

Abstract
Although hypoxia and transforming growth factor-β (TGF-β) inhibit differentiation of adipocytes from preadipocytes and bone marrow-derived cells in several species, the relationship between hypoxia and TGF-β signaling in adipocytogenesis is unknown. In this study, we evaluated the mechanisms of inhibition of adipocyte differentiation by hypoxia and TGF-β in human and murine marrow stromal cells (MSCs) and the role of TGF-β/Smad signaling in the inhibition of adipocytogenesis by hypoxia. Both hypoxia-mimetic deferoxamine mesylate (DFO) and TGF-β1 inhibited adipocyte differentiation (1.0% versus the control at 15 μm DFO and 1.4% versus the control at 1 ng/ml TGF-β1) and adipocyte gene expression (peroxisome proliferator-activated receptor-γ2 and lipoprotein lipase) in human MSCs after 21 days of treatment. Hypoxia (2% O2) and DFO (but not TGF-β1) increased hypoxia-inducible factor-1α as shown by Western blotting. Macroarrays and Western and Northern blot analyses showed that hypoxia activated the TGF-β/Smad signaling pathway and that both hypoxia and TGF-β1 modulated adipocyte differentiation pathways such as the insulin-, peroxisome proliferator-activated receptor-γ-, phosphatidylinositol 3-kinase-, and MAPK-associated signaling pathways. Studies with mouse marrow stromal cell lines derived from Smad3+/+ or Smad3−/− mice revealed that the TGF-β type I receptor (ALK-5) and its intracellular signaling molecule Smad3 were necessary for the inhibition of adipocyte differentiation by both TGF-β and hypoxia-mimetic DFO. Thus, the TGF-β/Smad signaling pathway is required for hypoxia-mediated inhibition of adipocyte differentiation in MSCs.
Oxygen homeostasis represents an important organizing principle for human development and physiology. Dysregulation of oxygen homeostasis is found in inflammatory and cardiovascular diseases, cancer, cerebrovascular disease, and chronic obstructive pulmonary disease (1, 2). One of the immediate sequelae of bone fracture is regional hypoxia resulting from vasculature disruption (3). In response to hypoxia or low O2 tension, mammalian tissues show increased expression of a wide variety of genes that stimulate erythropoiesis, angiogenesis, and glycolysis (4). Most of the hypoxia-regulated genes are transcriptionally up-regulated by hypoxia-inducible factor-1α (HIF-1α)1 (5–7).
Adipocyte differentiation of murine 3T3-L1 preadipocytes is inhibited under hypoxic conditions (0.01–2% O2), and its inhibition is not observed in mouse embryonic fibroblasts deficient in HIF-1α (8). Low levels of O2 reduce in vitro adipocytogeneis of murine skeletal muscle satellite cells compared with 20% O2, a finding that has been corroborated by similar observations in the pluripotent mesenchymal cell line C3H10T1/2 and 3T3 cells (9). A recent report shows that hypoxia-dependent inhibition of adipocyte differentiation of murine 3T3-F442A preadipocytes can be mediated by mitochondrial reactive oxygen species generation (10). Transforming growth factor-β (TGF-β) was reported to strongly inhibit adipocytogenesis and the amount of fat in adipocytes that develops in cultures of human bone marrow-derived stromal cells (11). It has also been observed that TGF-β inhibits adipocytogenesis of the murine 3T3-F442A and NIH3T3 cell lines by signaling through Smad3 (12, 13). In several systems, various effects of hypoxia appear to be mediated by TGF-β signaling. In human peritoneal fibroblasts, hypoxia increases TGF-β1 and TGF-β type I and II receptor mRNA levels, with no effect on TGF-β2 or TGF-β3 (14). In human umbilical vein endothelial cells, hypoxia results in phosphorylation and nuclear transportation of Smad2 and Smad3 proteins as well as stimulation of the transcriptional activities of Smad3 and HIF-1α and up-regulation of TGF-β2 and TGF-β type II receptor gene expression (15, 16). In human dermal fibroblasts, up-regulation of COL1A1 mRNA levels by hypoxia is blocked by a TGF-β1 antisense oligonucleotide and fails to occur in fibroblasts from TGF-β 1−/− mice (17). It is unknown whether TGF-β signaling has a role in the inhibition of adipocytogenesis by hypoxia.
This study was designed to test the hypothesis that the TGF-β/Smad signaling pathway is required for the inhibition of adipocyte differentiation of human marrow stromal cells (hMSCs) by hypoxia. hMSCs have the potential to differentiate to lineages of mesenchymal tissues, including bone, cartilage, fat, tendon, and muscle (18). Loss in bone volume (associated with osteoporosis and age-related osteopenia) is accompanied by an increase in bone marrow adipose tissue (reviewed in Ref. 19). Recently, we demonstrated cooperation between the TGF-β and Wnt signaling pathways in their stimulation of chondro-cytogenesis and inhibition of adipocytogenesis in hMSCs (20). The results presented here, together with those from our previous studies, demonstrate that several signaling pathways are involved in the inhibition of adipocyte differentiation of hMSCs and thus provide a basis for possible approaches to prevent and/or treat obesity and osteopenia/osteoporosis.
EXPERIMENTAL PROCEDURES
Cell Culture
Adherent hMSCs were prepared from femoral bone marrow that was obtained as discarded material from a 42-year-old woman undergoing total hip replacement for osteoarthritis (21). Low density mononuclear cells were isolated by density centrifugation with Ficoll/Histopaque 1077 (Sigma). This procedure enriches for undifferentiated cells, including a fraction of non-adherent hematopoietic cells and a stromal fraction capable of adherence and differentiation into various connective tissue cells (21). The adherent fraction was expanded in monolayer culture with phenol red-free α-minimal essential medium (Invitrogen), 10% heat-inactivated fetal bovine serum (FBS-HI; Atlanta Biologicals, Inc., Norcross, GA), and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin; Invitrogen). Mouse Smad3+/+ and Smad3−/− MSCs were derived from bone marrow cultures of wild-type and Smad3-null mice, respectively (22). The Smad3-null mice were provided by Dr. K. C. Flanders (National Institutes of Health, Bethesda, MD). The murine MSCs were maintained in McCoy’s 5A medium (Invitrogen) with 10% FBS until they reached confluence.
Conditions for Adipocyte Differentiation of hMSCs
After hMSCs reached confluence, the medium was changed to α-minimal essential medium (phenol red-free), 1% FBS-HI, and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin) with different combinations of the following adipocytogenic supplements: 10 μg/ml insulin (referred to as I), 1 μm dexamethasone (referred to as D), 0.5 mm 3-isobutyl-1-methylxanthine (referred to as M), and 100 μm indomethacin (referred to as Y) (Sigma). After 6 days of treatment, cultures were inspected by light microscopy for the presence of lipid-containing cells. RNA was extracted with TRIzol (Invitrogen) and analyzed by reverse transcription (RT)-PCR for expression of the peroxisome proliferator-activated receptor-γ2 (PPARγ2) and lipoprotein lipase (LPL) adipocyte marker genes. Lipid-containing cells were observed in four of the supplement combinations: DM, IDM, DMY, and IDMY. PPARγ2 expression was detected in cultures treated with DM or IDM and was up-regulated in hMSC cultures treated with DY and IDY, with which adipocytes were not observed. The greatest LPL expression was detected in cells treated with IDM compared with DM, DMY, and IDMY. In cultures without visible lipid-containing cells, LPL was up-regulated only in the group treated with IDY. Neither PPARγ2 nor LPL was detected by RT-PCR under basal conditions. These data indicate that up-regulation of PPARγ2 and LPL was achieved with the combination of insulin, dexamethasone, and 3-isobutyl-1-methylxanthine and that addition of indomethacin to the treatment did not increase gene expression or apparent accumulation of adipocytes in hMSCs. For most experiments, IDM supplementation was used as the standard adipocytogenic condition.
