

Abstract
The extracellular domain of mouse Notch1 contains 36 tandem epidermal growth factor like (EGF) repeats, many of which are modified with O-fucose. Previous work from several laboratories has indicated that O-fucosylation plays an important role in ligand mediated Notch activation. Nonetheless, it is not clear whether all, or a subset, of the EGF repeats need to be O-fucosylated. Three O-fucose sites are invariantly conserved in all Notch homologues with 36 EGF repeats (within EGF repeats 12, 26 and 27). In order to investigate which O-fucose sites on Notch1 are important for ligand-mediated signaling, we mutated the three invariant O-fucose sites in mouse Notch1, along with several less highly conserved sites, and evaluated their ability to transduce Jagged1 and Delta1 mediated signaling in a cell-based assay. Our analysis revealed that mutation of any of the three invariant O-fucose sites resulted in significant changes in both Delta1 and Jagged1 mediated signaling, but mutations in less highly conserved sites had no detectable effect. Interestingly, mutation of each invariant site gave a distinct effect on Notch function. Mutation of the O-fucose site in EGF repeat 12 resulted in loss of Delta1 and Jagged1 signaling, while mutation of the O-fucose site in EGF repeat 26 resulted in hyperactivation of both Delta1 and Jagged1 signaling. Mutation of the O-fucose site in EGF repeat 27 resulted in faulty trafficking of the Notch receptor to the cell surface and a decreased S1 processing of the receptor. These results indicate that the most highly conserved O-fucose sites in Notch1 are important for both processing and ligand mediated signaling in the context of a cell-based signaling assay.
The Notch protein is a transmembrane receptor involved in a wide variety of cell fate decisions in metazoans (1,2) Importantly, mutations of the Notch protein and components of its signaling pathway have been implicated in an array of human diseases (e.g. CADASIL, T-cell leukemia, multiple sclerosis)(3–5). Notch becomes activated upon binding of its extracellular domain to its ligands, members of the Delta and Serrate/Jagged families, which are present on the surface of apposed cells. The extracellular domain of Notch contains 36 tandem Epidermal Growth Factor-like (EGF)1 repeats many of which contain consensus sequences for modification by O-fucose (6,7). Some of the O-fucose moieties on EGF repeats of Notch can be further elongated by the action of Fringe, a fucose-specific β1,3-N-acetylglucosaminyltransferase (8,9). Modification of Notch by Fringe modulates its response to ligands, inhibiting signaling from Serrate/Jagged ligands but potentiating signaling from Delta ligands (8,10–14).
Work from several laboratories has established that protein O-fucosyltransferase-1 (O-FucT-1), which catalyzes O-fucosylation of EGF repeats, is essential for Notch function. Experiments carried out in mice and Drosophila have demonstrated that deletion or reduction in levels of O-FucT-1 results in embryonic lethality, and more importantly, that the phenotypes observed are consistent with those observed due to loss of Notch signaling (15–17). The mechanism by which O-fucose exerts its effects on Notch signaling is not entirely clear. Several studies have demonstrated that lack of O-fucose alters the interaction of Notch with its ligands (17,18). With regard to specific O-fucose sites, a previous study revealed that Drosophila Notch bearing a mutation of a highly conserved O-fucose site within EGF repeat 12 is still able to support neurogenesis while showing defects in wing development (19). This mutant also demonstrated increases in both Delta and Serrate binding to Notch in in vitro binding assays. This effect was specific to the EGF repeat 12 mutant, as mutation of other O-fucose sites did not result in changes in Notch activity. The role of individual O-fucose sites in mammalian Notch signaling has not yet been established. The recent demonstration that O-FucT-1 is localized to the endoplasmic reticulum where it could function in quality control or possibly even as a molecular chaperone raises the possibility that specific O-fucose sites may be necessary for the processing and maturation of the Notch receptor (20,21). Thus, evaluation of the role of O-fucose at various sites is essential to dissect the mechanism by which these sugars affect Notch function.
Notch contains multiple sites for O-fucose modification scattered throughout its EGF repeats. An important clue in the evaluating which sites are most important for the regulation of Notch is their degree of conservation (Figure 1) (7). Three sites, found in EGF repeats 12, 26 and 27, are invariantly conserved in all known Notch homologues with 36 EGF repeats. In this work, we have sought to determine the role these three O-fucose sites in mouse Notch1 by analyzing the effects of mutation on ligand-mediated signaling, S1-processing, and cell-surface expression. As well, we have analyzed less-highly conserved O-fucose sites scattered throughout the extracellular domain. This analysis has revealed that mutation of two highly conserved O-fucose sites results in altered ligand-mediated Notch signaling, and in a third site alteration of the processing of the Notch receptor.
Figure 1. Sites of O-fucosylation on mouse Notch1.

The extracellular domain of mouse Notch1 contains multiple sites for O-fucosylation based on the consensus sequence C2X4-5(S/T)C3, most of which appear to be modified (6). These sites are conserved to varying degrees when compared to Notch receptors with 36 EGF repeats (indicated by the number of * from a total of 15 homologues) (adapted from (7)). The O-fucose sites mutated in this study are indicated by number. The location of the ligand binding domain (24), the negative regulatory Abruptex region (26), and a putative region of flexibility (25) of the Notch protein are also indicated.
Experimental Procedures
Site Directed Mutagenesis
A full-length construct of mouse Notch1 where with C-terminal PEST domain is replaced by six tandem MYC epitopes in pCS2+ (a kind gift of Dr. Raphael Kopan, Washington University School of Medicine) was used as a template for site-directed mutagenesis using the Quikchange site-directed mutagenesis kit (Stratagene). All glycosylation sites (T) were mutated to A. All constructs were sequenced to confirm the presence of the site-mutant. Revertant mutations were generated from constructs bearing site-mutants to ensure that no inadvertent mutations were introduced during the mutagenesis reaction.
