leica TCS-SP confocal microscope equipped with 63× (NA 1.3) oil immersion objective. Spots were bleached with the 488-nm argon laser line (Bis-Bodipy FL C11-PC; 0.2s) or 568-nm krypton laser line (DiI, 0.2 s), and recovery was sampled at 10 Hz. Data were corrected for slight (<5%) background bleaching and fitted with single exponents using Clampfit software (Axon Instruments, Union City, CA).
RESULTS
Cortical Actin Motility Correlates with Agonist-induced PIP2 Breakdown
We recently reported the use of a FRET-based assay to monitor the kinetics of receptor-mediated PIP2 breakdown in single cells (van der Wal et al., 2001). We showed that distinct G protein–coupled receptors (GPCRs) induce PIP2 hydrolysis with their own characteristic kinetic profiles. Representative examples are depicted in Figure 1, B–F (left traces). To establish the relationship between membrane [PIP2] and actin dynamics, we set out to study the actin cytoskeleton under identical conditions. N1E-115 cells do not possess actin stress fibers, but the cortical actin cytoskeleton is well developed, and it mediates agonist-induced cell shape changes (Jalink et al., 1993; van Leeuwen et al., 1999). Cortical actin dynamics were studied by in vivo time-lapse imaging of cells that express GFP-tagged actin. GFP-actin at the cell cortex displays an inhomogeneous, patchy distribution (Figure 1A, top panel), and it is highly dynamic, with individual structures showing seemingly random as well as directed movements. Strikingly, after addition of PIP2-hydrolyzing agonists, such as endothelin and neurokinin A, actin movements are inhibited within a minute.
To study these effects in more detail, we set up an assay for the quantification of GFP-actin dynamics. Essentially, in this assay motility is expressed as change (lack of correlation) between successive images in a time-lapse series (see the legend to Figure 1 and MATERIALS AND METHODS). We compared the kinetics of actin dynamics to those of the concomitant PIP2 decreases after agonist addition. The strong PLC activator endothelin (ET, Figure 1B) causes transient retardation of actin dynamics that correlates well with the decrease in membrane PIP2 content (representative result of 5 experiments). Agonists that induce weaker PIP2 hydrolysis, such as histamine, LPA, and thrombin (van der Wal et al., 2001), caused less pronounced or undetectable inhibition of actin motility (n = 9; Figure 1C). Bradykinin, which induces a short-lived drop in [PIP2] caused a minor and transient drop in actin motility (n = 6; Figure 1D), whereas the sustained PIP2 hydrolysis evoked by a desensitization-defective mutant of the NKA receptor (Alblas et al., 1995; van der Wal et al., 2001) correlates with prolonged immobilization of cortical actin (Figure 1E, n = 6). These results show that cortical actin dynamics correlate well with membrane PIP2 content, providing evidence for the causal relationship that was hypothesized in the recent literature. To further address that actin motility changes are secondary to PIP2 hydrolysis, we inhibited PIP2 resynthesis by blocking PIP kinases with a low dose (1–4 μM) of phenylarsine oxide (PAO). As shown in Figure 1F, pretreatment with this drug did not influence basal [PIP2] or actin motility, but it completely blocked recovery of PIP2 to basal levels after BK-induced PLC activation (van der Wal et al., 2001). This was paralleled by persistent reduction of actin dynamics. Similar observations were made with the PIP kinase inhibitor quercetin, which blocks ATP binding to the kinase domain, and with H2O2 (Mesaeli et al., 2000; J. Halstead and N. Divecha, personal communication), which is thought to disrupt PIPkinase function by modifying critical thiol groups outside the ATP binding domain.
GFP-PH Distribution at the Plasma Membrane Is Not Homogenous
It has been hypothesized (DiNubile and Huang, 1997; Honda et al., 1999; Lanier and Gertler, 2000; Tall et al., 2000) that local differences in membrane PIP2 content may transmit extracellular signals into local cytoskeletal changes. Do such local enrichments or depletions in PIP2 content (PIP2 patches; note that although for brevity we will often mention “PIP2 enrichments” our analyses were equally focused on PIP2 enrichments and decreases) at the plasma membrane exist? To study the spatial distribution of PIP2, cells expressing a GFP-tagged pleckstrin homology domain derived from PLCδ1 were imaged on the confocal microscope. We noted that GFP fluorescence along the plasma membranes does not appear to be homogenous. Rather, in several cell types, including N1E-115 mouse neuroblastoma cells (Figure 2A), NIH-3T3 mouse fibroblasts and HEK293 human embryonic kidney cells, GFP fluorescence shows distinct bright patches that are two- to threefold more intense than the rest of the membrane. Bright patches can be observed in medial sections through the cells (top left panel), where they often colocalize with membrane ruffles and lamellae. Patches are also particularly apparent in the basal membranes of cells grown on coverslips, usually displaying a slender, elongated shape (bottom left panel). Similar observations were recently reported by Tall et al. (2000).