Regulation of Adipocyte Differentiation in Human and Murine MSCs
hMSCs were cultured in 12-well tissue culture plates; after they reached confluence, the medium was changed to α-minimal essential medium containing 1% FBS-HI and supplemented with IDM, which had been shown to promote PPARγ2 and LPL gene expression and development of lipid-containing cells as described above. hMSCs were treated with 15 μm hypoxia-mimetic deferoxamine mesylate (DFO; 15 mm stock solution in tissue culture-grade sterile phosphate-buffered saline (PBS); Sigma), with 1 ng/ml TGF-β1 (R&D Systems, Minneapolis, MN), or with the vehicle control (0.1% bovine serum albumin in PBS). Three weeks later, lipid accumulation in cells was visualized by staining with 0.3% oil red O (Sigma) (20). The lipid-containing cells in three to six random areas of 1 mm2 were enumerated in each culture well (n = 9) per group. The means ± S.D. are presented. The phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002, the p38 MAPK inhibitor SB203580, the p42/44 MAPK inhibitor PD098059, and the TGF-β type I receptor ALK-5 (activin receptor-like kinase-5) inhibitor SB431542 were purchased from Sigma, and the stock solutions in Me2SO were kept at −20 °C. For murine MSCs, after reaching confluence, the medium was changed to McCoy’s 5A medium containing 1% FBS-HI and supplemented with IDM for 4 days, and then the adipocytogenic supplements were changed to 10 μg/ml insulin only. Seven days after treatment with the vehicle control, 15 μm DFO, or 1 ng/ml TGF-β1 with or without 10 μm SB431542, lipid accumulation in cells was visualized by staining with 0.3% oil red O. The lipid-containing cells in three to six random areas of 1 mm2 were enumerated in each culture well (n = 3) per group.
Cell Viability Assay
Confluent cultures of hMSCs (from a 42-year-old woman) were exposed to increasing concentrations of DFO (1–1,000 μm). Cells were collected after 24 h by trypsinization, and, including cells floating in the medium, cell viability was assessed using the trypan blue exclusion assay. The percentage of viable cells was calculated as the number of viable cells (unstained) divided by the sum of dead (stained) and viable cells.
Western Blotting
hMSCs (from a 42-year-old woman) were cultured in 100-mm dishes. After reaching confluence, the cells were cultured in α-minimal essential medium containing 1% FBS-HI and supplemented with IDM under hypoxic (2% O2) or standard (19% O2) conditions with or without DFO (15 μm) or TGF-β1 (1 ng/ml) for 16 h. Whole cell lysates were prepared with urea denaturing buffer containing 6.7 m urea, 10 mm Tris-HCl (pH 6.8), 5 mm dithiothreitol, 1% SDS, 10% glycerol, and a mixture of protease inhibitors (Roche Diagnostics). They were homogenized with a Kontes Pellet Pestle and separated from insoluble cell materials by centrifugation at 16,000 × g in an Eppendorf bench-top centrifuge at 4 °C. Protein concentration was determined using the BCA system (Pierce). Western immunoblotting was performed as described (20, 23). In brief, proteins were resolved electrophoretically through 4–12% SDS-polyacrylamide gel (NuPAGE® BisTris gel, Invitrogen) and transferred to polyvinylidene difluoride membranes (Amersham Biosciences). The membranes were blocked with 5% nonfat milk in PBS containing 0.1% Tween 20 (PBST) for 2–3 h at room temperature and then incubated overnight with primary antibodies at 4 °C. The anti-HIF-1α primary antibody was purchased from BD Biosciences; the rabbit anti-phospho-Smad2/3 (Ser433/Ser435) IgG antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); and the anti-β-actin antibody was purchased from Sigma. After removal of the unbound primary antibodies by three 10-min washes with PBST, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature and washed three times for 10 min with PBST. The horseradish peroxidase-conjugated anti-mouse IgG secondary antibody was purchased from Amersham Biosciences, and the horseradish peroxidase-conjugated anti-rabbit IgG antibody was purchased from Santa Cruz Biotechnology, Inc. The antibody-associated protein bands were visualized with the ECL Plus Western blotting system (Amersham Biosciences).
RNA Isolation and RT-PCR
Total RNA was isolated from MSCs with TRIzol. For RT-PCR, 2 μg of total RNA was reverse-transcribed into cDNA with SuperScript II (Invitrogen) following the manufacturer’s instructions. One-tenth of the cDNA was used in each 50-μl PCR (30–35 cycles of 94 °C for 1 min, 55–60 °C for 1 min, and 72 °C for 2 min) with gene-specific primers. The human LPL forward primer is 5′-GAGATTTCTCTGTATGGCACC-3, and the reverse primer is 5′-CTGCAAATGAGACACTTTCTC-3′ (1261–1536 bp; GenBankTM accession number NM_000237.1). Primers for the human PPARγ2 (24) and mouse adipsin (25) genes were used for amplification as described previously.
Gene Macroarray
GEArrayTM human TGF-β/bone morphogenetic protein signaling pathway and insulin pathway macroarrays (Super-Array Bioscience Corp., Bethesda, MD) were performed as described previously (20, 26). In brief, 3 μg of total RNA was reverse-transcribed with [α-32P]dCTP (PerkinElmer Life Sciences) and the reagents provided by the manufacturer. The labeled cDNAs were hybridized overnight to the macroarrays; washed; and exposed to x-ray film for 5, 16–24, 48, and 72 h. Digital images were obtained by scanning autoradiographs with an Epson transparency adapter. Data were extracted from the images with ScanAlyze software as described in detail elsewhere (26). Data were organized by functional category of the genes, and a qualitative measure of relative expression under control conditions is indicated. Genes whose expression levels under control conditions were measured during short film exposures (5–24 h) were classified as high expression (+++); 48-h film exposures were classified as low expression (++); and 72-h film exposures were classified as very low expression (+). Undetectable or absent calls (−) were made for pixel intensity equal to the background level. The expression value for each gene on the array was calculated with GEArrayAnalyzerTM software provided by the manufacturer and was verified by visual comparison with the autoradiographs. The usable linear range of pixel intensity was established as 9,000–65,000 as described previously (26). The quantitative magnitude of change evoked by treatment is expressed as fold change upon treatment relative to the control. A qualitative increase or decrease or no change evoked by treatment relative to the control is indicated for genes that were expressed at levels too low for quantification or where a neighboring strong spot bled into the feature of interest.
Northern Blot Analysis
Northern blot analysis was performed with modification as described previously (26). In brief, 10 μg of total RNA was electrophoretically resolved on formaldehyde-containing 1% aga-rose gel and blotted onto a positively charged nylon membrane (Roche Diagnostics) by downward capillary transfer in 20× SSC. The nylon membrane was prehybridized in formamide prehybridization/hybridization buffer (5× SSC, 5× Denhardt’s solution, 1% SDS, 50% formamide, and 0.1 mg/ml denatured salmon sperm DNA) for 3 h at 42 °C. Hybridization was performed overnight in formamide prehybridization/hybridization buffer with random-labeled probes at 42 °C. The probes for human TGFBI, plasminogen activator inhibitor-1 (PAI-1), PKM2, and INSR (insulin receptor) were labeled with a random-primed DNA labeling kit (Roche Diagnostics) with 25 ng of RT-PCR products of each gene and purified using the QIAquick nucleotide removal kit (Qiagen Inc.). The primers for human TGFBI (26), PAI-1 (27), PKM2 (28), and INSR (29) were described previously. The internal control 18 S oligonucleotide probe (Ambion Inc.) was labeled with a DNA 5′-end labeling kit (Roche Diagnostics). For removal of nonspecifically bound probes, membranes were washed twice with 2× SSC with 0.1% SDS (5 min each) and twice with 0.2× SSC and 0.1% SDS (5 min each) at room temperature and twice with 0.2× SSC and 0.1% SDS (15 min each) at 42 °C. Hybridization was visualized by autoradiography at −80 °C with Eastman Kodak X-Omat Blue XB-1 film.
Statistical Analysis
All experiments were performed two to five times. Data are presented as the means ± S.D. Quantitative data were analyzed by one-way analysis of variance. A p value <0.05 was considered significant.
RESULTS
Hypoxia and Its Mimetic DFO Stabilize HIF-1α and Activate the TGF-β Signal in hMSCs
The effects of hypoxia, hypoxia-mimetic DFO, and TGF-β 1 on HIF-1α and TGF-β intracellular signaling molecule Smad proteins in hMSCs (from a 42-year-old woman) were assessed by Western immunoblot analysis. Confluent hMSCs were cultured in α-minimal essential medium containing 1% FBS-HI and supplemented with IDM for 16 h under hypoxic (2% O2) or standard (19% O2) conditions with or without 15 μm hypoxia-mimetic DFO or 1 ng/ml TGF-β1. Hypoxia (2% O2) and its mimetic DFO (15 μm) up-regulated HIF-1α protein (120 kDa) compared with normoxia and the vehicle control (Fig. 1). Under these conditions, TGF-β1 (1 ng/ml) did not modulate HIF-1α protein level in hMSCs. Similar results were obtained in the human bone marrow stromal cell line KM101 (data not shown). In addition, hypoxia, its mimetic DFO, and TGF-β1 activated the TGF-β signal as shown by increased phosphorylated Smad2/3 in hMSCs by Western blotting (Fig. 1).