Cell Lines
Cos-7 cells or Cos-7 cells stably transfected with empty vector (pCDNA4) were maintained in DMEM (Gibco) supplemented 10% Bovine Calf Serum and 250 μg/ml Zeocin (Invitrogen). L-cells were obtained from American Type Culture Collection and maintained in DMEM supplemented with 10% Bovine Calf Serum. L-cells stably expressing rat Jagged1 and rat Delta1 (Dl-19) (generously provided by Dr. Gerry Weinmaster) were maintained in DMEM supplemented with 10% Bovine Calf Serum.
Co-Culture Assay
These assays were adapted from previously described co-culture assays (11). Cos-7 cells (1 x 106) stably transfected with empty vector (pCDNA4) were plated in a 10 cm tissue culture dish, and the next day transiently transfected with 12.8 μg of wildtype or mutant Notch1-pCS2+, 2.98 μg TP-1 Luciferase reporter construct (Ga981-6, a kind gift of Dr. Georg Bornkamm, Munich, Germany), and 1.49 μg gWIZ beta-galactosidase construct (GTS) to normalize transfection efficiency using Lipofectamine 2000 (Invitrogen) according to manufacturer’s specifications. Four hours after transfection, Lipofectamine reagent was removed, and cells were allowed to recover in fresh DMEM for 1.5 hours. L-cells (1.0x 105) or L-cells expressing Jagged1 or Delta1 were then plated in each well of a 12-well tissue culture plate. Co-culture was established by overlaying L-cells with the transfected Cos-7 cells (7.5 x104). After 42.5 hours of co-culture, cell lysates were prepared in Reporter Lysis Buffer (Promega). Luciferase assays were performed as previously described (28). Each co-culture was performed in triplicate, and co-cultures resulting in significant changes as compared to co-culture of wildtype Notch were performed at least twice.
Metabolic Labeling
Cos-7 cells were transiently transfected with pSecTag2 (Invitrogen) constructs encoding EGF repeats 11–15, 26, or 27 of mouse Notch1 using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The production of the EGF repeat 11–15 and 26 constructs have been previously described (6). The EGF repeat 27 construct was produced by amplifying EGF repeat 27 from full-length Notch1 template using primers: (5′) GCTAAAGCTTCGGATGTCAATGAGTGTGAT, (3′) CGATCTCGAGTCACAAGGTTCTGGCAG. The products were digested with HindIII and XhoI, and subcloned into pSecTag2 and sequenced. Following transfection, cells were incubated with 20 μCi/ml [6-3H]fucose for 48 hours. Proteins were purified from media, and O-glycans were released by alkali-induced β-elimination and analyzed by gel-filtration chromatography as previously described (8,29).
Determination of percent fucitol
Saccharide species obtained from gel-filtration chromatography were pooled and dried in a Speed-Vac evaporator (Savant). Samples were then hydrolyzed for 2 hours in 2 M trifluoroacetic acid at 100 °C. The hydrolysates were dried in a Speed-Vac, resuspended in water, and dried again to insure complete removal of acid. Samples were then analyzed by high pH anion-exchange chromatography (HPAEC) on a Dionex DX300 using an MA-1 column (Dionex) as previously described (30). Samples were mixed with internal standards (1nmol each of fucitol, fucose, and glucose) prior to injection. Fractions (0.5 min) were collected and radioactivity corresponding to fucose and fucitol peaks was determined by liquid scintillation counting. The percentage of radioactivity corresponding to fucitol in each sample was determined by dividing cpms in fucitol by the total cpms in both fucitol and fucose. These percentages were then used to calculate the amount of O-fucose in each pooled peak by multiplying the total cpm in the peak by the percent fucitol.
Cell-surface Biotinylation
Cos-7 cells (5x 105) were plated in a 35 mm tissue culture dish and transfected the next day with 4 μg wildtype or mutant Notch1-pCS2+ using Lipofectamine 2000. Cells were incubated for 24 hours. Cells were washed three times with Hank’s Balanced Salt Solution (HBSS) (Gibco), and then incubated with 2.5 mM EZ-link sulfo-NHS-Biotin (Pierce Chemical Company), or with HBSS (as a control) two times for 10 minutes each time. The biotinylation reagent was then removed, and cells were incubated with 100 mM Tris-HCl (pH 8.0) for 15 minutes to quench the biotinylation reaction. Cells were lysed in 1.0 ml RIPA buffer (50 mM Tris-HCL, 150 mM NaCl, 1% NP40, 0.5% Deoxycholic Acid, 0.1% SDS) containing protease inhibitor cocktail (Roche). Lysates were cleared by centrifugation. ImmunoPure avidin-agarose (25 μl) (Pierce) was added to 100 μl of lysate and incubated for 16 hours at 4 °C. The avidin-agarose was pelleted by brief centrifugation and the lysate removed. Avidin-agarose was washed three times with RIPA buffer, and then boiled in 100 μl SDS sample buffer. Avidin-bound biotinylated and non-biotinylated samples along with their respective lysates were then run on a 10% SDS-PAGE, transferred to nitrocellulose, and detected using a mouse anti-myc epitope antibody (generously provided by Dr. Jen-Chih Hsieh, Stony Brook University). Cadherin expression was determined by blotting samples with mouse anti-PAN cadherin antibody (Sigma). A mouse anti-β-actin antibody (Abcam) was used to analyze β-actin expression. A horseradish peroxidase-conjugated goat anti-mouse antibody (Jackson ImmunoResearch Laboratories) was used as a secondary antibody. Quantification of immunoblots was performed using NIH image software.
S1 Cleavage Analysis
Cos-7 cells (5x 105) were plated in a 35 mm tissue culture dish and transfected the next day with 0.5 μg wildtype or mutant Notch1-pCS2+ plus 3.5 μg empty vector (pCS2+) using Lipofectamine 2000. Following a 24-hour incubation, cells were washed three times in HBSS, and then lysed in 1.0 ml RIPA buffer containing protease inhibitor cocktail (Roche), and a proteosome inhibitor (50μM Z-Leu-Leu-Leu-al) (Sigma). Lysates were cleared by centrifugation. A portion (10 μl) of each sample was then resolved on an 8% SDS-PAGE, transferred to nitrocellulose, and detected with mouse anti-myc epitope antibody (generously provided by Dr. Jen-Chih Hsieh, Stony Brook University). To exclude the possibility that the S1-processing was influenced by the amount of DNA transfected, variable amounts of DNA (0.5–4 μg) were used in transfection and S1-processing evaluated. While there was an increase in the amount of S1 cleaved Notch, there was no detectable change in the proportion of cleaved versus uncleaved Notch1 in either the wildtype or mutant samples (data not shown).