Figure 2.

Colocalization of GFP-PH and DiI. Living N1E-115 cells expressing GFP-PH were stained with the lipophilic membrane dye DiI. (A) Images of the fluorescent protein and DiI were collected from both medial (top panels) and basal (bottom panels) sections, using a confocal microscope. Scale bar, 5 μm. (B) Pixel intensity for DiI was plotted against GFP-PH intensity in a scatter plot. Note the difference from the scatter plots in Figure 1, where one fluorophore is imaged at two points in time.
When imaged in living cells these patches appear highly dynamic: over time, individual patches may disappear, show directed movements, and occasionally branch. These structures further colocalize with F-actin but not with vinculin or other components of focal adhesions. Because many actin-binding proteins can interact with PIP2 in vitro, GFP-PH patches were interpreted to represent local concentrations of PIP2 (Tall et al., 2000). However, alternative explanations for the local concentration of GFP-PH have not been addressed.
GFP-PH Staining Pattern Reflects Membrane Content
We set out to address the possibility that bright GFP-PH patches reflect local increases in membrane area, due to local membrane folding. Cells expressing GFP-PH were stained with the lipophilic membrane dye diphenyl-DiI and simultaneously imaged for GFP and dye fluorescence on a confocal microscope. Images were collected at the basal membrane and at medial sections through the cell. Strikingly, we observed strong colocalization of GFP and DiI fluorescence in all sections in N1E-115 cells (Figure 2A), NIH-3T3 cells, and several other cell types. A colocalization analysis was carried out by constructing scatter plots to compare pixel intensities of GFP-PH and DiI (Figure 2B). In these plots, any structures present in the GFP-PH, but not in the DiI image, will be apparent as off-diagonal clusters of dots. No evidence was found for GFP-PH enrichment beyond the level predicted by lipid mass as detected by DiI fluorescence (>35 images analyzed). Conceivably, however, the DiI dye might localize preferentially to places enriched in PIP2 or F-actin. We therefore used a panel of different membrane markers with widely different physicochemical properties, including lipophilic dyes that intercalate in the lipid doublelayer, phospholipids with fluorescently labeled acyl chain or headgroup, and fluorescent proteins, targeted to the membrane with lipid anchors (Table 1). All membrane markers colocalized with GFP-PH, strongly suggesting that patches represent sites with increased membrane content.
Table 1.
Membrane markers used in colocalization studies
| Membrane anchor | Dye | Fluorophore | Colocalization with GFP-PHa |
|---|---|---|---|
| Octadecyl (C18) | Diphenyl DiI | Diphenyl indocarbocyanines | + |
| SP-DiIC18(3) | Sulfophenyl indocarbocyanines | + | |
| SP-DiOC18(3) | Sulfophenyl oxacarbocyanine | +b | |
| DiIC18(5) | Long λ-ex. Indocarbocyanine | + | |
| Fatty Acid (C11) | BODIPY 564/570 C11 | Bodipy 564/570 | + |
| Lipid, Headgroup labeled | TRITC DHPE | TRITC | + |
| Lipid, Acyl labeled | Bis-BODIPY FL C11-PC | Bodipy FL | +b |
| NBD-PA | NBD | +b | |
| Palmitate | YFP-MEM | Yellow fluorescent protein | +b |
| Geranyl-geranyl | GFP-CAAX | Green fluorescent protein | +b |
Cells were loaded with indicated dyes or transfected with membrane-targeted GFP or YFP as detailed in MATERIALS AND METHODS.
Colocalization was assessed directly by simultaneous imaging of the indicated marker and GFP-PH (+) or, for dyes with green emission, indirectly by comparing the pattern of the indicated marker to that of DiI (+b).
