

Abstract
Mitochondrial DNA (mtDNA) deletions are a common cause of mitochondrial disorders and have been found to accumulate during normal aging. Despite the fact that hundreds of deletions have been characterized at the molecular level, their mechanisms of genesis are unknown. We tested the effect of double-strand breaks of muscle mtDNA by developing a mouse model where a mitochondrially-targeted Restriction Endonuclease (PstI) was expressed in skeletal muscle of mice. Because mouse mtDNA harbors two PstI sites, transgenic founders developed a mitochondrial myopathy associated with mtDNA depletion. The founders showed a chimeric pattern of transgene expression and their residual level of wild-type mtDNA in muscle was ~ 40% of controls. We were able to identify the formation of large mtDNA deletions in muscle of transgenic mice. A family of mtDNA deletions was identified, and most of these rearrangements involved one of the PstI sites and the 3′-end of the D-loop region. The deletions had no or small direct repeats at the breakpoint region. These features are essentially identical to the ones observed in humans with multiple mtDNA deletions in muscle, suggesting that doublestrand DNA breaks mediate the formation of large mtDNA deletions.
Introduction
MtDNA deletions can present as a single, clonal species or as multiple deletions. Multiple mtDNA deletions have been associated with mutations in the mitochondrial DNA polymerase gamma, adenine nucleotide translocator and the helicase twinkle (reviewed in (1)). Low levels of multiple mtDNA deletions have also been associated with normal aging (2). The molecular features of multiple mtDNA deletions include the presence of very small (or no) direct repeats at the deletion breakpoint and a bias towards involving the end of the D-loop control region (3, 4). Although slip-misparing has been proposed to mediate mtDNA deletions (5, 6), more recent findings suggest that doublestrand breaks could be involved in the formation of deletions with small homologies at the breakpoint region (4). To better understand the consequence of double-strand breaks in animal muscle mtDNA, we targeted PstI to mouse skeletal muscle mitochondria. Our results provided clues to the formation of large-scale deletions in mammals.
Expression of Mito-PstI in transgenic mouse muscle.
Because mouse mtDNA harbors two sites for the restriction endonuclease PstI, we attempted to cause double-strand breaks in muscle mtDNA by producing transgenic mice expressing PstI specifically in muscle mitochondria. A construct harboring a mammalianized (i.e. codon usage optimized for mammalian ribosomes) version of the bacterial PstI gene downstream of a mitochondrial targeting sequence and the human skeletal muscle actin promoter was used to produce transgenic mice (Fig. 1a). Three male PstI transgenic founder mice (Fd1, Fd2 and Fd3) were obtained. Both Fd1 and Fd2 mice showed growth defects, while Fd3 did not. At approximately 3 months of age, both founders were growing slower than a control sibling of the same age (Fig. 1b). Fd1 and Fd2 failed to transmit the transgene to offsprings (Fig. 1c), whereas Fd3 transmitted the transgene to 50% of F1 generation (Fig. 1c). However, the Fd3 progeny did not express the PstI protein in muscle mitochondria, whereas Fd1 and Fd2 did (Fig. 1d). At 6 months (Fd1) and 7 months (Fd2) of age these mice developed a myopathy and had difficulties moving (Supplemental movie S1). When the myopathy became severe, the mice were sacrificed for skeletal muscle analysis.
Figure 1. Production of transgenic mice expressing a mitochondrial-targeted PstI in skeletal muscle.
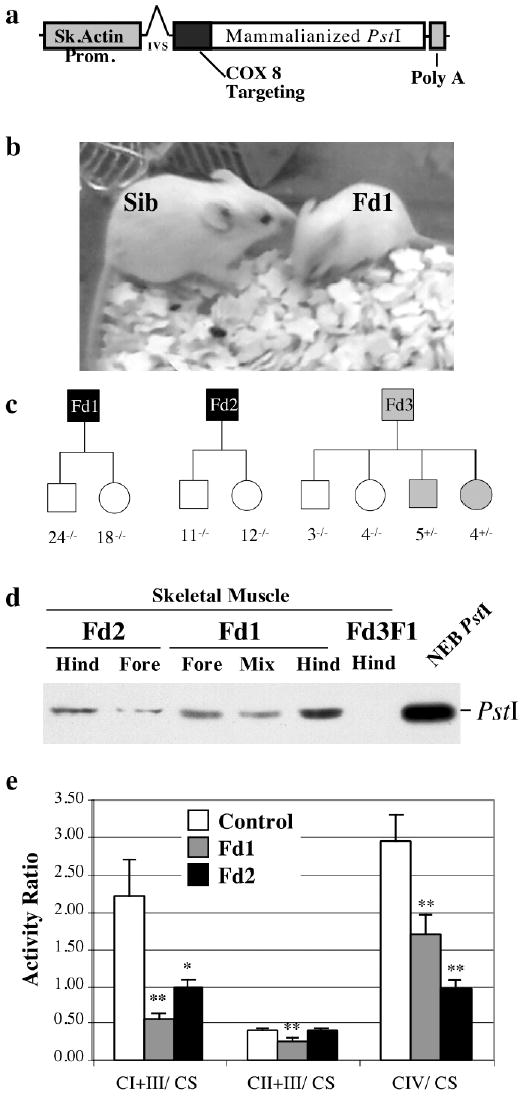
Panel a depicts the structure of the transgene construct containing a mammalianized version of the bacterial PstI gene fused in frame with an N-terminal mitochondrial targeting sequence of COX VIII. Panel b shows founder 1 (Fd1) and a non-transgenic sibling. Extensive breeding of Fd1 and Fd2 failed to produce pups containing the transgene, whereas 50% of Fd3 pups had the transgene (Panel c). Western blot analysis showed that mitochondria isolated from Fd1 and Fd2 skeletal muscle (fore limb and hind limb muscles) contained the transgene product. Muscle mitochondria from a Fd3 pup harboring the transgene did not express PstI (panel d). Muscle mitochondria isolated from Fd1, Fd2 and non-transgenic littermate controls were analyzed for the activity of complex I+III, II+III and IV by spectrophotometric assays (panel e). The activities were expressed as a ratio to citrate synthase activity to normalize the results to mitochondrial abundance. Complexes I and IV were significantly decreased in the transgenic Fd1 and Fd2. *p<0.05; **p<0.005; n=3 independent assays of each mitochondrial preparation.