Fig. 1. Effects of hypoxia, hypoxia-mimetic DFO, or TGF-β on HIF-1α and phospho-Smad2/3.
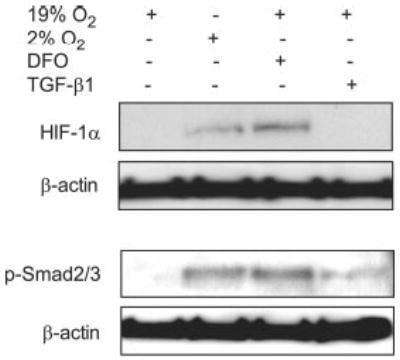
The HIF-1α and phospho-Smad2/3 (p-Smad2/3) proteins were evaluated by Western immunoblotting. hMSCs obtained from a 42-year-old woman were cultured for 16 h under hypoxic (2% O2) or normoxic (19% O2) conditions with or without 15 μm DFO or 1 ng/ml TGF-β1. Five μg of protein from whole cell lysates was loaded in each sample. β-Actin was used as a control.
Hypoxia-mimetic DFO and TGF-β Inhibit Adipocytogenesis and Adipocyte Gene Expression in hMSCs
hMSCs (from a 42-year-old woman) were used to assess the effects of hypoxia and TGF-β on adipocyte differentiation. After 3 weeks of culture in adipocytogenic supplements with or without hypoxia-mimetic DFO (2.5–15 μm) or TGF-β1 (1 ng/ml), development of lipid-containing cells was visualized by staining with oil red O (Fig. 2A). DFO significantly blocked adipocyte differentiation in a dose-dependent manner (38.8, 20.4, and 1.0% versus the control at 2.5, 5.0, and 15.0 μm DFO, respectively) (Fig. 2B). TGF-β1 significantly decreased adipocyte number (1.4% versus the control; p < 0.001) (Fig. 2B). Both DFO (50 μm) and TGF-β1 (1 ng/ml) blocked expression of the PPARγ2 and LPL adipocyte genes in hMSCs as shown by RT-PCR (Fig. 2C).
Fig. 2. Effects of hypoxia-mimetic DFO or TGF-β on adipocyte differentiation in hMSCs.
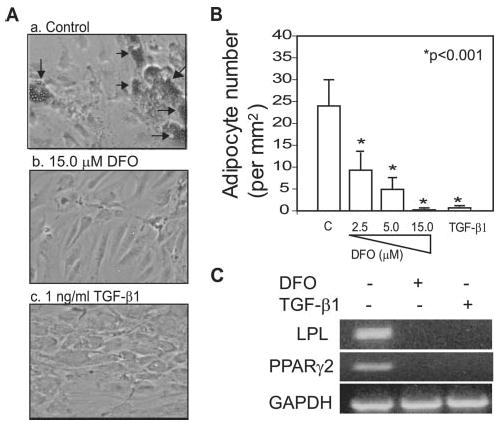
A, shown is the morphology of hMSCs (from a 42-year-old woman) cultured in adipocytogenic medium for 21 days (oil red O staining; magnification ×100). Arrows indicate lipid-containing cells. B, the number of lipid-containing cells was determined in three to six random areas of 1 mm2 in each of nine culture wells. *, p < 0.001 (treated versus the control). C, RT-PCR results show that 1 ng/ml TGF-β1 or 50 μm DFO blocked PPARγ2 and LPL gene expression. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene.
Hypoxia-mimetic DFO Does Not Alter Cell Viability
The effect of DFO on cell viability was determined with the trypan blue exclusion assay. After 24 h of treatment with 0, 1, 10, 50, 100, and 1000 μm DFO in α-minimal essential medium containing 1% FBS-HI, the viability of hMSCs (from a 42-year-old woman) was 97, 96, 97, 95, 95, and 92%, respectively. Following 3 weeks of treatment with 2.5, 5.0, 15.0, and 50 μm DFO in α-minimal essential medium containing 1% FBS-HI and supplemented IDM, the viability of hMSCs was similar for DFO-treated and control groups.
Hypoxia and TGF-β Alter Gene Expression in Adipocyte-associated Pathways
Because both hypoxia and TGF-β inhibited adipocyte differentiation in hMSCs, we assessed whether hypoxia and TGF-β have similar effects on adipocyte-associated signaling pathways. We surveyed effects on gene expression with a cDNA macroarray. Many categories of adipocyte-associated genes were coordinately up- or down-regulated upon 48 h of exposure to hypoxia (2% O2) (Table I). Hypoxia down-regulated PPARγ and its target genes, including adipsin (43% versus the normoxic control), AEBP1 (70%), and SLC27A4 (38%). Among the insulin receptor-associated genes, hypoxia down-regulated CAP (adenylyl cyclase-associated protein; 53%), INSR (84%), NCK2 (32%), CRK (63%), and PPP1CA (protein phosphatase-1, catalytic subunit, alpha isoform). Expression of the primary insulin target gene v-jun was decreased by hypoxia (64%). In the PI3K pathways, hypoxia altered gene expression of PI3K pathway components, including EIF4EBP1 (up-regulation), PIK3R1 (12%), PIK3R2 (25%), and GSK3A and GSK3B (down-regulation), and PI3K pathway target genes, including PAI-1 (426%), vascular endothelial growth factor (VEGF; 210%), PKM2 (310%), the low density lipoprotein receptor, and ACACB (acetyl-coenzyme A carboxylase beta; down-regulation). In the MAPK pathway, hypoxia down-regulated MAPK pathway components, including MAP2K2 (55%), K-ras, and c-raf, and MAPK pathway target genes, including angiogenin (88%) and bcl-x (48%). Under these conditions, there was no effect on insulin-activated transcription factor SREBP1/ADD1 gene expression, but there was up-regulation of the SREBP1 target genes PKM2 (310%) and PCK2 (313%) and down-regulation of ACACB (acetyl-coenzyme A carboxylase beta) in hMSCs.
Table I.
Adipocyte-associated signaling and target gene expression profiles of hMSCs after exposure to hypoxia (2% O2) or treatment with TGF-β 1 (1 ng/ml) for 48 h
| Fold change byb |
||||
|---|---|---|---|---|
| Category and gene | GenBankTM accession no. | Level of expression (control)a | Hypoxia | TGF-β 1 |
| PPAR γ and its target genes | ||||
| PPAR γ | NM_015869 | + | ↓ | ↓ |
| Adipsin | NM_001928 | +++ | 0.43 | 0.36 |
| AEBP1 | NM_001129 | +++ | 0.70 | 3.46 |
| C/EBP-α | NM_004364 | − | ↔ | ↔ |
| C/EBP-β | NM_005194 | ++ | ↓ | ↔ |
| SLC27A4 | NM_005094 | ++ | 0.38 | 0.76 |
| SREBP1 and its target genes | ||||
| SREBP1 | NM_004176 | + | ↔ | ↔ |
| PKM2 | NM_002654 | ++ | 3.10 | 1.89 |
| PCK2 | NM_004563 | ++ | 3.13 | 1.22 |
| ACACB | NM_001093 | + | ↓ | ↓ |
| Insulin receptor-associated genes | ||||
| CAP | BC017196 | +++ | 0.53 | 2.04 |
| INSR | NM_000208 | +++ | 0.84 | 0.49 |
| NCK2 | NM_003581 | +++ | 0.32 | 0.67 |
| PTPRF (LAR) | NM_002840 | ++ | 1.13 | 1.49 |
| CRK | NM_016823 | ++ | 0.63 | 0.41 |
| SHC1 | U73377 | ++ | 1.06 | 1.63 |
| PTPN11 | NM_002834 | + | 0.96 | 1.64 |
| PPP1CA | AB028949 | + | ↓ | ↓ |
| Insulin signaling target gene | ||||
| v-jun | NM_002228 | ++ | 0.64 | 0.33 |
| PI3K pathway | ||||
| Components | ||||
| AKT (PKBβ)c | M77198 | ++ | ↔ | ↔ |
| EIF4EBP1 | NM_004095 | ++ | ↑ | 2.52 |
| PIK3R1 | M61906 | ++ | 0.12 | 27 |
| PIK3R2 | NM_005027 | + | 0.25 | ↔ |
| GLUT1 | NM_006516 | + | ↔ | 1.71 |
| GSK3B | NM_002093 | + | ↓ | ↔ |
| GSK3A | NM_019884 | + | ↓ | ↓ |
| Target genes | ||||
| PAI-1 | M16006 | ++ | 4.26 | 4.51 |
| VEGF | NM_003376 | + | 2.10 | 1.22 |
| LDLR | NM_000527 | + | ↓ | ↔ |
| MAPK pathway | ||||
| Components | ||||
| RRAS | NM_006270 | ++ | 1.35 | 0.82 |
| MAP2K1 | NM_002755 | + | ↔ | 0.28 |
| MAP2K2 | L11285 | + | 0.55 | 0.31 |
| SOS2 | L13858 | + | 1.07 | 1.18 |
| K-ras | M54968 | + | ↓ | ↔ |
| c-raf | X03484 | + | ↓ | ↔ |
| Target genes | ||||
| Angiogenin | M11567 | +++ | 0.88 | 0.64 |
| bcl-x | Z23115 | ++ | 0.48 | 0.47 |
| TIEG | U21847 | + | ↔ | 1.10 |
| ERCC1 | M28650 | + | ↔ | ↑ |
Symbols indicate the relative level of expression in controls: +++, high expression, normalized to the housekeeping gene peptidylprolyl isomerase A or β-actin (16–24-h exposure); ++, low expression, normalized to the housekeeping gene peptidylprolyl isomerase A (48-h exposure); +, very low, normalized to the housekeeping gene peptidylprolyl isomerase A (3-day exposure); −, undetectable, no normalized data (3-day exposure).