Strong colocalization of GFP-PH with membrane markers was not restricted to the PH domain from PLCδ1, because similar observations were obtained using the PIP2-specific PH domain derived from PLCδ4, and a PIP2-specific mutant (E41K) derived from the PH domain of Bruton's tyrosine kinase (Btk; Varnai et al., 1999). Furthermore, bradykinin-induced PLC activation caused GFP-PH patches to disappear, whereas the DiI staining pattern remained unaltered. As shown for medial and basal confocal sections in Figure 3, GFP-PH returned to the exact same sites to colocalize again with the DiI patches after resynthesis of PIP2. Taken together, these data demonstrate that patches enriched in GFP-PH are the consequence of locally increased membrane area (i.e., folds and ruffles), rather than of local PIP2 enrichment.
Figure 3.

Localization of GFP-PH and DiI during PIP2 hydrolysis. N1E-115 cells expressing GFP-PH were stained with DiI and imaged on the confocal microscope in medial (A) and basal (B) sections. Bradykinin (BK, 1 μM) induced PIP2 breakdown, resulting in translocation of GFP-PH. After resynthesis of PIP2 (at 120 s), GFP-PH returned to the membrane. In contrast, DiI patches (see arrow) remain at the membrane during PIP2 hydrolysis. Scale bar: (A) 5 μm, (B) 1 μm.
Membrane Folds at GFP-PH Patches Can Be Directly Visualized
Although the above results indicate that GFP-PH patches are sites of membrane folding, detailed confocal imaging studies failed to directly visualize folds in a considerable subset of the patches. This is perhaps not surprising, because the resolution of the confocal microscope is limited by the objective point spread function (PSF), this is the Gaussian intensity profile that is detected when a true point source is imaged with an objective. The PSF of the best objectives are close to two orders of magnitude larger than the thickness (5 nm) of the lipid bilayer. Two types of experiments were performed to investigate whether subresolution membrane folds cause the bright fluorescence in all membrane patches.
First, cells expressing GFP-PH were swollen by a hypotonic shock. The medium was diluted from 350 mOsmol to a final value of 120 mOsmol, while confocal images were continuously collected (Figure 4A). This caused an increase in cell volume of 40–50% that led to straitening out of the membrane with consequent disappearance of bright GFP-PH patches. Disappearance of bright patches was not due to swelling-induced PIP2 hydrolysis, because [PIP2] in these cells remained constant (Figure 4B). These experiments strongly argue that bright fluorescent patches are in fact folds in the lipid bilayer.
Figure 4.

GFP-PH patches disappear after osmotic swelling. (A) Confocal images of NIH-3T3 cells expressing GFP-PH, subjected to hypotonic swelling. Osmolarity of the medium was adjusted from a basal value of 350 mOsmol (1) to 235 (2) and 120 mOsmol (3). Ionomycin, 5 μM, was added (4) to cause complete translocation of the fluorescent proteins. (B) PIP2 degradation was essayed by FRET in a single cell, subjected to the same protocol. Note that although swelling eliminates the patches, total PIP2 in the cell is virtually unaltered. Scale bar, 5 μm.
Second, we set out to directly visualize subresolution membrane folding, using a numerical approach to increase the axial resolution. In these studies, we recorded stacks of images at 40- or 80-nm axial distance, thus oversampling the axial resolution (PSF) of the objective up to 25 times (see Figure 5). The intensity of a small region of interest (ROI) was plotted against the axial position for the images in this stack. Because the thickness of the membrane (∼5 nm) can be ignored, the resulting curve basically reconstructs the axial PSF (Figure 5C). When such reconstructed axial PSFs are compared for ROIs inside (blue mask) and just outside (red mask) of the GFP-CAAX patches, the normalized curves consistently show small offsets, indicating differences in Z-position of the fluorescent membrane. It should be noted that by fitting the PSFs, the Z-axis offset can in fact be estimated with a precision considerably higher that the axial step size (see MATERIALS AND METHODS). By applying this technique on a pixel-by-pixel basis to the image stack, the three-dimensional surface profile of the basal membrane could be visualized with ∼15-nm axial resolution (Figure 5D). In these images, upward and downward protrusions measuring between 15 and 150 nm in the basal membrane are observed (Figure 5D) that correspond to the bright GFP-CAAX patches. Therefore, these data directly demonstrate that patches in effect represent submicrometer peaks and valleys in the landscape of the basal membrane.
Further analysis of these structures is described in the Supplementary Data section, online. It is demonstrated that GFP-PH patches do not represent sites of cell adhesion, such as focal adhesions. In addition, we show that motility of GFP-PH patches is potently inhibited by cytochalasin D and myosin light chain kinase blockers, indicating that patches are the result, rather than the cause, of local actin-dependent forces.