Skeletal muscle of Mito-PstI transgenic mice showed mitochondrial respiratory chain defects.
Isolated mitochondria were used to measure the respiratory chain enzyme complex activities. Muscle mitochondria from both Fd1 and Fd2 mice showed a significant defect in the complex I+III and complex IV activity (P< 0.005, Figure 1d). Morphological analyses showed mitochondrial abnormalities in muscle of Fd1 by electron microscopy and by histochemical staining. EM of Fd1 muscle showed mitochondrial proliferation, swelling and cristae disorganization (supplemental figure S2). Gomori trichrome stain showed the presence of ragged-red fibers, which were more evident in Fd1 than in Fd2 (figure 2a). Cytochrome oxidase (COX) deficiency was evident in both mice, but was not observed in all the fibers, suggesting chimerism (figure 2a). COX deficient fibers had increased succinate dehydrogenase (SDH) activity staining, confirming that mitochondria with mtDNA defects were present at increased amounts. Skeletal muscle sections from control, Fd1 and Fd2 mice were also analyzed for the levels of PstI and the mtDNA-coded CoxIp by immunocytochemistry. PstI immunostaining in both Fd1 and Fd2 mice muscle revealed a chimeric pattern of PstI expression (Fig. 2b), which is compatible with the chimeric COX deficiency. This observation may also explain why Fd1 and Fd2 failed to transmit the transgene. It is likely that, if expressed in all fibers after germline transmission, Mito-PstI would cause an embryonic lethal phenotype. Muscle fibers expressing PstI had a significant decrease in CoxIp (Fig. 2b), implying damage to mtDNA molecules.
Figure 2. Chimeric expression of the Mito-PstI in muscle of Fd1 and Fd2.
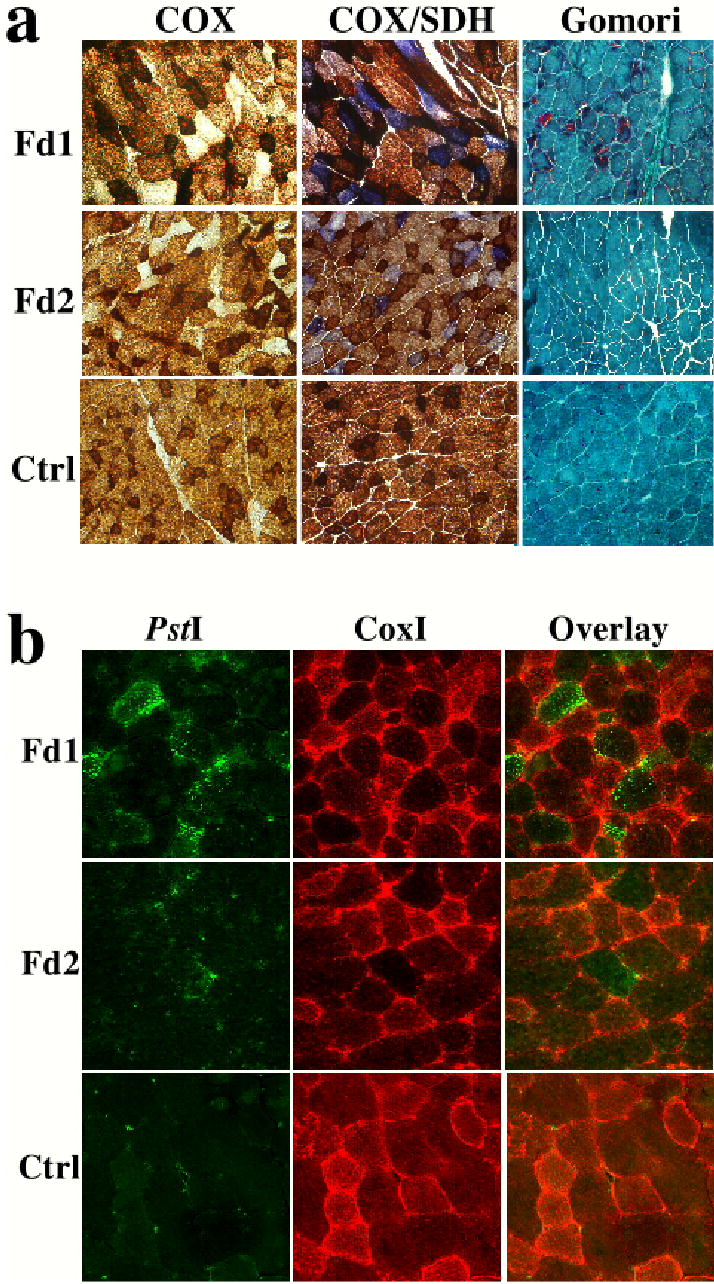
Panel a shows histochemical staining for cytochrome oxidase (COX) and succinate dehydrogenase (SDH) activities. Both Fd1 and Fd2 had COX deficiency but normal or elevated SDH activity. The COX deficiency was present in a mosaic pattern. The mosaic pattern of expression of Mito-PstI was also observed by immunocytochemistry using an anti-PstI antibody (Panel b). Fibers expressing Mito-PstI had decreased levels of CoxIp, a mtDNA-coded polypeptide (panel b).