Arrows indicate qualitative change by hypoxia or TGF-β1 in genes for which quantification was not possible: ↑, increase; ↓, decrease; ↔, no change.
PKBβ, protein kinase Bβ; LDLR, low density lipoprotein receptor.
RNA obtained from hMSCs treated for 48 h with TGF-β1 (1 ng/ml) or the vehicle control was also evaluated using the signaling pathway macroarray (Table I). TGF-β1 decreased expression of PPARγ and its target genes adipsin (36% versus the control) and SLC27A4 (76%) and increased expression of AEBP1 (346%). TGF-β1 did not affect expression of CAAT/enhancer-binding protein (C/EBP)- β, and C/EBP-α was not detected in this analysis. Among the insulin receptor-associated genes, TGF-β1 down-regulated INSR (49%), NCK2 (67%), CRK (41%), and PPP1CA and up-regulated CAP (204%), LAR (149%), SHC1 (163%), and PTPN11 (164%). TGF-β1 inhibited the insulin signaling target gene v-jun (33%). Among the components of the PI3K pathway, TGF-β1 increased expression of EIF4EBP1 (252%) and GLUT1 (171%) and decreased GSK3A, whereas some genes were unaffected (AKT, GSK3B, and PIK3R2). Among the PI3K target genes, TGF-β1 up-regulated expression of PAI-1 (451%) and VEGF (122%), down-regulated ACACB, and had no effect on SREBP1 and the low density lipoprotein receptor. In the MAPK pathway, TGF-β1 down-regulated MAPK pathway components, including RRAS (82%), MAP2K1 (28%), and MAP2K2 (31%); decreased MAPK target genes, including angiogenin (64%) and bcl-x (47%); and increased MAPK target genes, including TIEG (110%) and ERCC1. Under these experimental conditions, TGF-β1 had no effect on insulin-activated transcription factor SREBP1/ADD1 gene expression, but up-regulated the SREBP1 target genes PKM2 (189%) and PCK2 (122%) and down-regulated ACACB.
Changes in adipocyte-associated signaling and target genes induced by hypoxia were compared with changes induced by TGF-β (Table I). First, there were very similar effects on most genes, such as decreased expression of PPARγ and its target genes adipsin and SLC27A4; the insulin receptor-associated genes INSR, NCK2, CRK, and PPP1CA; the insulin target gene v-jun, PI3K pathway-associated genes such as PIK3R1 and GSK3A; MAPK pathway-associated genes such as MAP2K2, angiogenin; and bcl-x; and the SREBP1 target gene ACACB. In addition, both hypoxia and TGF-β1 increased expression of the SREBP1 target genes PKM2 and PCK2 and the PI3K component gene EIF4EBP1 and its PAI-1 and VEGF target genes. Second, there were different effects on some genes, including the PPARγ target gene AEBP1; the PI3K pathway component genes PIK3R2, GLUT1, and GSK3B; the PI3K low density lipoprotein receptor target gene; the MAPK pathway component genes RRAS, MAP2K1, K-ras, and c-raf; and the MAPK target gene ERCC1.
Inhibition of PPARγ2 and LPL Gene Expression in hMSCs by Hypoxia-mimetic DFO, TGF-β1, a PI3K Inhibitor, or MAPK Inhibitors
Specific inhibitors were used to determine the effects of hypoxia, TGF-β, PI3K, and MAPK signaling on PPARγ2 adipocyte master gene and LPL adipocyte marker gene expression. After confluence, hMSCs were cultured with or without IDM and treated with the vehicle control, hypoxia-mimetic DFO (50 μm), TGF-β1 (1 ng/ml), the PI3K inhibitor LY294002 (40 μm), the p38 MAPK inhibitor SB203580 (10 μm), or the p42/44 MAPK inhibitor PD098059 (50 μm). Analysis by RT-PCR showed that hMSCs expressed the PPARγ2 and LPL genes when cultured with IDM for 7 days (Fig. 3A). Hypoxia-mimetic DFO, TGF-β1, the PI3K inhibitor LY294002, and the p38 MAPK inhibitor SB203580 significantly down-regulated PPARγ2 and LPL gene expression compared with the IDM control (n = 3; p < 0.001) (Fig. 3, A and B). In contrast, the p42/44 MAPK inhibitor PD098059 appeared to up-regulate the PPARγ2 gene (p < 0.05, n = 3) and did not modulate LPL gene expression compared with the IDM control (Fig. 3, A and B).
Fig. 3. Effects of hypoxia-mimetic DFO, TGF-β, or PI3K or MAPK inhibitors on PPARγ2 and LPL gene expression in hMSCs.
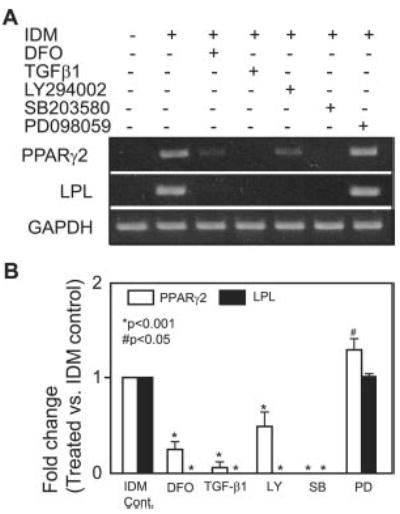
A, the effects of hypoxia-mimetic DFO (50 μm), TGF-β1 (1 ng/ml), the PI3K inhibitor LY294002 (LY; 40 μm), the p38 MAPK inhibitor SB203580 (SB; 10 μm), or the p42/44 MAPK inhibitor PD098059 (PD; 50 μm) after 7 days of treatment on PPARγ2 and LPL gene expression in hMSCs (from a 42-year-old woman) were determined by semiquantitative RT-PCR. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. B, quantitative data (n = 3) indicate the -fold difference of treatment versus IDM control (Cont.) after normalization to the glyceraldehyde-3-phosphate dehydrogenase housekeeping gene. Data analysis was performed by one-way analysis of variance.
Hypoxia Alters Gene Expression in the TGF-β/Smad Signaling Pathway
A specific macroarray was used to assess the effects of hypoxia on TGF-β/Smad signaling in hMSCs cultured with IDM for 48 h (Table II). Under control normoxic conditions (19% O2), hMSCs expressed TGF-β type II and III receptors Smad2, Smad5, and Smad9; their expression was not modulated by hypoxia (2% O2). Hypoxia up-regulated TGF-β1 and its intracellular signaling molecule Smad3 and target genes, including TGFBI, PAI-1, TIMP1, COL1A2, p21WAF1/CIP1, etc. The genes altered most by hypoxia in hMSCs were TGFBI (310% versus the normoxic control), PAI-1 (361%), and inhibin-α (12%). Hypoxia moderately up-regulated COL1A2 (142%), TIMP1 (185%), p21, ALK-1, ALK-6, and RUNX1/AML1 and down-regulated STAT1, RUNX2/CBFA1 (71%), endoglin (70%), and v-jun (83%) gene expression. These data demonstrate that hypoxia activates the TGF-β/Smad signaling pathway.
Table II.