Diffusion Limits the Establishment and Maintenance of PIP2 Gradients
Summarizing the above results, GFP-PH labeling indicates that spatial differences in the concentration of unbound PIP2, at least at a micrometer scale, do not exist in our cells. However, there might be [PIP2] differences on a more global scale, e.g., between the leading and trailing edge of polarized cells or in organelles such as lamellipodia. For 3′-phosphorylated phosphatidylinositols, it has been shown that such gradients can exist in chemotactic and phagocytic cells (Haugh et al., 2000; Servant et al., 2000; Marshall et al., 2001). At what scale can spatial differences in [PIP2] be induced in the plasma membrane of living cells?
Gradients in [PIP2] are the result of local synthesis and breakdown, combined with lateral diffusion of the lipid in the membrane. We studied the diffusion kinetics of bodipy-labeled PIP2 in N1E-115 cells using laser-induced photobleaching (see MATERIALS AND METHODS). As can be seen in Figure 6A, the fluorescence in micrometer-sized photobleached spots in the plasma membrane completely recovered within seconds. Thus, it is likely that at this scale, induced gradients in [PIP2] rapidly dissipate by diffusion. The observed recovery rate is similar to those of other freely diffusible membrane labels, including DiI and bodipy-labeled phosphatidylcholine (Figure 6A). Published diffusion coefficients (D) for these labels average ∼1–2 μm2/s (Yechiel and Edidin, 1987; Fulbright et al., 1997), in reasonable agreement with the value reported recently for PIP3 (0.5 μm2/s; Haugh et al., 2000). These D values therefore predict that gradients in PIP2 at a larger scale may occur. To test whether PIP2 concentration differences can be induced and detected on this scale, we used focal stimulation with Neurokinin A from a micropipette (Figure 6). This caused a rapid initial translocation of GFP-PH at the stimulus site that subsequently spread to the neighboring membrane. In particular in cases where diffusion is restricted, such as in neurites (Figure 6A), sustained local decreases in [PIP2] could reliably be evoked (Figure 6B). Taken together, these observations show that agonists can induce PIP2 differences along the membrane, whereas at a micrometer-scale lateral diffusion limits maintenance of such gradients.
Figure 6.

Local PIP2 gradients are limited by diffusion. (A) Cells were labeled with DiI, bodipy-TR PtdIns(4,5)P2, or bis-bodipy-FL C11-phosphatidylcholine. Spots were bleached at the plasma membrane in neurites (red trace) or the soma (black trace) with a 0.2-s pulse of 488- or 568-nm laser light, and recovery was quantified from confocal images. Note that recovery upon spot photobleaching in neurites (red lines) was reduced by orders of magnitude. (B) N1E-115 cell expressing GFP-PH and a desensitization-defective NK2 receptor was imaged at the indicated time points using a confocal microscope. PLC was stimulated locally in a developing neurite by a precisely confined stream of neurokinin A (NKA) generated by means of an application (A) and a suction pipette (S). The resulting PIP2 degradation was monitored as GFP-PH translocation (see arrow). Similar results were obtained with bradykinin. The intensity of the images is color-coded with the “glow” color look-up table of Leica TCS software. Scale bar, 5 μm.
DISCUSSION
In this study, we used GFP-PH as in vivo PIP2 tag to investigate the hypothesis that local pools of free PIP2 at the plasma membrane (patches) spatially regulate cytoskeletal remodeling. The existence of PIP2 patches and their involvement in the local regulation of cellular physiology (e.g., actomyosin remodeling, vesicle budding, etc.) has been hypothesized by several groups (DiNubile and Huang, 1997; Raucher et al., 2000; Rozelle et al., 2000; Brown et al., 2001; for review see Caroni, 2001; Gillooly and Stenmark, 2001). In addition, some data have been presented that seems to support this notion (Honda et al., 1999; Botelho et al., 2000; Tall et al., 2000; Micheva et al., 2001). At odds with these reports, the detailed spatial analysis presented here indicates, within the limits of optical microscopy (∼200 nm), that in all cell lines checked the GFP-PH labeling pattern simply reflects the amount of membrane rather than local PIP2 enrichment. We have previously shown (van der Wal et al., 2001) that the PH domain from PLCδ1 detects PIP2 (rather than IP3) in vivo and that membrane-bound GFP-PH is in rapid (∼1 s) equilibrium with a cytosolic pool of approximately equal size. Because in the present article it was shown that gradients in [PIP2] could be induced and detected, it is fair to argue that GFP-PH is capable of reporting, at least qualitatively, the local free PIP2 concentration in single, living cells. From our failure to observe micrometer-sized PIP2 patches, we conclude that spatial differences in PIP2 regulation of the cytoskeleton do not exist at this scale.