Results
Mutation of conserved O-fucose sites results in changes in Delta1 and Jagged1 mediated Notch1 signaling
In order to evaluate the effects of individual O-fucose site mutants on the ability of Notch1 to signal via Delta and Jagged ligands, a previously reported cell-signaling system (11) was adapted for use in Cos-7 cells. In this system, Cos-7 cells were transiently transfected with a plasmid encoding mouse Notch1 along with a luciferase reporter and a β-galactosidase reporter (for normalization of transfection efficiency). The Notch1 expressing Cos-7 cells were then co-cultured with L-cells, or L-cells stably expressing Delta1- or Jagged1-ligand. The cells were subsequently lysed, and luciferase and β-galactosidase were assayed to determine relative-fold activation as previously described (11). Co-culture of Notch1-expressing Cos-7 cells with either Delta1 or Jagged1 expressing L-cells resulted in a statistically significant increase in Relative-Fold Activation (RFA) when compared to co-culture with L-cells expressing no ligand (Figure 2A, B). Interestingly, in contrast to what has been observed using the same assay system in other cells (11,14) the activation of Notch1 in Cos-7 cells by Delta1-expressing L-cells was greater than that from the Jagged1-expressing cells. This suggests the Cos-7 cells may express an endogenous Fringe homologue (see below). Co-culture of control Cos-7 cells (transfected with empty vector, pCS2+) and ligand-expressing L cells resulted in a slight increase over controls, presumably due to activation of endogenous Notch in Cos-7 cells. Thus, activation of exogenous mouse Notch1 by both Delta1 and Jagged1 can be assayed using the Cos-7 cell system.
Figure 2. Activation of mouse Notch1 can be assayed using transiently transfected Cos-7 cells.
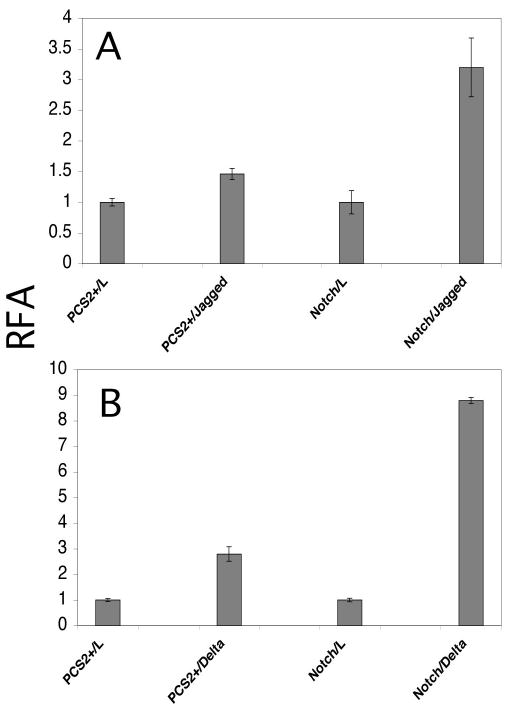
Cos-7 cells were transiently transfected with empty vector (pCS2+) or wildtype mouse Notch1 along with a luciferase reporter and a β-galactosidase reporter for normalization of transfection. Transfected cells were then co-cultured for 42.5 hours with control L-cells, L-cells expressing Jagged1 (A), or L-cells expressing Delta1 (B). Luciferase values were divided by β-galactosidase activity values to normalize for transfection efficiency. The resulting values were then normalized to activity observed with co-culture of L-cells, yielding a Relative Fold Activation (RFA).
To examine which of the O-fucose sites on mouse Notch1 are important for mediating Delta1 and/or Jagged1 activation, glycosylation site mutants were prepared (T to A). Haines and Irvine (7) showed that the O-fucose sites found in the extracellular domain of Notch are conserved to varying degrees (7) (Figure 1). Based on their analysis, we mutated the sites that are conserved in all Notch homologues (EGF repeats 12, 26, 27). In addition, O-fucose sites with lesser degrees of conservation in various regions of the extracellular domain were similarly mutated (EGF repeats 9, 16, 20, 24 and 30, see Figure 1). Notch1 constructs bearing each of these mutations were then used in the Cos-7 cell co-culture assay, in parallel with a co-culture assay of the wildtype Notch1 construct, as described above. Analysis of the relative activation in this assay revealed that mutation of the O-fucose sites in EGF repeats 12 and 27 resulted in a nearly complete loss in Jagged1- (Figure 3A) and Delta1- (Figure 3B) mediated Notch1 signaling when compared to wildtype. In contrast, mutation in the O-fucose site of EGF 26 resulted in a four-fold increase in Jagged1-mediated signaling (Figure 3A) and a twofold increase in Delta1-mediated signaling (Figure 3B). The O-fucose site mutations in EGF repeats 9, 16, 20, 24, and 30 did not result in any significant changes with either Jagged1 or Delta1. Thus, mutation of the most highly conserved O-fucose sites resulted in significant changes in both Delta1 or Jagged1 mediated Notch1 activation, while single mutants at other sites had no apparent effect. In order to confirm that the observed effects on Notch1 signaling were due to mutation of O-fucose sites (and not to inadvertent mutations introduced by the PCR-mediated mutagenesis), revertant mutations, which restored the threonine residue at the site of O-fucosylation, were generated in Notch constructs bearing mutations in EGF-repeats 12, 26, and 27. These revertant mutations effectively restored Delta and Jagged mediated signaling to wildtype levels (Figure 3C).
Figure 3. Mutation of O-fucose sites at EGF repeats 12, 26, and 27 in Notch1 alters Notch activation.