TGF-β/bone morphogenetic protein signaling and target gene expression profiles of hMSCs exposed to hypoxia (2% O2) for 48 h
| Category and gene | GenBankTM accession no. | Level of expression (normoxia)a | Fold change by hypoxiab |
|---|---|---|---|
| Cytokines | |||
| Inhibin-α | NM_002191 | ++ | 0.12 |
| TGF-β1 | X02812 | + | ↑ |
| Signaling molecules | |||
| RUNX2/CBFA1 | L40992 | +++ | 0.71 |
| RUNX1/AML1 | NM_001754 | + | ↑ |
| Endoglin | NM_000118 | ++ | 0.70 |
| Smad1 | HSU59912 | − | ↔ |
| Smad2 | NM_005901 | + | ↔ |
| Smad3 | NM_005902 | + | ↑ |
| Smad4 | U44378 | − | ↔ |
| Smad5 | NM_005903 | + | ↔ |
| Smad6 | NM_005585 | − | ↔ |
| Smad7 | NM_005904 | − | ↔ |
| Smad9 | NM_005905 | + | ↔ |
| Smad target genes | |||
| TGFBI | NM_000358 | +++ | 3.10 |
| COL1A2 | NM_000089 | +++ | 1.42 |
| COL3A1 | NM_000090 | +++ | 0.95 |
| TIMP1 | NM_00325 | +++ | 1.85 |
| v-jun | NM_002228 | +++ | 0.83 |
| PAI-1 | M16006 | + | 3.61 |
| IGFBP3 | M31159 | + | 1.03 |
| STAT1 | M97935 | + | ↓ |
| p21WAF1/CIP1 | L47233 | + | ↑ |
| Receptors | |||
| TGF-β type II | D50683 | ++ | ↔ |
| TGF-β type III | NM_003242 | ++ | ↔ |
| ALK-1 | NM_000020 | + | ↑ |
| ALK-2 | NM_001105 | + | ↔ |
| ALK-3 (BMPR1A) | NM_004329 | + | ↔ |
| ALK-4 | NM_004302 | − | ↔ |
| ALK-5 (TGF-β type I) | L11695 | − | ↔ |
| ALK-6 (BMPR1B) | NM_001203 | − | ↔ |
| BMPR2 | Z48923 | + | ↑ |
| ACVR2 | NM_001616 | + | ↔ |
| AMHR2 | NM_020547 | + | ↔ |
Symbols indicate the relative level of expression with normoxia: +++, high expression, normalized to the housekeeping gene RP13A (5–6-h exposure); ++, low expression, normalized to the β-actin housekeeping gene (16-h exposure); +, very low, normalized to the housekeeping gene peptidylprolyl isomerase A (3-day exposure); −, undetectable (3-day exposure).
Arrows indicate the qualitative change by hypoxia in genes for which quantification was not possible: ↑, increase; ↓, decrease; ↔, no change.
Confirmation of Macroarray Data by Northern Blot Analysis
The expression levels of selected genes that were shown by macroarrays to be affected by hypoxia and/or TGF-β were evaluated by Northern blotting (Fig. 4A). This showed changes similar to those in the arrays, but with different magnitudes (Fig. 4B). Hypoxia (2% O2 for 48 h) up-regulated TGFBI/BIGH3 (383% versus the normoxic control), PAI-1 (686%), and PKM2 (135%) and down-regulated INSR (35%) gene expression. TGF-β1 (1 ng/ml for 48 h) had similar effects compared with hypoxia on expression of these genes. TGF-β1 up-regulated TGFBI/BIGH3 (537% versus the control), PAI-1 (1343%), and PKM2 (66%) and down-regulated INSR (44%) gene expression.
Fig. 4. Confirmation of macroarray data by Northern blot analysis.
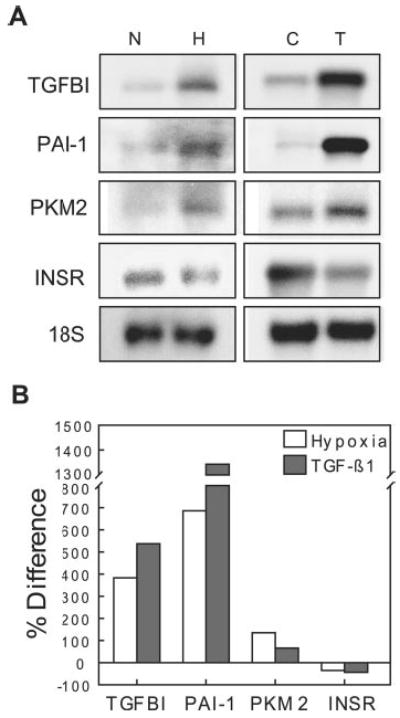
A, Northern blot analysis was performed on selected genes that were shown to be altered by hypoxia (H; 2% O2 for 48 h) versus normoxia (N) or TGF-β1 (T) (1 ng/ml for 48 h) versus the vehicle control (C) in macroarrays. B, expression levels are presented as the percent difference of band density for hypoxia versus normoxia or for TGF-β1-treated versus the vehicle control after normalization to 18 S ribosomal RNA.
Smad3 Is One of the Mediators of Inhibition of Adipocyte Differentiation in Murine MSCs by Hypoxia or TGF-β
Murine MSCs were used to assess the role of Smad3 in mediating the inhibition of adipocytogenesis. Smad3−/− MSCs were derived from bone marrow cultures of neonatal Smad3-null mice; Smad3+/+ MSCs were from wild-type mice. There were two notable differences in adipocyte development in bone marrow cultures from Smad3−/− and Smad3+/+ mice. First, there was more extensive (37-fold) adipocyte differentiation in bone marrow cultures from Smad3−/− mice than from Smad3+/+ mice cultured under adipocytogenic conditions (Fig. 5, A and B); this finding suggests that Smad3 is a critical inhibitor of adipocyte differentiation. Second, there was a 7-fold attenuation of TGF-β inhibition and a 2-fold attenuation of DFO inhibition of adipocytogenesis in cells lacking Smad3 (Fig. 5B). Hypoxia-mimetic DFO (15 μm) decreased adipocyte number in Smad3+/+ MSCs (32.9% versus the control; p < 0.05) compared with Smad3−/− MSCs (67.7% versus the control; p < 0.05). TGF-β1 (1 ng/ml) decreased adipocyte number in Smad3+/+ cells (5.5% versus the control; p < 0.05) compared with Smad-3−/− cells (39.2% versus the control; p < 0.05). These data support the conclusion that Smad3 is required for the inhibition of adipocyte differentiation by either TGF-β or hypoxia. The effect of Smad3 on lipid accumulation was reflected in the expression pattern of the adipocyte marker gene adipsin in Smad3+/+ and Smad3−/− MSCs. After 7 days of treatment, 15 μm DFO or 1 ng/ml TGF-β1 down-regulated adipsin gene expression in Smad3+/+ MSCs, but not in Smad3−/− MSCs (Fig. 5C). SB431542 (10 μm), a specific inhibitor of the TGF-β type I receptor (ALK-5), antagonized the inhibitory effects of DFO and TGF-β1 on adipsin gene expression in Smad+/+ MSCs (Fig. 5C). This is confirmation that TGF-β/Smad is required for the inhibition of adipocyte differentiation by either TGF-β or hypoxia.
Fig. 5. Smad3 mediation of the inhibition of adipocytogenesis in MSCs by hypoxia-mimetic DFO or TGF-β.
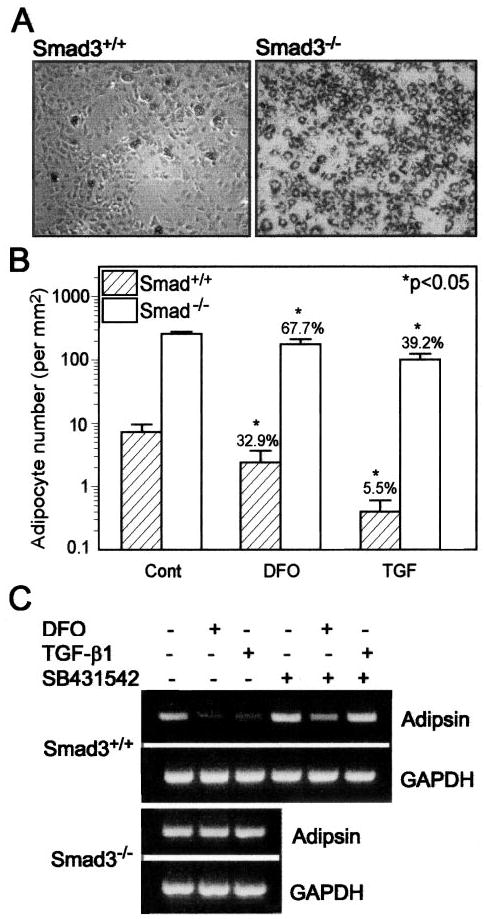
A, the micrographs show the morphology of murine Smad3+/+ and Smad3−/− MSCs cultured in adipocytogenic medium for 7 days (oil red O staining; magnification ×50). B, the number of lipid-containing cells in control (Cont) cultures and in cultures treated with either 15 μm DFO or 1 ng/ml TGF-β1 was determined in three to six random areas of 1 mm2 in each of three culture wells. *, p < 0.05 (treated versus the control, one-way analysis of variance). The percent change of the treated group versus the control group is shown on the top of each bar. C, the effects of 15 μm DFO or 1 ng/ml TGF-β1 with or without the ALK-5 inhibitor SB431542 (10 μm) after 7 days of treatment on adipsin adipocyte marker gene expression in Smad3+/+ and Smad3−/− MSCs were determined by RT-PCR. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
DISCUSSION
Although hypoxia and TGF-β both inhibit adipocyte differentiation, the relationship between their signaling pathways in adipocytogenesis was unknown. In this study, we investigated the mechanisms of inhibition of adipocyte differentiation of MSCs by hypoxia and TGF-β as well as the role of the TGF-β/Smad signaling pathway in this process.