MtDNA levels were decreased in the skeletal muscle of transgenic Mito-PstI mice.
To determine the mtDNA abundance, Southern blot membranes with muscle and liver DNA samples from control and transgenic founder mice were hybridized to mtDNA (COXI region) and a nuclear 18S rDNA probes (figure 3b-i). This analysis showed a significant depletion in the mtDNA levels in the skeletal muscle of both Fd1 and Fd2 mice. Fd1 and Fd2 mice showed 66% and 60% reduction of mtDNA levels when compared to an age-matched control (Fig. 3j).
Figure 3. Detection of mtDNA depletion and mtDNA deletions in muscle of Mito-PstI transgenic animals.
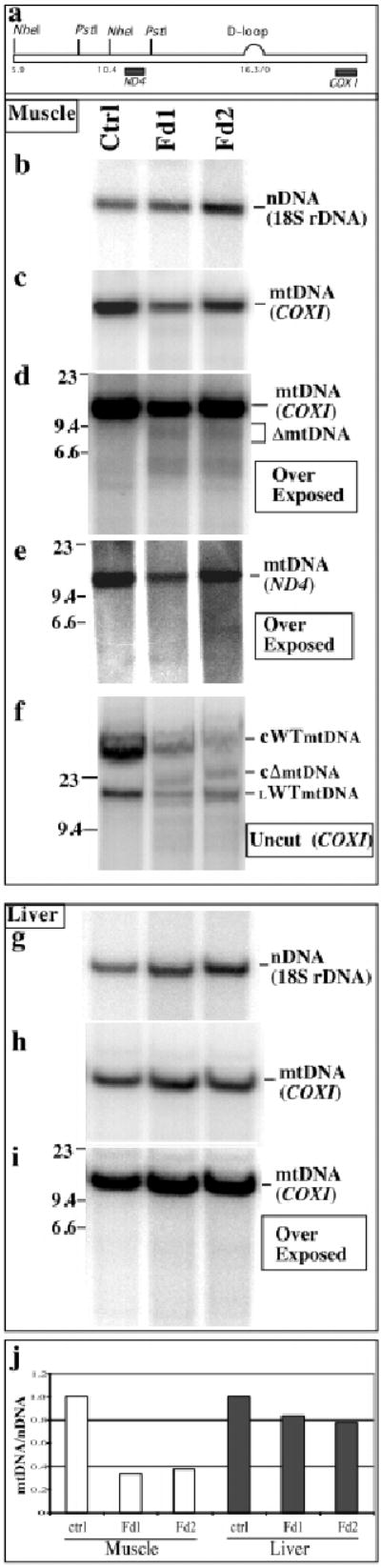
Southern blot analyses of DNA extracted from skeletal muscle of Fd1 and Fd2 was performed with mtDNA probes [COXI (panels c, d and f) and ND4 regions (panel e)] and the nuclear 18S rDNA (panel b). A similar analysis was also performed for DNA extracted from liver of transgenic founders (panels g-i). Total muscle DNA was either digested with NheI (most panels), or analyzed undigested (panel f). These studies showed a muscle specific mtDNA depletion (an approximate 60–70% reduction) in the founders (panel j). The Southern analysis also showed the presence of molecules migrating faster than the wild-type mtDNA in muscle of Fd1 and Fd2 (panel a, COXI probe), which could correspond to partially-deleted mtDNAs (ΔmtDNA). The undigested sample (panel f), showed the presence of bands that were tentatively assigned as, wild-type circles (cWT); linearized “broken” circles (lWT); and circular partially-deleted molecules (cΔ). A PstI-digested fragment would migrate at 12.5 kbp, and could be present at low levels.
Detection of large-scale mtDNA rearrangements in the skeletal muscle of transgenic Mito-PstI mice
In addition to the band corresponding to the wild-type mtDNA, a relatively smaller size band (~ 9.3 Kb) of weaker intensity was detected in the muscle samples from the transgenic mitochondrial PstI founders with the COXI probe. Such a band was absent from the control muscle sample (Fig. 3d) or liver samples (Fig. 3i). By re-probing the blot with the ND4 probe, the ~9.3 Kb mtDNA species was no longer detected in the muscle samples from transgenic founders (Fig. 3e) suggesting that the free double-strand ends generated at the site of PstI digestion participated in recombination events, creating large-scale deletions. The COXI probe also detected a “broad hybridization signal” in the 5–6 kbp region (figure 3d). Although this signal was also observed in the control sample, it appeared slightly stronger in muscle of Fd1 and Fd2. At this point we do not know if this signal corresponds to additional mtDNA rearrangements. Undigested DNA showed the presence of abnormal bands in the Fd1 and Fd2 DNA, which could be relaxed circles of partially deleted mtDNAs. PstI-digested mtDNA should migrate as a 12.5 kbp fragment, which also could be present at low levels, even though band identity of undigested DNA is difficult to prove (figure 3f).