The above-mentioned contrast between our findings and literature reports deserves further attention. Because this study addressed the hypothesized spatial regulation of the cytoskeleton by PIP2, we used detection by decoration with GFP-PH to visualize the distribution of the free (unbound) pool of this lipid along the membrane. PIP2 that is bound to proteins at the plasma membrane constitutes a second pool that, likely, may display local molar enrichment or gradients due to, respectively, clustering at specific sites or gradients of the PIP2-interacting proteins. In this case, PIP2 enrichments are the result, rather than the cause of protein clustering, and because such enrichments are not available for decoration with GFP-PH, our studies will fail to detect them. However, lipid-biochemical approaches, such as those used to study rafts, obviously would. These rafts, which are detergent-insoluble microdomains in the membrane, contain signal-transducing proteins and may have roles in membrane trafficking and signaling (reviewed by Simons and Ikonen, 1997). The resolution of the here used imaging techniques is not sufficient to investigate rafts (<70 nm; Varma and Mayor, 1998; Pralle et al., 2000). Nevertheless, the observed highly homogeneous distribution of GFP-PH along the membrane suggests that hypothetical rafts are either very abundant and evenly distributed along the membrane or they do not contain excess free PIP2. Reports that rely on labeling of PIP2 with specific antibodies require fixation and permeabilization of the cells (Laux et al., 2000). However, this procedure may well have caused artifacts (Mayor et al., 1994; Kenworthy and Edidin, 1998; Laux et al., 2000). For example, it was observed by Laux and colleagues that the size of PIP2 clusters observed in monkey kidney epithelial cells depends on the fixation method. Furthermore, in a recent electron microscopy study (Watt et al., 2002) that used immunogold labeling of PLCδ1-PH-GST to visualize PIP2 at the membrane, it was shown that lipids are not efficiently fixed with aldehyde at room temperature. Interestingly, these authors showed good labeling results at 0°C but did not observe PIP2 patches at the membrane.
The published literature generally lacks rigorous colocalization analysis to exclude the possibility of “apparent PIP2 enrichment” by membrane folding. In our studies, we used a panel of 10 different membrane stains with widely different physicochemical properties, and we have used three different GFP-tagged PIP2-specific pleckstrin homology domains. Strong colocalization of GFP-PH with lipid dyes was observed in all cell types tested, irrespective of the phase of the cell cycle and of growth conditions (i.e., both when cultured in serum-free and in serum-containing medium). Furthermore, because our data directly contradict the interpretation put forward by Tall et al. (2000), we have addressed this issue by additional, independent approaches. These included experiments that involve hypotonic cell swelling, interference reflection microscopy, and the here-introduced technique of reconstructed axial PSF imaging. Together, the experiments show conclusively that GFP-PH patches represent submicroscopic folds and protrusions in the membrane that are caused by actomyosin-based forces, rather than local PIP2 enrichments. In addition, in vivo FRAP experiments were carried out using fluorescently labeled lipids that incorporated in the plasma membrane after addition to the culture medium. These experiments should be interpreted with some caution, because membrane insertion and possible binding to proteins of the exogenously added PIP2 has not been characterized extensively. With this caveat, the analysis revealed that diffusion of PIP2 occurs freely at a rate comparable to that of phosphatidylcholine, showing that micrometer-sized patches will dissipate within seconds. The recently reported gradients in 3′ phosphorylated phosphoinositides (3′ PI) in chemotactic and phagocytic cells (Haugh et al., 2000; Servant et al., 2000; Marshall et al., 2001) were analyzed by Haugh and coworkers. In a model of local synthesis and breakdown of 3′ PI, the experimentally revealed unhindered diffusion along the membrane (D = ∼0.5 μm2/s) and the lifetime of ∼40 s of 3′ PI were consistent with the gradients observed in vivo. Our FRAP experiments indicate very similar diffusion rates for PIP2. Furthermore, from the observation that agonist-induced PIP2 breakdown and subsequent resynthesis can take place well within 2 min, we conclude that under these conditions the PIP2 lifetime may be short enough to enable buildup of gradients. Taken together, our results challenge the view that PIP2 regulation of the actin cytoskeleton is localized at a micrometer scale. However, on a larger scale and in cases where diffusion is limited, sustained PIP2 gradients may exist for prolonged periods of time.