Hypoxia-induced gene expression is mediated by HIF-1α. Hypoxia stabilizes the HIF-1α protein, which is otherwise (under normoxic conditions) degraded by a ubiquitin-dependent proteasome (5–7). Our results show that hypoxia and its mimetic DFO stabilized the HIF-1α protein in hMSCs and that continued treatment with DFO inhibited adipocyte differentiation and PPARγ2 and LPL adipocyte gene expression in hM-SCs. It has been reported that many growth factors, cytokines, and circulatory factors, including insulin, insulin-like growth factor-1 and -2, platelet-derived growth factor, epidermal growth factor, epidermal growth factor-2, TGF-β1, hepatocyte growth factor, tumor necrosis factor-1, interleukin-1β, etc., stimulate HIF-1α through pathways distinct from that employed by the classical hypoxic pathway (30). Our results show that 1 ng/ml TGF-β1 blocked adipocyte differentiation and down-regulated PPARγ2 and LPL adipocyte gene expression, but did not change HIF-1α protein levels in hMSCs. These data indicate that hypoxia may inhibit adipocytogenesis of hMSCs by stabilizing HIF-1α, but that, under these conditions, TGF-β does not require the stabilization of HIF-1α for its inhibition of adipocytogenesis of hMSCs.
To compare the effects of hypoxia and TGF-β on adipocyte-associated signaling, we used a targeted macroarray that included several insulin receptor-associated genes and insulin signaling target genes, PI3K pathway components and target genes, MAPK pathway components and target genes, insulin-activated transcription factor SREBP1 target genes, and PPARγ target genes. The macroarray data were confirmed by Northern blot analysis of several highly expressed genes.
PPARγ appears to function as both a direct regulator of many fat-specific genes and as a master regulator that can trigger the program of adipocytogenesis (31). Hypoxia inhibits PPARγ2 expression in murine 3T3-L1 preadipocytes (8). TGF-β/Smad3 inhibits induction of C/EBP-α and PPARγ in murine 3T3-F442A preadipocytes (12). Our RT-PCR and macroarray results show that both hypoxia and TGF-β1 inhibited PPARγ2 expression in hMSCs and down-regulated several PPARγ target genes. Another transcription factor that promotes adipocytogenesis is the insulin-activated transcription factor SREBP1c (sterol regulatory element-binding protein-1c)/ADD1 (32, 33). It is a basic helix-loop-helix protein expressed abundantly in adipose tissue (34) and has been shown to increase fatty acid and fat synthesis; this has been attributed in part to its proposed influence on PPARγ activity (35). Expression of SREBP1/ADD1 is also increased during osteoblast differentiation; however, this indicates that increased expression of this protein is not specific to adipocyte differentiation (36). Our results show that hypoxia and TGF-β had no effect on SREBP1 gene expression in hMSCs.
Insulin, the major anabolic hormone, promotes in vivo accumulation of adipose tissue. Insulin is a potent inducer of adipocytogenesis, and differentiation of adipocytes requires many components of the insulin signaling pathway, including the insulin receptor, insulin receptor substrate-1, and PI3K (37). There is growing interest in the effects of age on redistribution of body fat deposits, on insulin actions on site-specific adipocytes and their progenitors, and on dysdifferentiation of mesenchymal precursors into mesenchymal adipocyte-like default cells (38). Our macroarray results show strikingly similar effects of hypoxia and TGF-β in decreasing expression of the insulin receptor-associated genes INSR, NCK2, CRK, and PPP1CA and the insulin target gene v-jun. These findings suggest that both treatments block insulin signaling as part of their inhibition of adipocyte differentiation in MSCs.
PI3K is important in a wide variety of cellular processes, including intracellular trafficking; organization of the cytoskeleton; cell growth, differentiation, and transformation; and prevention of apoptosis (39, 40). Pharmaceutical inhibition of PI3K or overexpression of dominant-negative mutant subunits abrogates differentiation of adipocytes in vitro (41, 42); this suggests that the insulin/PI3K/AKT signaling pathway is required for adipocytogenesis. Our RT-PCR results show that the PI3K inhibitor LY294002 down-regulated PPARγ2 and LPL adipocyte gene expression; this demonstrates that the PI3K pathway promotes adipocyte differentiation in hMSCs. Our macroarray results show that hypoxia down-regulated PI3K pathway-associated genes and increased expression of PI3K target genes such as PAI-1 and VEGF. TGF-β1 had similar effects on PI3K signaling gene expression compared with hypoxia. Although PAI-1 and VEGF were named as PI3K pathway target genes in this commercial macroarray, the induction of the PAI-1 (43, 44) and VEGF (45) genes by hypoxia involves HIF-1, hypoxia response elements in the PAI-1 and VEGF gene promoters, and PI3K/AKT (protein kinase B). Therefore, we cannot conclude whether hypoxia activates the PI3K pathway through up-regulation of the PAI-1 and VEGF genes by hypoxia. The effects of hypoxia and TGF-β on the PI3K pathway in adipocyte differentiation in hMSCs need to be further evaluated. Down-regulation of PI3K pathway components, including PIK3R1, PIK3R2, GSK3A, and GSK3B, etc., may contribute to the inhibition of adipocytogenesis in hMSCs by hypoxia and TGF-β because the PI3K inhibitor down-regulated PPARγ2 and LPL gene expression.
Both hypoxia and TGF-β inhibited the p42/44 MAKP signaling pathway during adipocyte differentiation of hMSCs. Reports in the literature concerning the role of the p42/44 MAPK pathways in adipocyte differentiation have been contradictory (46). p42/44 MAPK is required (47, 48) or has no effect or inhibits (49) adipocyte differentiation of preadipocytes under different conditions. Under our experimental conditions (1% FBS-HI, IDM), the p42/44 MAPK inhibitor PD98059 up-regulated the PPARγ2 gene and had no effect on LPL gene expression in hMSCs; this indicates that p42/44 MAPK may inhibit adipocyte differentiation in hMSCs. Thus, the inhibition of the p42/44 MAPK signaling pathway by both hypoxia and TGF-β is unlikely to mediate their inhibition of adipocyte differentiation in hMSCs. It has been reported that JNK suppresses the process of adipocyte differentiation (50), and p38 MAPK has been shown to promote adipocyte differentiation (51). It was unknown whether p38 MAPK and JNK play roles in the effects of hypoxia or TGF-β on adipocyte differentiation. It has been reported that hypoxia activates JNK and p38 MAPK in carcinoma cells (52). Like hypoxia, TGF-β has also been reported to activate the JNK pathway in skeletal muscle cells (53) and the p38 MAPK signaling pathway in murine mesangial cells (54). The effects of hypoxia and TGF-β1 on JNK and p38 MAPK need to be evaluated further in our system. Nevertheless, our RT-PCR data show that the p38 MAPK inhibitor blocked PPARγ2 and LPL gene expression in hMSCs and imply that p38 MAPK is an activator of adipocytogenesis in hMSCs. Thus, the inhibition of adipocyte differentiation is not through activation of p38 MAPK by hypoxia or TGF-β in hMSCs.