To define the main mtDNA regions involved in recombination, we performed PCR with various sets of widely-spaced primers distributed along the mouse mtDNA (see methods). In standard PCR conditions, the primer regions are too far apart to generate a product. However, if deletions occur between the primers, smaller amplicons are obtained. As templates, we used total DNA isolated from skeletal muscle of control and transgenic founder mice. Most of the reactions did not yield a product, including reactions using primers flanking the two PstI sites (not shown). However, PCR analysis with a forward primer flanking one of the PstI sites (8,300-F) and the reverse primer in the mtDNA D-loop region (15,982-B) amplified a ~ 650 bp fragment in the skeletal muscle from both Fd1 and Fd2 mice, that was absent from the control skeletal muscle. This result suggested the presence of a large size deletion in the mtDNA of both transgenic founders (Figs. 4a and 4b). Cloning and sequencing analysis of individual ~650 bp inserts revealed the presence of a family of 6.9–7.0 Kb size deletions in the skeletal muscle of both Fd1 and Fd2 mice (Fig. 4c), which are compatible with the size of the smaller band observed by Southern blot. Interestingly, the deleted molecules showed one of the breakpoints exactly at, or close to, the PstI site located at nucleotide position 8,420 (i.e. distal from the D-loop). Some of the partially deleted molecules contained 2–4 bp homology repeats and the deletion breakpoints involved the nucleotide positions 8,346–8420 on one end and nucleotide positions 15,368–15,433 on the other, suggesting that the free double-stranded ends generated by PstI cleavage recombined with the 3′ end of the D-loop region.
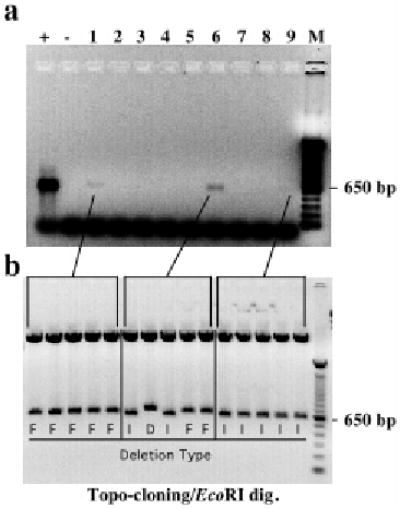
Figure 4. Characterization of mtDNA deletions associated with a double-strand break.

Using oligonucleotide primers flanking a PstI site and the D-loop region (arrows in panel a), we amplified an approximate 650 bp PCR fragment only in DNA extracted from muscle of Fd1 and Fd2 (panel b). The PCR products were cloned into plasmids and sequenced. Sequence analysis showed the presence of a family of mtDNA deletions involving the PstI site region and the end of the D-loop (panel c). Most deletions had small homologies at the breakpoint region (underlined nucleotides in panel c).
An additional primer pair combination (8,300-F and 13,125-B) yielded a faint band after PCR (not shown). After nested re-amplification, cloning and sequencing, we determined that the amplification corresponded to another group of deletions involving the same PstI site (at position 8,420) and a mtDNA region between positions 12,957–13,094. The features of the breakpoints were essentially identical to the ones observed at the D-loop region (i.e. 0–3 bp homology repeats involved in the breakpoint). Therefore, although prevalent, deletions involving the free end and the D-loop are not the only ones, a finding that also parallels observations in muscle of patients with multiple mtDNA deletions.
Clonal expansion of mtDNA deletions in skeletal muscle.
To test if deletions formed by double-strand breaks expand in aging muscle, we isolated single muscle fiber segments (20 μm) of COX-stained sections and PCR amplified the breakpoint of the previously observed mtDNA deleted region. Isolated fiber segments were subjected to amplification of both a mtDNA region outside the observed deleted region (i.e. outside the nt 8,420-nt15,500 region) and the deletion breakpoint region (using the same primers shown in figure 4). COX-positive fiber segments showed amplification of the wild-type region, whereas they did not show amplification of the deleted mtDNA breakpoints. Amplification of the deleted mtDNAs was observed in 4/20 of COX-negative fibers. Using the same conditions that allowed amplification of wild-type mtDNA in COXpositive fibers, only 11/20 COX-negative fibers showed amplification of the wild-type mtDNA region, and the amplification product was weaker than the ones observed in COX-positive fibers (not shown). These findings suggest that most COX deficient fibers have a mtDNA depletion, which would be compatible with the Southern blot results. COX-positive fiber segments did not show amplification of the deleted mtDNA breakpoints, whereas a product was observed in four COX-negative fibers (figure 5a). DNA fragments from three fibers were cloned into plasmids and restriction digestion and sequencing of the inserts showed that two of the fiber segments had only one type of deletion (type F or I, see figure 4), whereas one fiber had a mixture of three different types of deletions (figure 5b). These findings indicate that individual muscle fiber segments were enriched for one subtype of mtDNA deletion, suggesting clonal expansion during mitochondrial proliferation.
Figure 5. Clonal expansion of mtDNA deletions in muscle.