We also observed that agonist-induced PIP2 changes as detected using the FRET assay correlate well with the dynamics of cortical actin as quantified using the assay that was introduced in this article, both with respect to the magnitude and the time course of the response. A possible delay of the actin response was not detected, although it should be noted that the temporal resolution of the assay is limited to ∼30 s (see MATERIALS AND METHODS). These observations provide experimental evidence for the actin-modulatory role of PIP2 that has been widely hypothesized (e.g., Lassing and Lindberg, 1988; Sun et al., 1997; Andra et al., 1998; Wang et al., 1999), based on the PIP2-binding properties displayed by many actin-binding proteins. Furthermore, this correlation held true when agents were used that circumvent receptor activation to cause PIP2 decrease (the PIPkinase inhibitors PAO and quercetin, and H2O2; Mesaeli et al., 2000; J. Halstead and N. Divecha, personal communication), suggesting that the observed actin changes are not due to signaling events independent from PIP2 metabolism. In support of this viewpoint, a recent report from Cunningham and coworkers showed that incubation of cells with a membrane permeable peptide with potent PIP2-binding activity inhibited cell migration (Cunningham et al., 2001). Indeed, we observed that the transient drop in F-actin dynamics during agonist-induced PIP2 breakdown was mirrored in loss of motility of the leading edge in fibroblasts. Thus, our data show, for the first time, that the PIP2 decreases triggered by agonist-induced PLC activation suffice to influence cortical actin motility in vivo.
Finally, a notable observation is the existence of highly dynamic, actin-rich structures in the basal membrane of nonmigratory cells. These structures represent places of close membrane-substrate proximity but do not appear to relate to sites of cell adhesion. Whereas it is striking that their dynamic behavior appears PIP2 dependent, a possible function awaits future investigations.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Division of Cell Biology and Drs. W. Moolenaar, N. Divecha, and J. Hallstead (Division of Cellular Biochemistry) for stimulating discussions and critical reading of the manuscript. We also thank Drs. T. Balla, T. Meyer, J. Neefjes, and W. Moolenaar for plasmids. This work was supported by NWO grant 901–02-236.
Abbreviations used:
- CFP
cyan fluorescent protein
- FRAP
fluorescence recovery after photobleaching
- FRET
fluorescence resonance energy transfer
- GFP
green fluorescent protein
- GPCR
G protein-coupled receptor
- PH
pleckstrin homology
- PLC
phospholipase C
- PIP2
phosphatidylinositol(4,5) bisphosphate
- PSF
pointspread function
- YFP
yellow fluorescent protein
Footnotes
Online version of this article contains supplementary video and data materials. Online version is available at www.molbiolcell.org.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E02–04–0231. Article and publication date are at www.molbiolcell.org/cgi/doi/10.1091/mbc.E02–04–0231.
REFERENCES
- Alblas J, van Etten I, Khanum A, Moolenaar WH. C-terminal truncation of the neurokinin-2 receptor causes enhanced and sustained agonist-induced signaling. Role of receptor phosphorylation in signal attenuation. J Biol Chem. 1995;270:8944–8951. doi: 10.1074/jbc.270.15.8944. [DOI] [PubMed] [Google Scholar]
- Andra K, Nikolic B, Stocher M, Drenckhahn D, Wiche G. Not just scaffolding: plectin regulates actin dynamics in cultured cells. Genes Dev. 1998;12:3442–3451. doi: 10.1101/gad.12.21.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho RJ, Teruel M, Dierckman R, Anderson R, Wells A, York JD, Meyer T, Grinstein S. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J Cell Biol. 2000;151:1353–1368. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown FD, Rozelle AL, Yin HL, Balla T, Donaldson JG. Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J Cell Biol. 2001;154:1007–1017. doi: 10.1083/jcb.200103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P. New EMBO members'review. Actin cytoskeleton regulation through modulation of PI(4,5)P(2) rafts. EMBO J. 2001;20:4332–4336. doi: 10.1093/emboj/20.16.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CC, Vegners R, Bucki R, Funaki M, Korde N, Hartwig JH, Stossel TP, Janmey PA. Cell permeant polyphosphoinositide-binding peptides that block cell motility and actin assembly. J Biol Chem. 2001;276:43390–43399. doi: 10.1074/jbc.M105289200. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Emr SD, McPherson PS, Novick P. Phosphoinositides as regulators in membrane traffic. Science. 1996;271:1533–1539. doi: 10.1126/science.271.5255.1533. [DOI] [PubMed] [Google Scholar]