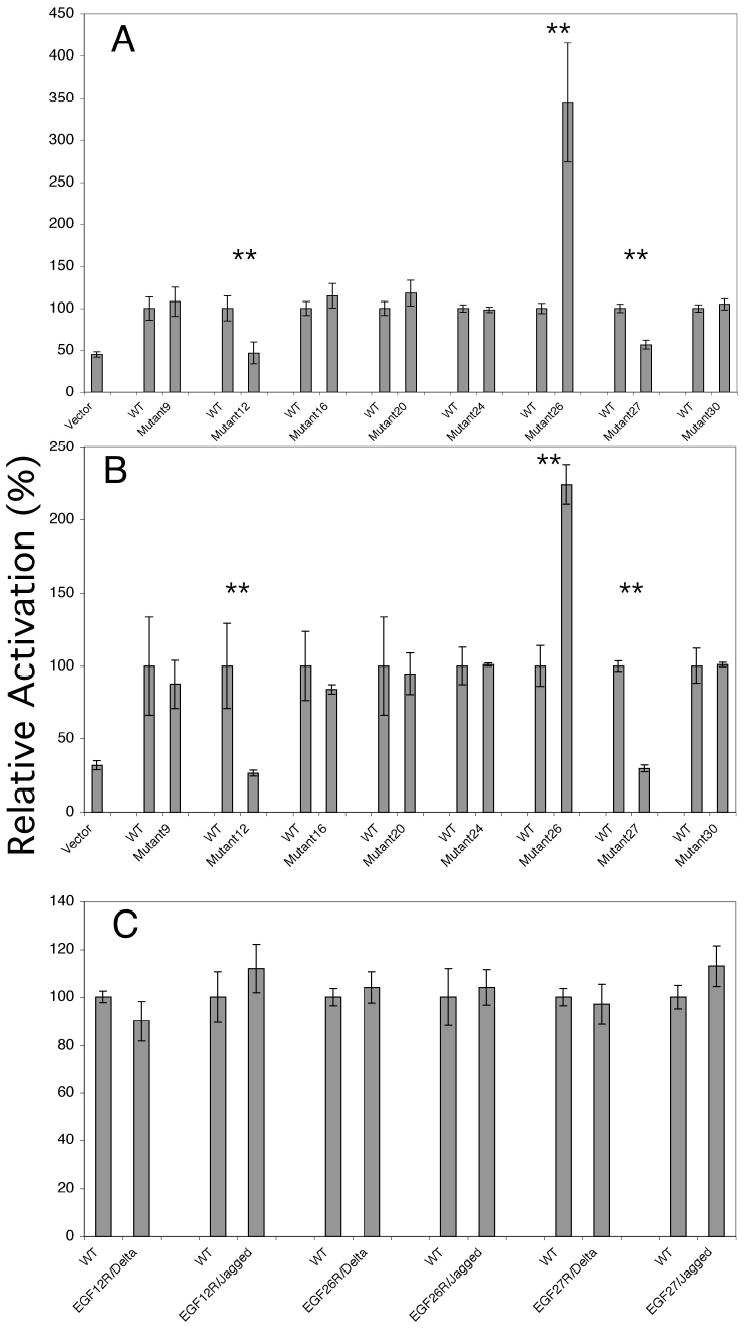
Wild type mouse Notch1 or Notch 1 containing O-fucose site mutants were evaluated in the co-culture assay described in Figure 2 with L-cells expressing Jagged1 (A), or Delta1 (B). For each set of assays, the RFA of the wildtype was normalized to 100% (see Figure 2 for actual RFA obtained with wildtype Notch1 with each ligand), and the RFA of the mutant normalized to that of the wildtype to allow for comparison of all sites analyzed. The symbol (**) indicates co-cultures in which Notch bearing an O-fucose site mutant resulted in a difference in RFA when compared to the wildtype that was statistically significant as determined by one-way ANOVA analysis. Mutations found to result in a statistical change in Notch signaling (mutations in EGF repeats 12, 26, and 27) were then reverted to wildtype and used in the co-culture assay with Delta1 and Jagged1 expressing L-cells (C). There was no statistical difference between the revertant mutants and wildtype Notch.
O-fucose on EGF repeats 26 and 27 is elongated by endogenous Fringe
The fact that Delta1 activates Notch1 to a greater extent than Jagged1 in the Cos-7 cell system suggests that Cos-7 cells contain endogenous Fringe. In most contexts, Fringe stimulates activation of Notch1 from Delta1 while inhibiting activation from Jagged1 (11,14). To examine whether any of the Fringes are modifying O-fucose on EGF repeats 12, 26, or 27, we examined the structure of the O-glycans on these EGF repeats. Although methods for analyzing O-fucose structures at individual glycosylation sites in the context of the native protein have not yet been developed, we have previously shown that individual EGF repeats encode determinants required for Fringe modification (6). Thus analysis of the state of elongation of O-fucose on smaller fragments of the Notch1 extracellular domain (or individual EGF repeats) can be used to approximate Fringe modification at specific sites in the context of the entire Notch molecule. To investigate whether endogenous Fringe modifies O-fucose on EGF repeats 12, 26, or 27, plasmids encoding these EGF repeats were transfected into Cos-7 cells. The cells were metabolically radiolabeled with [3H]-fucose, the proteins were purified, the O-fucose glycans released from the protein by alkali-induced β-elimination, and the released saccharides were analyzed by gel filtration chromatography (Figure 4). In the case of both EGF repeats 26 and 27, significant elongation of the monosaccharide to di-, tri- and tetrasaccharide species is observed (Figure 4B, C, D). This indicates that Cos-7 cells contain an endogenous Fringe activity. Immunoblot analysis shows the presence of Lunatic fringe in extracts of Cos-7 cells, indicating the presence of at least one endogenous Fringe (data not shown). This is consistent with the higher level of Delta1 than Jagged1 activation seen in Figure 2, and suggests that Fringe modification at EGF repeats 26 and 27 may contribute to this effect. Importantly, this also raises the possibility that the effects observed in the co-culture assay with regard to the mutations at EGF repeats 26 and 27 (Figure 3) may in part be attributable to Fringe modulation of Notch signaling. In contrast EGF repeat 12 is modified almost exclusively by O-fucose monosaccharide (Figure 4A, D), suggesting that this site is a poorer substrate for Fringe than EGF repeats 26 and 27. Similar results were obtained in analysis of the glycosylation of EGF repeat 12 in CHO (Chinese hamster ovary) cells, which also express endogenous Lunatic fringe (6). These results suggest that the effects of the mutation at EGF repeat 12 in the co-culture assay (Figure 3) are mediated largely by loss of the O-fucose monosaccharide.