Muscle sections of transgenic founders were stained for cytochrome oxidase activity and dissected by Laser Capture Microscopy. Panel A shows a nested PCR for the mtDNA deleted region breakpoint of representative COX-negative fiber segments. Positive reactions were cloned into plasmids and digested with EcoRI to release the insert (panel B). Sequencing reactions identified the deletion breakpoints, which were coded as in figure 4.
Discussion
Mito-PstI expressing muscle showed the presence of low levels of mitochondrial genomes with large-scale deletions. The free ends generated by the endonuclease were recombined with a region at the 3′ end of D-loop, deleting a large fragment of mtDNA. The size of the deletions ranged from 6.9–7.0 Kb and many deleted molecules contained a 2–4 bp homologies at the breakpoint region, suggesting the participation of homologous recombination. It was surprising to observe that most double-strand breaks preferentially recombined with a specific region at the end of the D-loop. This could be due to the fact that the D-loop region has a loose triple strand conformation that might allow an easy access for single strand annealing, exchange and recombination (7). It may also be related to recent observations, which suggested that the D-loop region mediates replication fork arrest (8). Replication fork arrest is known to stimulate recombination events (9). Such polymerase pauses may also contribute to recombination at the less abundant nt 13,000 region.
Although the presence of direct repeats at the breakpoint region of some mtDNA deletions suggests a slip-mispairing mechanism (5), the mechanism of genesis of mtDNA deletions without direct repeats at the breakpoint has been puzzling. Samuels and colleagues studied the molecular features of 263 mtDNA deletions in humans and found that among deletions flanked by small direct repeats (<5 bp) or not flanked by direct repeats (also known as Class II deletions (10)), there was a remarkable bias for the 3′ end of the deletion to be located at the very end of the D-loop region (6) (positions 16000–16100 in the human mtDNA, which correspond approximately to positions 15400–15500 in the mouse mtDNA). If double-strand breaks are a major cause of Class II mtDNA deletions in human, how could they be generated? In the case of sporadic mtDNA deletions, double-strand breaks could be caused by ionizing radiation or the collapse of replication forks when the replication machinery encounters single-stranded breaks in the template DNA (11). Reactive oxygen species, which are abundant in the vicinity of mtDNA molecules could cause single-strand or double-strand breaks through the formation of hydroxyl radicals (12). Double-strand breaks could also occur during normal mtDNA replication. Replication of the lagging strand (in a coupled replication model) could be influenced by polymerase gamma pauses in the D-loop region (4), triggering a replication-induced break/recombination event.
There are 3 known mechanisms associated with double-strand break repair: Homologous recombination (HR), Single-strand annealing (SSA) and Non-homologous end-joining (NHEJ). HR and SSA are more prevalent in prokaryot cells, whereas NHEJ is commonly associated with repair of eukaryot cells (13). HR differs from SSA in that in the latter two internal repeats (present in a single-strand segments) recombine, deleting the distal segments. Moreover, continuing replication is not necessary in the SSA model (13). In the HR or SSA models, after a double-strand break, exonucleases produce an unpaired free 3′ end, which has been shown by Szostak and colleagues to be an initial step in double-strand break repairs (14). The free 3′ end of the double-strand break, through small homologies, could anneal with the end of the unpaired D-loop strand and serve as a primer for replication (figure 6a), or simply as templates for repair by single strand annealing (13). Single strand regions can also be created during coupled replication (figure 6b). In either case, repair could be achieved simply by extending the annealed 3′ end. However, if elongation does not proceed around the circle, a more complex mechanism, involving the resolution of a “holliday junction-type” structure would be required.
Figure 6. A model for the formation of Class II deletions.
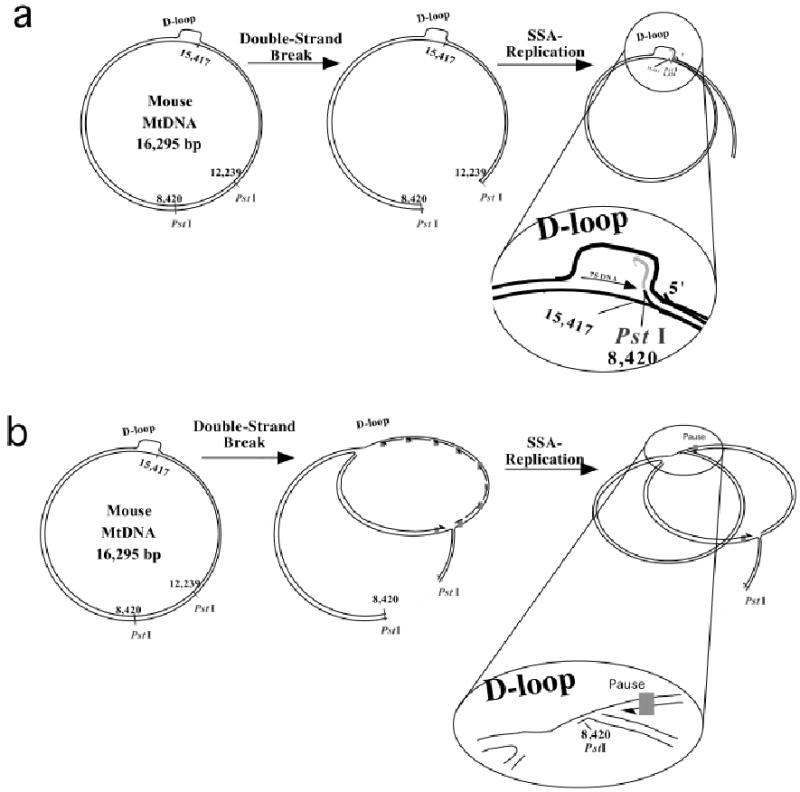
Because of the molecular similarities between the PstI generated deletions and class II deletions in humans, we propose that double-strand breaks could be a generator of these type of rearrangements. There are at least two potential mechanisms: The free 3′ end produced by PstI could anneal at the end of the D-loop region with a the participation of small repeats (Single-Strand Annealing, SSA) and function as a replication primer, extending the strand back into the D-loop region and beyond (panel a). Alternatively, polymerase pauses during replication would expose single-strand regions that could be annealed to the free ′ end of the cut DNA (panel b).