Based on these new data, we propose that hypoxia inhibition of adipocytogenesis in hMSCs requires TGF-β/Smad signaling. Hypoxia up-regulated TGF-β1, its intracellular signaling molecule Smad3, and many of its target genes such as TGFBI, PAI-1, TIMP1, COL1A2, and p21WAF1/CIP1 in hMSCs. The experiments with mouse Smad3 cell lines showed that Smad3 was required for inhibition of adipocytogenesis by either DFO or TGF-β1. In addition, SB431542 abrogated the inhibitory effects of DFO on adipocyte gene expression. SB431542 has been identified as an inhibitor of ALK-5 (TGF-β type I receptor) (55). These data demonstrate that hypoxia inhibition of adipocyte differentiation in MSCs requires activation of the TGF-β/Smad signaling pathway. There is no information about bone marrow or body fat in Smad3−/− mice or about the mechanism of their substantial perinatal mortality. Recently, however, we reported that long-term bone marrow cultures established from neonatal Smad3−/− mice show 16.5-fold more adipocytes in the adherent layer and prolonged hematopoiesis (>20 weeks) compared with Smad3+/+ mice (56). Those data confirm the negative regulation by endogenous TGF-β of adipocytogenesis and long-term hematopoiesis in vitro.
Hypoxia activation through TGF-β/Smad signaling is known in other systems. It has been reported that hypoxia stimulates collagen synthesis and COL1A1 transcription through the action of TGF-β1 in human dermal fibroblast (17). Cooperation between the hypoxia and TGF-β pathways is required for regulation of several genes, including endoglin (57), erythropoietin (58), VEGF (59), etc. Hypoxia results in a significant increase in TGF-β1 and TGF-β type I and II receptor mRNA levels in human peritoneal fibroblasts (14). Exposure of human umbilical vein endothelial cells to hypoxia results in phosphorylation and nuclear transportation of Smad2 and Smad3 proteins as well as stimulation of the transcriptional activities of Smad3 and HIF-1α and culminates in up-regulation of TGF-β2 gene expression (16). Thus, Smad proteins may play an important role in vascular responses to hypoxia and ischemia (15). It has been reported that reactive oxygen species mediate TGF-β-induced expression of PAI-1 in rat mesangial cells (60) and TIMP3 in human and bovine primary articular chondrocytes (61) and apoptosis in rat fetal hepatocytes (62). It is unknown whether reactive oxygen species have a role in the TGF-β/Smad-mediated inhibition of adipocytogenesis by hypoxia in hMSCs.
Understanding the balance between positive and negative regulators of adipogenesis has important health-related implications for anti-obesity medical therapy and lipodystrophy (63). TGF-β has been shown to be increased in adipose tissue in obese mice (64). However, overexpression of TGF-β1 in adipose tissue in transgenic mice results in a dramatic reduction in total body fat (65). Adipose tissue growth is angiogenesis-dependent (66). Hypoxia has been reported to increase the expression of pro-angiogenic factors in adipocytes and fat tissues (67, 68). Our results show that hypoxia and TGF-β modulated expression of pro-angiogenic genes such as VEGF and angiogenin and inhibited adipocyte differentiation in hMSCs. The relationship between the effects of hypoxia and TGF-β on adipocyte differentiation of MSCs and adipose tissue in obesity merits further investigation. Because of the relationship between bone marrow fat and skeletal aging, these results may also have implications for the regulation of MSC differentiation into adipocytes and osteoblasts.
In summary, hypoxia and TGF-β inhibited adipocyte differentiation and modulated adipocyte-associated signaling pathways in MSCs. Hypoxia activated the TGF-β/Smad signaling pathway, and the TGF-β intracellular signaling molecule Smad3 was necessary for the inhibition of adipocytogenesis by both TGF-βand hypoxia. These findings indicate that TGF-β/Smad signaling is required for the inhibition of adipocyte differentiation by hypoxia.
Acknowledgments
We greatly appreciate help from Drs. K. C. Flanders, M. Kikuchi, S. Mizuno, X. Wang, C. Wykoff, K. E. Yates, and M. Epperly with different aspects of these experiments.
Footnotes
This work was supported by Grants AR45870 and AG 025015 from the National Institutes of Health (to J. G.). This work was presented as a plenary poster at the 26th Annual Meeting of the American Society for Bone and Mineral Research, October 1–5, 2004, Seattle, WA.
The abbreviations used are: HIF-1α, hypoxia-inducible factor-1α; TGF-β, transforming growth factor-β; hMSCs, human marrow stromal cells; FBS-HI, heat-inactivated fetal bovine serum; RT, reverse transcription; PPARγ2, peroxisome proliferator-activated receptor-γ2; LPL, lipoprotein lipase; DFO, deferoxamine mesylate; PBS, phosphate-buffered saline; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein kinase; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)-propane-1,3-diol; PAI-1, plasminogen activator inhibitor-1; VEGF, vascular endothelial growth factor; C/EBP, CAAT/enhancer-binding protein; JNK, c-Jun N-terminal kinase.
References
- 1.Semenza GL. Trends Mol Med. 2001;7:345–350. doi: 10.1016/s1471-4914(01)02090-1. [DOI] [PubMed] [Google Scholar]
- 2.Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernascono S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A. J Exp Med. 2003;18:1391–1402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komatsu DE, Hadjiargyrou M. Bone. 2004;34:680–688. doi: 10.1016/j.bone.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 4.Bunn HF, Poyton RO. Physiol Rev. 1996;76:839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- 5.Semenza GL, Wang GL. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang GL, Jiang BH, Rue EA, Semenza GL. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semenza GL. Cell. 2001;107:1–3. doi: 10.1016/s0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 8.Yun Z, Maecker HL, Johnson RS, Giaccia AJ. Dev Cell. 2002;2:331–341. doi: 10.1016/s1534-5807(02)00131-4. [DOI] [PubMed] [Google Scholar]
- 9.Csete M, Walikonis J, Slawny N, Wei Y, Korsnes S, Doyle JC, Wold B. J Cell Physiol. 2001;189:189–196. doi: 10.1002/jcp.10016. [DOI] [PubMed] [Google Scholar]
- 10.Carriere A, Carmona MC, Fernandez Y, Rigoulet M, Wenger RH, Penicaud L, Casteilla L. J Biol Chem. 2004;279:40462–40469. doi: 10.1074/jbc.M407258200. [DOI] [PubMed] [Google Scholar]
- 11.Locklin RM, Oreffo RO, Triffitt JT. Cell Biol Int. 1999;23:185–194. doi: 10.1006/cbir.1998.0338. [DOI] [PubMed] [Google Scholar]
- 12.Choy L, Skillington J, Derynck R. J Cell Biol. 2000;149:667–682. doi: 10.1083/jcb.149.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choy L, Derynck R. J Biol Chem. 2003;278:9609–9619. doi: 10.1074/jbc.M212259200. [DOI] [PubMed] [Google Scholar]
- 14.Saed GM, Collins KL, Diamond MP. Am J Reprod Immunol. 2002;48:387–393. doi: 10.1034/j.1600-0897.2002.01090.x. [DOI] [PubMed] [Google Scholar]
- 15.Akman HO, Zhang H, Siddiqui MA, Solomon W, Smith EL, Batuman OA. Blood. 2001;98:3324–3331. doi: 10.1182/blood.v98.12.3324. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Akman HO, Smith EL, Zhao J, Murphy-Ullrich JE, Batuman OA. Blood. 2003;101:2253–2260. doi: 10.1182/blood-2002-02-0629. [DOI] [PubMed] [Google Scholar]