Figure 4. O-fucose is elongated by Fringe on EGF repeats 26 and 27.
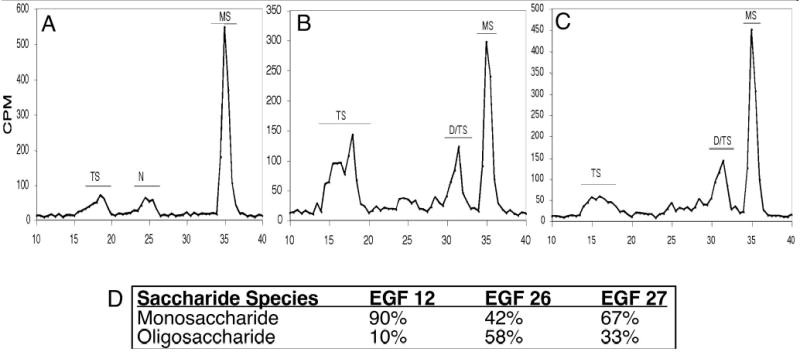
Cos-7 cells were transiently transfected with constructs encoding mouse Notch1 EGF repeat 11–15 (where EGF repeat 12 contains the only O-fucosylation site (6)) (A), EGF repeat 26 (B), or EGF repeat 27 (C). Cells were then metabolically radiolabeled with [3H]-fucose, proteins were purified from the medium, and saccharide structures were analyzed as described in Materials and Methods. The migration position of O-fucose saccharides is indicated: MS (monosaccharide, fucitol); D/TS (disaccharide, GlcNAc-β1,3-fucitol, and trisaccharide, Gal-β1,4-GlcNAc-β1,3-fucitol, migrate close together in this system making them difficult to differentiate); TS (tetrasaccharide, Sia-α2,3/6-Gal-β1,4-GlcNAc-β1,3-fucitol) (30). In order to confirm the presence of O-fucose containing saccharide in each peak, the indicated material was pooled, acid hydrolyzed, and analyzed by HPAEC as described in Materials and Methods. The percent of fucitol (relative to fucitol plus fucose) was determined for each peak. Fucitol is derived from O-fucose, whereas fucose is from contaminating glycans bearing terminal fucose (most likely N-glycans. See (30) for a more complete description of this method). Peaks containing no fucitol (presumably from contaminating N-glycans) are indicated by “N”. The percent fucitol in each peak is as follows: EGF repeat 12 (panel A): TS (83.7%), N (0%), and MS (98.5%); EGF repeat 26 (panel B): TS (45%), D/TS (20%), MS (50%); EGF repeat 27: TS (63%), D/TS (85%), and MS (97%). These values were used to calculate the amount of O-fucose saccharide elongated by Fringe (Oligosaccharide, combination of fucitol in TS and D/TS), and unelongated O-fucose (Monosaccharide, fucitol in MS) on each EGF repeat (panel D).
Mutation of O-fucose site in EGF repeat 27 results in a reduction of cell-surface expression and S1 cleavage of Notch1
The variations in Delta1- and Jagged1-mediated Notch1 signaling observed in the co-culture assays may be attributable to changes in the cell surface expression of the mutated Notch1 proteins. To assess this possibility, Cos-7 cells were transfected with either wildtype Notch1 or Notch1 bearing O-fucose site mutants in EGF repeats 12, 26, or 27. Cell surface biotinylation was then carried out, followed by streptavidin precipitation (Figure 5A). Controls with a cell surface protein (cadherin) and a cytoplasmic protein (β-actin) were performed with each sample to demonstrate the efficacy of the biotinylation procedure. A ratio of cell surface Notch1 to cell-surface cadherin was performed to normalize for any differences in sample loading. This analysis showed that Notch1 bearing O-fucose site mutations in either EGF repeats 12 or 26 were expressed on the cell surface similar to wild type. In contrast, there was decreased cell-surface expression of Notch bearing an O-fucose mutation in EGF repeat 27 compared to wildtype. Thus, mutation of the O-fucose site in EGF repeat 27 results in reduced cell-surface expression of the Notch protein.
Figure 5. O-fucose site mutation in EGF repeat 27 causes a reduction in cell-surface expression and S1-processing of Notch1.

(A) To evaluate cell-surface expression, Cos-7 cells were transiently transfected with wildtype Notch1 or Notch1 bearing an O-fucose site mutation in EGF repeats 12, 26, or 27. Cells were subjected to the cell-surface biotinylation procedure described in Materials and Methods. Immunoblot analysis of cell lysate and avidin-bound material was performed using an anti-myc epitope antibody (to detect transfected Notch1). As a positive control, lysate and avidin-bound material were immunoblotted with an anti-cadherin antibody, and as negative control with an anti-βactin antibody. Densitometry analysis of immunoblot indicates that there is no difference in the ratio of Notch to cadherin between wildtype Notch and Notch with O-fucose site mutants in EGF repeat 12 (1.04 versus 1.05 +/− 0.01 respectively) and EGF repeat 26 (0.40 +/− 0.001 versus 0.35 +/− 0.01 respectively). However, there is an approximately two-fold difference in the ratio of wildtype Notch to cadherin versus Notch bearing an EGF repeat 27 site mutant and cadherin (0.72 +/− 0.06 versus 0.37 +/−0.01 respectively). Thus, there is a reduction in cell surface expression of Notch bearing an EGF repeat 27 O-fucose site mutant. (B) To analyze S1 cleavage of wildtype Notch and Notch bearing O-fucose site mutants, Cos-7 cells were transfected with wildtype Notch, or a Notch construct bearing an O-fucose site mutant in EGF repeats 12, 26, or 27. Cells were lysed and lysate was resolved by SDS-PAGE on an 8% gel and analyzed by immunoblot with an anti-myc epitope antibody. The migration positions of S1 cleaved fragment and full-length uncleaved Notch are indicated.