In the case of mtDNA multiple deletions, there is emerging evidence for the participation of double-strand breaks. The disease known as Mitochondrial NeurogastroIntestinal Encephalomyopathy (MNGIE) is caused by mutations in the Thymidine Phosphorylase gene, leading to a defect in the thymidine nucleotide pool. Patients present with mtDNA depletion, point mutations and multiple deletions in muscle (15, 16). A nucleotide pool unbalance has been hypothesized to lead to replication fork stalling (16). Wanrooij et al. have proposed that a double-strand break mechanism could be associated with autosomal dominant multiple deletions (which are mostly Class II deletions). They based their suggestion on the molecular features of mtDNA deletions observed in muscle of patients with mutations in the helicase twinkle and the polymerase gamma (4). They found that replication stalling at homopolymeric runs, which are frequently present at deletion breakpoints associated with polymerase gamma mutations, are likely to promote double-strand breaks. They also noted that the higher proportion of spontaneously linearized full-length muscle mtDNA molecules in patients with polymerase gamma mutations supports the double-strand break model. Mutations in the helicase twinkle, which has been shown to control mtDNA copy number (17), could be directly involved in double-strand break repair, as has been shown for other helicases (18). Finally, we obtained direct evidence that double-strand breaks of mtDNA lead to the formation of Class II mtDNA deletions, with molecular features that are remarkably similar to the ones caused by mutations in the mtDNA replication/repair apparatus in humans. These findings suggest that double-strand breaks mediate the formation of class II mtDNA deletions in mammals.
MtDNA molecules linearized by restriction endonucleases are not stable and only traces of such molecules can be observed in Southern blots [(19) and this study]. It is likely that most of them are rapidly degraded by exonucleases, whereas a small fraction undergoes recombination. The presence of low levels of mtDNA deletions in muscle of an animal with homoplasmic levels of a PstI-susceptible mtDNA haplotype, suggests that mitochondria targeted restriction endonucleases could still be safe to shift mtDNA haplotypes in an heteroplasmic environment, as the repopulation with non-susceptible mtDNA haplotypes should staunch the accumulation of partially deleted mtDNA.
Material and Methods
Production of transgenic PstI mice.
A mammalian version (synthetic and codon optimized) of the bacterial PstI gene (19) with a 5′ mitochondrial targeting sequence from human COX VIII (cytochrome c oxidase subunit VIII) and a 3′ PolyA signal was cloned in a plasmid downstream of a human skeletal muscle actin promoter (pSMA), kindly donated by Dr. Jeffrey Chamberlain, University of Washington. The skeletal muscle actin promoter was originally cloned in Dr. Larry Kedes’ lab (20). The promoter fragment was characterized in Dr. Hardeman’s lab in transgenic mice. She reported that in newborn animals the SMA promoter was expressed in fiber types in the following order: IIB>IIX>IIA>I (21). In adult animals, the expression of dystrophin under this promoter did not show fiber-type variation. It also did not show heart expression between 2 weeks and 2 years (J. Chamberlain, personal communication).
Briefly, the PstI gene along with the targeting sequence was excised with EagI from the plasmid pCDNA3PstI, described elsewhere and was cloned into a NotI site of pSMA (22). The final construct pSMAPstI was linearized with enzymes ClaI and PvuI and the fragment was purified using elutip-D minicolumn (Schleicher and Schuell, Keene, N.H). The purified fragment was used for transgenic microinjection. Microinjection into B6/SJLF1 fertilized oocytes was performed by the transgenic core facility of the University of Miami School of Medicine. Out of 29 mice born from implantation of injected fertilized oocytes into pseudo-pregnant mothers, 3 were positive for the Transgene by PCR of tail DNA. The sequence of the mouse mtDNA was obtained from GenBank # AJ512208.
Enzymes activity analyses.
For measuring the specific activity of the respiratory chain complexes, mitochondria were isolated, and assays were performed spectrophotometrically (DU-640 spectrophotometer, Beckman Instruments Inc., Fullerton, CA) as described elsewhere (23). The activities of NADH cytochrome c oxidoreductase (complex I + III), succinate-cytochrome c reductase (complex II + III), cytochrome c oxidase (complex IV), and citrate synthase were determined. Assays were performed at 37°C in 1ml medium (except the citrate synthase at 30°C).
Electron microscopy analysis.
Muscle tissue was fixed overnight in phosphate buffer containing 2% paraformaldehyde and 2.5% gluteraldehyde. The tissue was rinsed in phosphate buffer, treated with 1% osmium tetroxide and processed for transmission electron microscopy (TEM) as described (24). TEM images were captured using JEOL CX 100 at the EM core facility of the University of Miami.
Histochemistry.