- DiNubile MJ, Huang S. High concentrations of phosphatidylinositol-4,5-bisphosphate may promote actin filament growth by three potential mechanisms: inhibiting capping by neutrophil lysates, severing actin filaments and removing capping protein-beta2 from barbed ends. Biochim Biophys Acta. 1997;1358:261–278. doi: 10.1016/s0167-4889(97)00076-1. [DOI] [PubMed] [Google Scholar]
- Fulbright RM, Axelrod D, Dunham WR, Marcelo CL. Fatty acid alteration and the lateral diffusion of lipids in the plasma membrane of keratinocytes. Exp Cell Res. 1997;233:128–134. doi: 10.1006/excr.1997.3553. [DOI] [PubMed] [Google Scholar]
- Gillooly DJ, Stenmark H. Cell biology. A lipid oils the endocytosis machine. Science. 2001;291:993–994. doi: 10.1126/science.291.5506.993. [DOI] [PubMed] [Google Scholar]
- Haugh JM, Codazzi F, Teruel M, Meyer T. Spatial sensing in fibroblasts mediated by 3′ phosphoinositides. J Cell Biol. 2000;151:1269–1280. doi: 10.1083/jcb.151.6.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiska L, Alfthan K, Gronholm M, Vilja P, Vaheri A, Carpen O. Association of ezrin with intercellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2). Regulation by phosphatidylinositol 4, 5- bisphosphate. J Biol Chem. 1998;273:21893–21900. doi: 10.1074/jbc.273.34.21893. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW, Ball R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science. 1996;273:956–959. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- Honda A, et al. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 1999;99:521–532. doi: 10.1016/s0092-8674(00)81540-8. [DOI] [PubMed] [Google Scholar]
- Hope HR, Pike LJ. Phosphoinositides and phosphoinositide-utilizing enzymes in detergent- insoluble lipid domains. Mol Biol Cell. 1996;7:843–851. doi: 10.1091/mbc.7.6.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalink K, Eichholtz T, Postma FR, van Corven EJ, Moolenaar WH. Lysophosphatidic acid induces neuronal shape changes via a novel, receptor-mediated signaling pathway: similarity to thrombin action. Cell Growth Differ. 1993;4:247–255. [PubMed] [Google Scholar]
- Kavran JM, Klein DE, Lee A, Falasca M, Isakoff SJ, Skolnik EY, Lemmon MA. Specificity and promiscuity in phosphoinositide binding by pleckstrin homology domains. J Biol Chem. 1998;273:30497–30508. doi: 10.1074/jbc.273.46.30497. [DOI] [PubMed] [Google Scholar]
- Kenworthy AK, Edidin M. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of >100 Å using imaging fluorescence energy transfer. J Cell Biol. 1998;142:69–84. doi: 10.1083/jcb.142.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koreh K, Monaco ME. The relationship of hormone-sensitive and hormone-insensitive phosphatidylinositol to phosphatidylinositol 4,5-bisphosphate in the WRK-1 cell. J Biol Chem. 1986;261:88–91. [PubMed] [Google Scholar]
- Lanier LM, Gertler FB. Actin cytoskeleton: thinking globally, actin'locally. Curr Biol. 2000;10:R655–R657. doi: 10.1016/s0960-9822(00)00685-0. [DOI] [PubMed] [Google Scholar]
- Lassing I, Lindberg U. Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature. 1985;314:472–474. doi: 10.1038/314472a0. [DOI] [PubMed] [Google Scholar]
- Lassing I, Lindberg U. Specificity of the interaction between phosphatidylinositol 4,5- bisphosphate and the profilin:actin complex. JCell Biochem. 1988;37:255–267. doi: 10.1002/jcb.240370302. [DOI] [PubMed] [Google Scholar]
- Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P. GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol. 2000;149:1455–1472. doi: 10.1083/jcb.149.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JG, Booth JW, Stambolic V, Mak T, Balla T, Schreiber AD, Meyer T, Grinstein S. Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fcγ receptor-mediated phagocytosis. J Cell Biol. 2001;153:1369–1380. doi: 10.1083/jcb.153.7.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S, Rothberg KG, Maxfield FR. Sequestration of GPI-anchored proteins in caveolae triggered by cross-linking. Science. 1994;264:1948–1951. doi: 10.1126/science.7516582. [DOI] [PubMed] [Google Scholar]