Notch is cleaved by a furin-like convertase in the Golgi, which is required for efficient translocation to the cell surface (22). Loss of multiple O-fucose sites in mouse Notch3 results in defective S1 cleavage (23). To investigate the role of O-fucose site mutations on Notch1 processing, Cos-7 cells were transfected with wildtype Notch1 or Notch1 bearing mutations at EGF repeats 12, 26, or 27. The cells were lysed and examined by immunoblot analysis. S1-processing of Notch1 bearing a mutation in EGF repeat 12 was similar to wildtype, while that with a mutation in EGF repeat 26 showed a slight decrease in efficiency (Figure 5B). However, the mutation in EGF repeat 27 caused a significant reduction in S1 cleaved Notch1. Thus, mutation of the O-fucose site at EGF repeat 27 results in decreased in S1-mediated processing and subsequent cell-surface expression of the Notch protein. These results provide an explanation for the reduction in Notch1 signaling caused by the mutation in EGF repeat 27.
Discussion
In this work we have sought to determine the role of highly conserved O-fucose sites on mouse Notch1. Mutation of any of the three invariant sites across species, located in EGF repeats 12, 26 and 27, resulted in changes in both Delta1- and Jagged1-mediated Notch1 signaling. In contrast, there was no detectable effect of mutation of less-conserved O-fucose sites. Interestingly, the mutations in each of the highly conserved sites resulted in a distinct effect on Notch1. The mutation at EGF repeat 12 caused a decrease in Notch signaling, that at EGF repeat 26 caused an increase in Notch signaling, and that at EGF repeat 27 caused a decrease in S1-processing can cell-surface expression resulting in decreased Notch signaling. In addition, the O-fucose moieties at both EGF repeats 26 and 27 are elongated by endogenous Fringe in Cos-7 cells, while that at EGF repeat 12 remains mainly a monosaccharide. This suggests that the effect of the mutation at EGF repeat 12 is due mainly to the loss of the monosaccharide form of O-fucose while the mutations at EGF repeats 26 and 27 may be due to the loss of elongated forms.
The mutation in EGF repeat 12 resulted in loss of activation by both Delta1 and Jagged1 but no change in S1-processing or cell-surface expression. The fact that it proceeds through endoplasmic reticulum quality control checkpoints and is properly processed suggests that the EGF repeat 12 mutation does not cause a global folding defect. EGF repeats 11 and 12 have been identified as necessary and sufficient for ligand interactions (see Figure 1) (24) suggesting that the signaling effects observed in the EGF repeat 12 mutant are mediated by direct effects on Notch-ligand interactions. Nonetheless, several other studies have suggested that the O-fucose on EGF repeat 12 is not essential for ligand binding, but may instead be modulatory. For instance, a recent report demonstrated that bacterially expressed (and therefore unfucosylated) fragment containing EGF repeats 11–13 from mouse Notch1 can bind to cells expressing Delta1 in a calcium dependent fashion, although the EGF 11–13 fragment needed to be aggregated on streptavidin to obtain significant binding activity (25). Similarly, Lei et al. (19) revealed that Notch bearing a mutation in the O-fucose glycosylation site at EGF 12 still functions during neurogenesis (Delta-mediated Notch signaling) in Drosophila. This same mutation results in alterations of Delta and Serrate binding and shows changes in Notch activation in developing wing discs. Taken together, these studies seem to suggest that the cellular context greatly affects the importance of O-fucose on EGF 12. Presence or absence of O-fucose on EGF repeat 12 appears to modulate Notch-ligand interactions, but it does not appear to be essential in all contexts (e.g. Delta-mediated activation of Notch during neurogenesis in Drosophila). The specific context in which Notch exists, with respect to ligands, Fringe, and other modulators of Notch activity, appear to affect the relative importance of the O-fucose at this site. The fact that mutation of the O-fucose site on EGF repeat 12 has such a profound effect on Notch activation in Cos-7 cells suggests that these cells do not have mechanisms to compensate for this loss.
The mutation in EGF repeat 26 results in an increase in Notch activation by both ligands: a two-fold increase in Delta1-mediated signaling and a four-fold increase in Jagged1 signaling. The hyperactivation caused by this mutation does not correlate with alterations in processing or cell-surface expression, but it is reminiscent of the Abruptex mutations. These are gain-of-function point mutations in Drosophila Notch which cluster in EGF repeats 24–29 (Figure 1). The Abruptex region of Notch is thought to act as a negative regulatory domain possibly mediated through cell-autonomous inhibition by ligands (26). It is possible that mutation of the O-fucose site in EGF repeat 26 in some manner mimics the effect of Abruptex mutations. Additionally, it has been observed that Abruptex mutations are refractory to Fringe in Drosophila, suggesting that the Abruptex mutations may induce a change in Notch similar to that induced by Fringe. Fringe modifies O-fucose residues in the Abruptex region (6). Taken together, these observations suggest that the O-fucose modification on EGF repeat 26 may in fact function as a site for Fringe to negatively regulate Notch activation. Loss of O-fucose at this site, and as a consequence, loss of the ability of Fringe to modify EGF repeat 26, may prevent negative regulation of Notch signaling by Fringe, thus resulting in a hyperactivatable form of the receptor. Hambleton and co-workers have proposed that the majority of the EGF repeats in the Notch extracellular domain are fairly rigid, due to the presence of calcium-binding motifs between EGF repeats. Interestingly, EGF repeats 26, 28 and 29 (overlapping the Abruptex region) do not contain calcium-binding motifs and are thus predicted to be more flexible (see Figure 1 (25). It is conceivable that the conformation of Notch may be altered by the extent of elongation on O-fucose moieties at this specific location (Figure 6). The fact that the O-fucose glycans on both EGF repeats 26 and 27 are elongated supports this idea. Thus, we propose that the change in Notch activation caused by mutation at EGF repeat 26 may be caused by a change in the overall conformation of the extracellular domain.