Gomori trichome staining, succinate dehydrogenase (SDH) and cytochrome c oxidase (COX) activities were determined using 6 μm-thick muscle sections as described by Sciacco and Bonilla (25).
Western blot and Immunocytochemistry.
For western blots, 30 μg of muscle mitochondrial protein was resolved on 15% SDS-PAGE and transferred to PVDF (polyvinylidene difluoride, Immobilon, Bio-Rad) membrane. The membrane was blocked overnight in 5% milk and after washings in PBS Tween, the blot was incubated with a polyclonal anti-PstI antibody (19) for 2hrs at RT. The blot was washed and incubated with secondary antibody conjugated to HRP for 1hr at RT. Chemiluminescent substrate was used to detect the signal. For immunostaining, 6 μm-thick muscle sections from transgenic mice were incubated overnight with a 1:100 dilution of a polyclonal anti-PstI (19) and monoclonal anti-CoxIp (Molecular Probes, Eugene, OR) antibody in a humid chamber at 4°C. Next day, the sections were washed with PBS Tween and incubated with 1:200 dilution of secondary antibodies (anti-rabbit Alexa488 and anti-mouse Alexa594) for 2 hrs at RT in humid chamber. After 2 hrs, the sections were washed and mounted with Antifade mounting medium (Molecular Probes, Eugene, OR).
Southern blot analyses.
To perform Southern blot analysis, total DNA (5 μg) was digested with NheI, electrophoresed on 0.7 % agarose gel and transferred to a zeta-probe membrane (Biorad, Hercules, CA). To detect mtDNA, [Δ-32P] dCTP labeled probes were synthesized using random priming method. Two mtDNA probes were synthesized, one against the COXI region (nt positions 5,227–5,794) and the other against the ND4 region (nt positions 10,176–10,730) of mouse mtDNA (26). The COXI probe allowed the detection of both intact and partially-deleted mtDNA molecules whereas the ND4 probe allowed detection of intact mtDNA molecules only. The radioactive signal was quantitated using a Cyclone Phosphoimager system (Perkin-Elmer, Boston, MA). To determine the relative mtDNA abundance, the blots were stripped and hybridized with a nuclear 18S rDNA probe, obtained by PCR amplification of mouse DNA (primers spanning positions 1–574 according to GeneBank X00686). The ratio of the mtDNA signal intensity to the respective 18S rDNA signal were obtained to estimate mtDNA abundance.
PCR analysis and sequencing.
To search for mtDNA deletions, muscle mtDNA from control and transgenic Mito-PstI mice were PCR amplified using oligonucleotides corresponding to mtDNA positions [8,300-8,320:15,962-15,982]. A PCR product of approximately 650-bp amplified from muscle samples of transgenic Mito-PstI mice (corresponding to deletion breakpoint regions) was gel purified and cloned in a plasmid ‘TOPOblunt’ (Invitrogen). After bacterial transformation of the plasmid, eight clones were picked and the PCR fragment was sequenced using T7 and M13 reverse primers. Additional PCR reactions were performed to search for mtDNA deletions. The primer pairs used were at the following mtDNA locations: [8,300-8,320:12,340-12,322], [8,300-8,320:13,125-13,101], [8,300-8,320:14,234-14,211], [8,300-8,320:15,198-15,176].
Single fiber analyses.
COX stained skeletal muscle sections (20 μm thick) were microdissected by laser capture microscope (LCM). Total DNA was isolated from the single fibers using alkaline lysis method as described elsewhere (27). For amplification of a non-deleted mtDNA region in the single fiber segment analyses we used primers [5,227-5,250:5,794-5,771]. Nested PCR was performed with primer pair [5,310-5,332:5,690-5,666]. PCR for the deleted region was performed using the primer pair [8,300-8,320:15,962-15,982]. Reactions were re-amplified with nested the primers 8,321-F and 15,960-B. PCR products from the amplified fibers were cloned in a TOPO Blunt Vector (Invitrogen) followed by bacterial transformation. Five colonies from each transformation were subsequently analyzed by plasmid isolation and sequencing of the PCR product to determine the mtDNA deletion breakpoints.
Figure S2.

Transmission electron microscopy showed mitochondrial proliferation and swelling in muscle fibers of Fd1 (right panel). Mitochondrial cristae disorganization can also be observed in Fd1 muscle.
Acknowledgments
We are grateful to Dr. Richard Myers for suggestions and critically revising the manuscript. We also thank Ms. Dayami Hernandez for help with mice maintenance. This work was supported by Public Health Service grants EY10804 and NS041777.