- 17.Falanga V, Zhou L, Yufit T. J Cell Physiol. 2002;191:42–50. doi: 10.1002/jcp.10065. [DOI] [PubMed] [Google Scholar]
- 18.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 19.Nuttall ME, Gimble JM. Bone. 2000;27:177–184. doi: 10.1016/s8756-3282(00)00317-3. [DOI] [PubMed] [Google Scholar]
- 20.Zhou S, Eid K, Glowacki J. J Bone Miner Res. 2004;19:463–470. doi: 10.1359/JBMR.0301239. [DOI] [PubMed] [Google Scholar]
- 21.Mueller SM, Glowacki J. J Cell Biochem. 2001;82:583–590. doi: 10.1002/jcb.1174. [DOI] [PubMed] [Google Scholar]
- 22.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao JH, Ghosn C, Hinchman C, Forbes C, Wang J, Snider N, Cordrey A, Zhao Y, Chandraratna RAS. J Biol Chem. 2003;278:29954–29962. doi: 10.1074/jbc.M304761200. [DOI] [PubMed] [Google Scholar]
- 24.Schiller PC, D’Ippolito G, Brambilla R, Roos BA, Howard GA. J Biol Chem. 2001;276:14133–14138. doi: 10.1074/jbc.M011055200. [DOI] [PubMed] [Google Scholar]
- 25.Shi X, Shi W, Li Q, Song B, Wan M, Bai S, Cao X. EMBO Rep. 2003;4:374–380. doi: 10.1038/sj.embor.embor805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou S, Glowacki J, Yates KE. J Bone Miner Res. 2004;19:1732–1741. doi: 10.1359/JBMR.040702. [DOI] [PubMed] [Google Scholar]
- 27.Kurisaki K, Kurisaki A, Valcourt U, Terentiev AA, Pardali K, Ten Dijke P, Heldin CH, Ericsson J, Moustakas A. Mol Cell Biol. 2003;23:4494–4510. doi: 10.1128/MCB.23.13.4494-4510.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoo BC, Ku JL, Hong SH, Shin YK, Park SY, Kim HK, Park JG. Int J Cancer. 2004;108:532–539. doi: 10.1002/ijc.11604. [DOI] [PubMed] [Google Scholar]
- 29.Persaud SJ, Asare-Anane H, Jones PM. FEBS Lett. 2002;510:225–228. doi: 10.1016/s0014-5793(01)03268-9. [DOI] [PubMed] [Google Scholar]
- 30.Bilton RL, Booker GW. Eur J Biochem. 2003;270:791–798. doi: 10.1046/j.1432-1033.2003.03446.x. [DOI] [PubMed] [Google Scholar]
- 31.Spiegelman BM, Flier JS. Cell. 1996;87:377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- 32.Rosen ED. Ann N Y Acad Sci. 2002;979:143–158. doi: 10.1111/j.1749-6632.2002.tb04875.x. [DOI] [PubMed] [Google Scholar]
- 33.Gregoire FM, Smas CM, Sul HS. Physiol Rev. 2001;78:783–809. doi: 10.1152/physrev.1998.78.3.783. [DOI] [PubMed] [Google Scholar]
- 34.Kim JB, Spiegelman BM. Genes Dev. 1996;10:1096–1107. doi: 10.1101/gad.10.9.1096. [DOI] [PubMed] [Google Scholar]
- 35.Brun RP, Kim JB, Hu E, Altiok S, Spiegelman BM. Curr Opin Cell Biol. 1996;8:826–832. doi: 10.1016/s0955-0674(96)80084-6. [DOI] [PubMed] [Google Scholar]
- 36.Sawada Y, Noda M. Mol Endocrinol. 1996;10:1238–1248. doi: 10.1210/mend.10.10.9121491. [DOI] [PubMed] [Google Scholar]
- 37.Entingh AJ, Taniguchi CM, Kahn CR. J Biol Chem. 2003;278:33377–33383. doi: 10.1074/jbc.M303056200. [DOI] [PubMed] [Google Scholar]
- 38.Kirkland JL, Tchkonia T, Pirtskhalava T, Han J, Karagiannides I. Exp Gerontol. 2002;37:757–767. doi: 10.1016/s0531-5565(02)00014-1. [DOI] [PubMed] [Google Scholar]
- 39.Toker A, Cantley LC. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 40.Vanhaesebroeck B, Leevers SJ, Peanayoyou G, Waterfield MD. Trends Biochem Sci. 1997;22:267–272. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- 41.Sakaue H, Ogawa W, Matsumoto M, Kuroda S, Takata M, Sugimoto T, Spiegelman BM, Kasuga M. J Biol Chem. 1998;273:28945–28952. doi: 10.1074/jbc.273.44.28945. [DOI] [PubMed] [Google Scholar]
- 42.Xia X, Serrero G. J Cell Physiol. 1999;178:9–16. doi: 10.1002/(SICI)1097-4652(199901)178:1<9::AID-JCP2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 43.Kietzmann T, Roth U, Jungermann K. Blood. 1999;94:4177–4185. [PubMed] [Google Scholar]
- 44.Kietzmann T, Samoylenko A, Roth U, Jungermann K. Blood. 2003;101:907–914. doi: 10.1182/blood-2002-06-1693. [DOI] [PubMed] [Google Scholar]
- 45.Mazure NM, Chen EY, Laderoute KR, Giaccia AJ. Blood. 1997;90:3322–3331. [PubMed] [Google Scholar]
- 46.Aubert J, Belmonte N, Dani C. CMLS Cell Mol Life Sci. 1999;56:538–542. doi: 10.1007/s000180050450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yarwood SJ, Sale EM, Sale GJ, Houslay MD, Kilgour E, Anderson NG. J Biol Chem. 1999;274:8662–8668. doi: 10.1074/jbc.274.13.8662. [DOI] [PubMed] [Google Scholar]
- 48.Sale EM, Atkinson PGP, Sale GJ. EMBO J. 1995;14:674–684P. doi: 10.1002/j.1460-2075.1995.tb07046.x. G. P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fonte de Mora J, Pottas A, Ahn N, Santos E. Mol Cell Biol. 1997;17:6068–6075. doi: 10.1128/mcb.17.10.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Camp HS, Tafuri SR, Leff T. Endocrinology. 1999;140:392–397. doi: 10.1210/endo.140.1.6457. [DOI] [PubMed] [Google Scholar]
- 51.Engelman JA, Lisanti MP, Scherer PE. J Biol Chem. 1998;273:32111–32120. doi: 10.1074/jbc.273.48.32111. [DOI] [PubMed] [Google Scholar]
- 52.Shemirani B, Crowe DL. Oral Oncol. 2002;38:251–257. doi: 10.1016/s1368-8375(01)00052-5. [DOI] [PubMed] [Google Scholar]
- 53.Meriane M, Charrasse S, Comunale F, Gauthier-Rouviere C. Biol Cell. 2002;94:535–543. doi: 10.1016/s0248-4900(02)00023-0. [DOI] [PubMed] [Google Scholar]
- 54.Wang L, Ma R, Flavell RA, Choi ME. J Biol Chem. 2002;277:47257–47262. doi: 10.1074/jbc.M208573200. [DOI] [PubMed] [Google Scholar]
- 55.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 56.Epperly M, Cao S, Goff J, Shields D, Zhou S, Glowacki J, Greenberger JS. Exp Hematol. 2005;33:353–362. doi: 10.1016/j.exphem.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 57.Sanchez-Elsner T, Botella LM, Velasco B, Langa C, Bernabeu C. J Biol Chem. 2002;277:43799–43808. doi: 10.1074/jbc.M207160200. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez-Elsner T, Ramirez JR, Rodriguez-Sanz F, Varela E, Bernabeu C, Botella LM. J Mol Biol. 2004;336:9–24. doi: 10.1016/j.jmb.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 59.Shih SC, Claffey KP. Growth Factors. 2001;19:19–34. doi: 10.3109/08977190109001073. [DOI] [PubMed] [Google Scholar]
- 60.Jiang Z, Seo JY, Ha H, Lee EA, Kim YS, Han DC, Uh ST, Park CS, Lee HB. Biochem Biophys Res Commun. 2003;309:961–966. doi: 10.1016/j.bbrc.2003.08.102. [DOI] [PubMed] [Google Scholar]
- 61.Li WQ, Qureshi HY, Liacini A, Dehnade F, Zafarullah M. Free Radic Biol Med. 2004;37:196–207. doi: 10.1016/j.freeradbiomed.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 62.Herrera B, Alvarez AM, Sanchez A, Fernandez M, Roncero C, Benito M, Fabregat I. FASEB J. 2001;15:741–751. doi: 10.1096/fj.00-0267com. [DOI] [PubMed] [Google Scholar]
- 63.Harp JB. Curr Opin Lipidol. 2004;15:303–307. doi: 10.1097/00041433-200406000-00010. [DOI] [PubMed] [Google Scholar]
- 64.Loskutoff DJ, Fujisawa K, Samad F. Ann N Y Acad Sci. 2000;902:272–281. doi: 10.1111/j.1749-6632.2000.tb06322.x. [DOI] [PubMed] [Google Scholar]
- 65.Clouthier DE, Comerford SA, Hammer RE. J Clin Investig. 1997;100:2697–2713. doi: 10.1172/JCI119815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rupnick MA, Panigrahy D, Zhang CY, Dallabrida SM, Lowell BB, Langer R, Folkman MJ. Proc Natl Acad Sci U S A. 2002;99:10730–10735. doi: 10.1073/pnas.162349799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lomede K, Durand de Saint Front V, Galitzky J, Bouloumie A. Int J Obes. 2003;27:1187–1195. doi: 10.1038/sj.ijo.0802407. [DOI] [PubMed] [Google Scholar]
- 68.Hausman GJ, Richardson RL. J Anim Sci. 2004;82:925–934. doi: 10.2527/2004.823925x. [DOI] [PubMed] [Google Scholar]