Figure 6. Proposed effects of progressive O-fucose elongation at EGF repeats 12, 26 and 27 on the Notch extracellular domain.
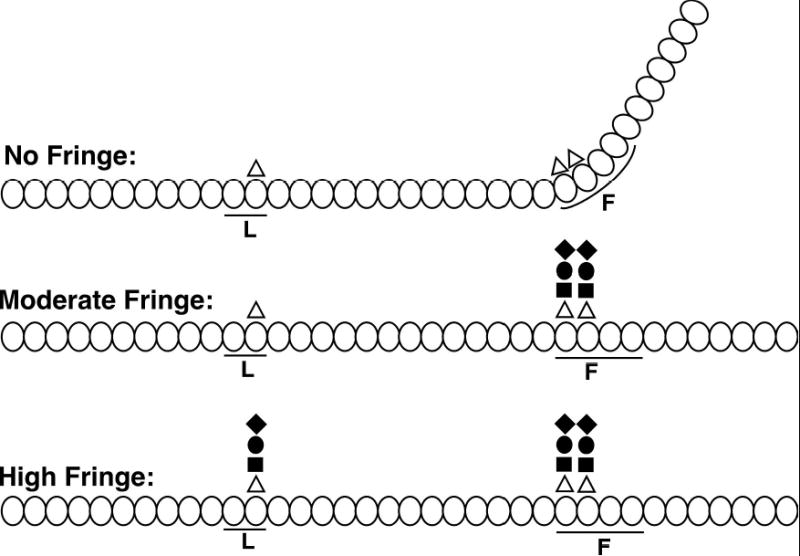
The 36 tandem EGF repeats of the Notch extracellular domain are represented by ovals, and those implicated in ligand binding (L) (24) and as a flexible region (F) (25) are indicated by lines. Fucose is indicated by a triangle, GlcNAc by a square, galactose by a circle, and sialic acid by a diamond (8). With moderate levels of Fringe, elongation occurs at EGF repeats 26 and 27, followed by elongation at EGF repeat 12 with high levels of Fringe. Modification at EGF repeats 26 and 27 is proposed to alter the flexibility at this region. The conformations shown (at “No Fringe” and “Moderate Fringe”) are only one example of many possible structures.
Mutation of the O-fucose in EGF repeat 27 results in a significant reduction in both Delta1-and Jagged1-mediated Notch1 activation that is most likely due to a reduction in S1 processing. It is possible that this highly conserved O-fucose site serves as a “regulatory checkpoint” for further processing of the Notch protein. Since EGF repeat 27 lies in the putative flexible region of Notch (Figure 6), alteration of its glycosylation state could alter the overall conformation of Notch, and this could affect the ability of the furin-like convertase to process Notch. Indeed, this would not be the first example of glycosylation altering the processing of a protein. Processing of the low density lipoprotein (LDL) receptor-related protein 1 (LRP1) is regulated by differential glycosylation (27). Alternatively, it is possible that loss of the O-fucose site in EGF repeat 27 causes retention of the Notch protein in the endoplasmic reticulum. Mutation of O-fucose sites has been observed to result in a decrease in the secretion of Drosophila Notch fragments or S1 processing of Notch3, suggesting they are trapped in the endoplasmic reticulum (20,23).
The finding that mutation of highly conserved O-fucose sites affects Notch processing and signaling supports the idea that O-fucosylation of individual sites is important for Notch function. Surprisingly, there appears to be heterogeneity in the roles of these highly conserved O-fucose sites. This may in part be explained by the fact some of these sites may also be important for Fringe regulation of Notch signaling. Indeed, our data suggests that the effects observed from the O-fucose on EGF repeat 26 might be most readily attributable to an alteration in the Fringe effect. In contrast, the effects of the O-fucose on EGF repeat 12 appear to be mediated mainly by the monosaccharide form of O-fucose. Interestingly, the O-fucose on EGF repeat 12 is mainly in the monosaccharide form in both CHO (6) and Cos-7 (Figure 4) cells, even though both cells express endogenous Lunatic fringe. In contrast, O-fucose on EGF repeat 26 is significantly elongated in both cell types. Overexpression of Lunatic or Manic fringe in CHO cells results in significant increases in O-fucose elongation at EGF repeat 12 (6) and loss of Jagged1-dependent activation (8,13). Thus, cells expressing low levels of Fringe (e.g. Cos-7 or CHO) preferentially modify EGF repeats 26 and 27 and have an intermediate effect on Delta1 activation and Jagged1 inhibition (see “Moderate Fringe”, Figure 6). Increased expression of Fringe causes O-fucose elongation at EGF repeat 12 and further inhibition of Jagged1-dependent activation (see “High Fringe”, Figure 6). This suggests that EGF repeat 12 may play a key role in the ability of Fringe to fully inhibit Serrate/Jagged-dependent signaling. This is consistent with the results reported by Lei and coworkers using the EGF repeat 12 mutant in Drosophila wing discs (19). Determination of the Fringe effect at specific sites, and determination of the glycosylation effect on the overall conformation of the Notch extracellular domain, is needed for further clarification of the roles of these O-fucosylation sites. Identification of specific amino acids within an EGF repeat necessary for recognition by Fringe will allow us to generate mutations that allow O-fucosylation but prevent Fringe elongation at a particular EGF repeat. This will allow us to differentiate between the effects of O-fucose monosaccharide and the elongated tetrasaccharide at a particular site (e.g. EGF repeat 26).
Acknowledgments
The authors would like to thank members of the Haltiwanger laboratory, Dr. Kenneth Irvine, and Dr. Pamela Stanley for many helpful discussions and comments on the manuscript. They would also like to thank Dr. Raphael Kopan for the pCS+ Notch1 plasmid, Dr. Georg Bornkamm for the TP-1 reporter plasmid, Dr. Gerry Weinmaster for the control, Delta1-expressing and Jagged1-expressing L-cells, and Jen-Chih Hsieh for anti-MYC antibody. R.R. was supported in part by an MSTP training grant (T32 GM008444). This work was supported by NIH grant GM 61126.