References
- 1.Zeviani M, Carelli V. Mitochondrial disorders. Curr Opin Neurol. 2003;16:585–594. doi: 10.1097/01.wco.0000093101.34793.c8. [DOI] [PubMed] [Google Scholar]
- 2.Ozawa T. Mitochondrial genome mutation in cell death and aging. J Bioenerg Biomembr. 1999;31:377–390. doi: 10.1023/a:1005479920097. [DOI] [PubMed] [Google Scholar]
- 3.Zeviani M, Bresolin N, Gellera C, Bordoni A, Pannacci M, Amati P, Moggio M, Servidei S, Scarlato G, DiDonato S. Nucleus-driven multiple large-scale deletions of the human mitochondrial genome: a new autosomal dominant disease. Am J Hum Genet. 1990;47:904–914. [PMC free article] [PubMed] [Google Scholar]
- 4.Wanrooij S, Luoma P, van Goethem G, van Broeckhoven C, Suomalainen A, Spelbrink JN. Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 2004;32:3053–3064. doi: 10.1093/nar/gkh634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schon EA, Rizzuto R, Moraes CT, Nakase H, Zeviani M, DiMauro S. A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science. 1989;244:346–349. doi: 10.1126/science.2711184. [DOI] [PubMed] [Google Scholar]
- 6.Samuels DC, Schon EA, Chinnery PF. Two direct repeats cause most human mtDNA deletions. Trends Genet. 2004;20:393–398. doi: 10.1016/j.tig.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Lee DY, Clayton DA. Initiation of mitochondrial DNA replication by transcription and R-loop processing. J Biol Chem. 1998;273:30614–30621. doi: 10.1074/jbc.273.46.30614. [DOI] [PubMed] [Google Scholar]
- 8.Bowmaker M, Yang MY, Yasukawa T, Reyes A, Jacobs HT, Huberman JA, Holt IJ. Mammalian mitochondrial DNA replicates bidirectionally from an initiation zone. J Biol Chem. 2003;278:50961–50969. doi: 10.1074/jbc.M308028200. [DOI] [PubMed] [Google Scholar]
- 9.Michel B, Flores MJ, Viguera E, Grompone G, Seigneur M, Bidnenko V. Rescue of arrested replication forks by homologous recombination. Proc Natl Acad Sci U S A. 2001;98:8181–8188. doi: 10.1073/pnas.111008798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mita S, Rizzuto R, Moraes CT, Shanske S, Arnaudo E, Fabrizi GM, Koga Y, DiMauro S, Schon EA. Recombination via flanking direct repeats is a major cause of large-scale deletions of human mitochondrial DNA. Nucleic Acids Res. 1990;18:561–567. doi: 10.1093/nar/18.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haber JE. Partners and pathways repairing a double-strand break. Trends Genet. 2000;16:259–264. doi: 10.1016/s0168-9525(00)02022-9. [DOI] [PubMed] [Google Scholar]
- 12.Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–1309. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
- 13.Dudas A, Chovanec M. DNA double-strand break repair by homologous recombination. Mutat Res. 2004;566:131–167. doi: 10.1016/j.mrrev.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- 15.Nishigaki Y, Marti R, Hirano M. ND5 is a hot-spot for multiple atypical mitochondrial DNA deletions in mitochondrial neurogastrointestinal encephalomyopathy. Hum Mol Genet. 2004;13:91–101. doi: 10.1093/hmg/ddh010. [DOI] [PubMed] [Google Scholar]
- 16.Nishigaki Y, Marti R, Copeland WC, Hirano M. Site-specific somatic mitochondrial DNA point mutations in patients with thymidine phosphorylase deficiency. J Clin Invest. 2003;111:1913–1921. doi: 10.1172/JCI17828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyynismaa H, Sembongi H, Bokori-Brown M, Granycome C, Ashley N, Poulton J, Jalanko A, Spelbrink JN, Holt IJ, Suomalainen A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum Mol Genet. 2004;13:3219–3227. doi: 10.1093/hmg/ddh342. [DOI] [PubMed] [Google Scholar]
- 18.Oshima J, Huang S, Pae C, Campisi J, Schiestl RH. Lack of WRN results in extensive deletion at nonhomologous joining ends. Cancer Res. 2002;62:547–551. [PubMed] [Google Scholar]
- 19.Srivastava S, Moraes CT. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum Mol Genet. 2001;10:3093–3099. doi: 10.1093/hmg/10.26.3093. [DOI] [PubMed] [Google Scholar]
- 20.Muscat GE, Kedes L. Multiple 5′-flanking regions of the human alpha-skeletal actin gene synergistically modulate muscle-specific expression. Mol Cell Biol. 1987;7:4089–4099. doi: 10.1128/mcb.7.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brennan KJ, Hardeman EC. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J Biol Chem. 1993;268:719–725. [PubMed] [Google Scholar]
- 22.Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol. 2000;150:1399–1410. doi: 10.1083/jcb.150.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrientos A, Kenyon L, Moraes CT. Human xenomitochondrial cybrids. Cellular models of mitochondrial complex I deficiency. J Biol Chem. 1998;273:14210–14217. doi: 10.1074/jbc.273.23.14210. [DOI] [PubMed] [Google Scholar]
- 24.Miller DL, Dougherty MM, Decker SJ, Bossart GD. Ultrastructure of the spermatozoa from a Florida manatee (Trichechus manatus latirostris) Anat Histol Embryol. 2001;30:253–256. doi: 10.1046/j.1439-0264.2001.00330.x. [DOI] [PubMed] [Google Scholar]
- 25.Sciacco M, Bonilla E. Cytochemistry and immunocytochemistry of mitochondria in tissue sections. Methods Enzymol. 1996;264:509–521. doi: 10.1016/s0076-6879(96)64045-2. [DOI] [PubMed] [Google Scholar]
- 26.Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26:167–180. doi: 10.1016/0092-8674(81)90300-7. [DOI] [PubMed] [Google Scholar]
- 27.Moraes CT, Schon EA. Detection and analysis of mitochondrial DNA and RNA in muscle by in situ hybridization and single-fiber PCR. Methods Enzymol. 1996;264:522–540. doi: 10.1016/s0076-6879(96)64046-4. [DOI] [PubMed] [Google Scholar]