- Mesaeli N, Tappia PS, Suzuki S, Dhalla NS, Panagia V. Oxidants depress the synthesis of phosphatidylinositol 4,5-bisphosphate in heart sarcolemma. Arch Biochem Biophys. 2000;382:48–56. doi: 10.1006/abbi.2000.2012. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Holz RW, Smith SJ. Regulation of presynaptic phosphatidylinositol 4,5-biphosphate by neuronal activity. J Cell Biol. 2001;154:355–368. doi: 10.1083/jcb.200102098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pralle A, Keller P, Florin EL, Simons K, Horber JK. Sphingolipid-cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells. J Cell Biol. 2000;148:997–1008. doi: 10.1083/jcb.148.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucher D, Stauffer T, Chen W, Shen K, Guo S, York JD, Sheetz MP, Meyer T. Phosphatidylinositol 4,5-bisphosphate functions as a second messenger that regulates cytoskeleton-plasma membrane adhesion. Cell. 2000;100:221–228. doi: 10.1016/s0092-8674(00)81560-3. [DOI] [PubMed] [Google Scholar]
- Rozelle AL, et al. Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr Biol. 2000;10:311–320. doi: 10.1016/s0960-9822(00)00384-5. [DOI] [PubMed] [Google Scholar]
- Sakisaka T, Itoh T, Miura K, Takenawa T. Phosphatidylinositol 4,5-bisphosphate phosphatase regulates the rearrangement of actin filaments. Mol Cell Biol. 1997;17:3841–3849. doi: 10.1128/mcb.17.7.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- Sun HQ, Lin KM, Yin HL. Gelsolin modulates phospholipase C. activity in vivo through phospholipid binding. J Cell Biol. 1997;138:811–820. doi: 10.1083/jcb.138.4.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenawa T, Itoh T, Fukami K. Regulation of phosphatidylinositol 4,5-bisphosphate levels and its roles in cytoskeletal re-organization and malignant transformation. Chem Phys Lipids. 1999;98:13–22. doi: 10.1016/s0009-3084(99)00014-6. [DOI] [PubMed] [Google Scholar]
- Tall EG, Spector I, Pentyala SN, Bitter I, Rebecchi MJ. Dynamics of phosphatidylinositol 4,5-bisphosphate in actin-rich structures. Curr Biol. 2000;10:743–746. doi: 10.1016/s0960-9822(00)00541-8. [DOI] [PubMed] [Google Scholar]
- van der Wal J, Habets R, Varnai P, Balla T, Jalink K. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J Biol Chem. 2001;276:15337–15344. doi: 10.1074/jbc.M007194200. [DOI] [PubMed] [Google Scholar]
- van Leeuwen FN, van Delft S, Kain HE, van der Kammen RA, Collard JG. Rac regulates phosphorylation of the myosin-II heavy chain, actinomyosin disassembly and cell spreading. Nat Cell Biol. 1999;1:242–248. doi: 10.1038/12068. [DOI] [PubMed] [Google Scholar]
- Varma R, Mayor S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 1998;394:798–801. doi: 10.1038/29563. [DOI] [PubMed] [Google Scholar]
- Varnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Rother KI, Balla T. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton's tyrosine kinase pleckstrin homology domain visualized in single living cells. J Biol Chem. 1999;274:10983–10989. doi: 10.1074/jbc.274.16.10983. [DOI] [PubMed] [Google Scholar]
- Wang T, Pentyala S, Rebecchi MJ, Scarlata S. Differential association of the pleckstrin homology domains of phospholipases C-beta 1, C-beta 2, and C-delta 1 with lipid bilayers and the beta gamma subunits of heterotrimeric G proteins. Biochemistry. 1999;38:1517–1524. doi: 10.1021/bi982008f. [DOI] [PubMed] [Google Scholar]
- Watt SA, Kular G, Fleming IN, Downes CP, Lucocq JM. Subcellular localization of phosphatidylinositol4,5-biphosphate using the pleckstrin homology domain of phospholipase Cδ1. Biochem J. 2002;363:657–666. doi: 10.1042/0264-6021:3630657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Hilgemann DH, Feng S, Bito H, Ishihara H, Shibasaki Y, Yin HL. Phosphatidylinositol 4,5-bisphosphate induces actin stress-fiber formation and inhibits membrane ruffling in CV1 cells. J Cell Biol. 2001;152:867–876. doi: 10.1083/jcb.152.5.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yechiel E, Edidin M. Micrometer-scale domains in fibroblast plasma membranes. J Cell Biol. 1987;105:755–760. doi: 10.1083/jcb.105.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.