Footnotes
Abbreviations: O-FucT-1, O-fucosyltransferase-1; CHO, Chinese Hamster Ovary; EGF, Epidermal Growth Factor-like; HPAEC, high-pH anion exchange chromatography; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; Gal, galactose; GlcNAc, N-acetylglucosamine; Sia, sialic acid.
References
- 1.Mumm JS, Kopan R. Dev Biol. 2000;228:151–165. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- 2.Artavanis-Tsakonas S, Rand MD, Lake RJ. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 3.Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 4.Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, Sklar J. Cell. 1991;66:649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 5.John GR, Shankar SL, Shafit-Zagardo B, Massimi A, Lee SC, Raine CS, Brosnan CF. Nat Med. 2002;8:1115–1121. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- 6.Shao L, Moloney DJ, Haltiwanger RS. J Biol Chem. 2003;278:7775–7782. doi: 10.1074/jbc.M212221200. [DOI] [PubMed] [Google Scholar]
- 7.Haines N, Irvine KD. Nat Rev Mol Cell Biol. 2003;4:786–797. doi: 10.1038/nrm1228. [DOI] [PubMed] [Google Scholar]
- 8.Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS, Vogt TF. Nature. 2000;406:369–375. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- 9.Bruckner K, Perez L, Clausen H, Cohen S. Nature. 2000;406:411–415. doi: 10.1038/35019075. [DOI] [PubMed] [Google Scholar]
- 10.Panin VM, Papayannopoulos V, Wilson R, Irvine KD. Nature. 1997;387:908–912. doi: 10.1038/43191. [DOI] [PubMed] [Google Scholar]
- 11.Hicks C, Johnston SH, DiSibio G, Collazo A, Vogt TF, Weinmaster G. Nature Cell Biology. 2000;2:515–520. doi: 10.1038/35019553. [DOI] [PubMed] [Google Scholar]
- 12.Fleming RJ, Gu Y, Hukriede NA. Development. 1997;124:2973–2981. doi: 10.1242/dev.124.15.2973. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Moloney DJ, Stanley P. ProcNatlAcadSci USA. 2001;98:13716–13721. doi: 10.1073/pnas.241398098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang LT, Nichols JT, Yao C, Manilay JO, Robey EA, Weinmaster G. Mol Biol Cell. 2005;16:927–942. doi: 10.1091/mbc.E04-07-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okajima T, Irvine KD. Cell. 2002;111:893–904. doi: 10.1016/s0092-8674(02)01114-5. [DOI] [PubMed] [Google Scholar]
- 16.Shi, S., and Stanley, P. (2003) Proc.Natl.Acad.Sci.USA IN PRESS
- 17.Sasamura T, Sasaki N, Miyashita F, Nakao S, Ishikawa HO, Ito M, Kitagawa M, Harigaya K, Spana E, Bilder D, Perrimon N, Matsuno K. Development. 2003;130:4785–4795. doi: 10.1242/dev.00679. [DOI] [PubMed] [Google Scholar]
- 18.Okajima T, Xu A, Irvine KD. J Biol Chem. 2003;278:42340–42345. doi: 10.1074/jbc.M308687200. [DOI] [PubMed] [Google Scholar]
- 19.Lei L, Xu A, Panin VM, Irvine KD. Development. 2003;130:6411–6421. doi: 10.1242/dev.00883. [DOI] [PubMed] [Google Scholar]
- 20.Okajima T, Xu A, Lei L, Irvine KD. Science. 2005;307:1599–1603. doi: 10.1126/science.1108995. [DOI] [PubMed] [Google Scholar]
- 21.Luo Y, Haltiwanger RS. J Biol Chem. 2005;280:11289–11294. doi: 10.1074/jbc.M414574200. [DOI] [PubMed] [Google Scholar]
- 22.Blaumueller CM, Qi HL, Zagouras P, Artavanis-Tsakonas S. Cell. 1997;90:281–291. doi: 10.1016/s0092-8674(00)80336-0. [DOI] [PubMed] [Google Scholar]
- 23.Arboleda-Velasquez, J. F., Rampal, R., Fung, E., Darland, D. C., M, L., Martinez, M. C., Donahue, C. P., Navarro-Gonzalez, M. F., Libby, P., D'Amore, P. A., Aikawa, M., Haltiwanger, R. S., and Kosik, K. S. (2005) Hum Mol Genet [DOI] [PubMed]
- 24.Rebay I, Fleming RJ, Fehon RG, Cherbas L, Cherbas P, Artavanis-Tsakonas S. Cell. 1991;67:687–699. doi: 10.1016/0092-8674(91)90064-6. [DOI] [PubMed] [Google Scholar]
- 25.Hambleton S, Valeyev NV, Muranyi A, Knott V, Werner JM, McMichael AJ, Handford PA, Downing AK. Structure (Camb) 2004;12:2173–2183. doi: 10.1016/j.str.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 26.De Celis JF, Bray SJ. Development. 2000;127:1291–1302. doi: 10.1242/dev.127.6.1291. [DOI] [PubMed] [Google Scholar]
- 27.May P, Bock HH, Nimpf J, Herz J. J Biol Chem. 2003;278:37386–37392. doi: 10.1074/jbc.M305858200. [DOI] [PubMed] [Google Scholar]
- 28.Nofziger D, Miyamoto A, Lyons KM, Weinmaster G. Development. 1999;126:1689–1702. doi: 10.1242/dev.126.8.1689. [DOI] [PubMed] [Google Scholar]
- 29.Moloney DJ, Lin AI, Haltiwanger RS. J Biol Chem. 1997;272:19046–19050. doi: 10.1074/jbc.272.30.19046. [DOI] [PubMed] [Google Scholar]
- 30.Moloney DJ, Shair L, Lu FM, Xia J, Locke R, Matta KL, Haltiwanger RS. J Biol Chem. 2000;275:9604–9611. doi: 10.1074/jbc.275.13.9604. [DOI] [PubMed] [Google Scholar]