

Abstract
We examined the utility of herpes simplex virus (HSV) vector-mediated gene transfer of vascular endothelial growth factor (VEGF) in a mouse model of diabetic neuropathy. A replication-incompetent HSV vector with VEGF under the control of the HSV ICP0 promoter (vector T0VEGF) was constructed. T0VEGF expressed and released VEGF from primary dorsal root ganglion (DRG) neurons in vitro, and following subcutaneous inoculation in the foot, expressed VEGF in DRG and nerve in vivo. At 2 weeks after induction of diabetes, subcutaneous inoculation of T0VEGF prevented the reduction in sensory nerve amplitude characteristic of diabetic neuropathy measured 4 weeks later, preserved autonomic function measured by pilocarpine-induced sweating, and prevented the loss of nerve fibers in the skin and reduction of neuropeptide calcitonin gene-related peptide and substance P in DRG neurons of the diabetic mice. HSV-mediated transfer of VEGF to DRG may prove useful in treatment of diabetic neuropathy.
Keywords: diabetic neuropathies, dorsal root ganglia, VEGF, HSV
Introduction
Peripheral neuropathy is a common and disabling complication of both type I and type II diabetes mellitus. No effective treatment to prevent the development or reverse the progression of diabetic neuropathy is currently available. Over the last decade, extensive studies in rodent models have demonstrated that systemic administration of neurotrophic factors, such as nerve growth factor1 or insulin-like growth factor,2 can prevent or reverse the manifestation of diabetic neuropathy in mice, but the side effects of systemically administered trophic factors limit the application of this approach in treatment of patients.3 We have shown that direct delivery of neurotrophin genes to the dorsal root ganglion (DRG) achieved by subcutaneous inoculation of a replication-defective herpes simplex virus (HSV)-based vector effectively prevents the development of streptozotocin (STZ)-induced neuropathy in mice.4 These vectors, deleted for one or more essential immediate-early (IE) HSV genes, are taken up by sensory nerve terminals innervating the skin and carried by retrograde axonal transport to the nucleus of the sensory neuron in the DRG.5 Expression of neurotrophic factors achieved by HSV-mediated gene transfer is also effective in preventing neuropathy in the pyridoxine and cisplatin models of polyneuropathy in rodents.6–8
Other peptides not conventionally considered to be neurotrophic have been shown to have substantial neuroprotective effects in a variety of models. Vascular endothelial growth factor (VEGF), originally identified by its ability to induce the growth of new blood vessels,9 can protect neurons from damage induced by ischemia10,11 and trauma.12,13 Intramuscular injection of a plasmid coding for VEGF prevents the development of diabetic neuropathy in rodents and rabbits.14 However, VEGF has substantial effects on tumor angiogenesis15 that may enhance tumor formation and tumor growth16 if the peptide is administered systemically. For that reason, local release of VEGF in peripheral nerve may be required in order to use this treatment safely in diabetic neuropathy. In this study, we examined whether gene transfer to DRG using an HSV vector engineered to express VEGF could be used to prevent progression of diabetic neuropathy in mice with STZ-induced diabetes.
Results
VEGF expression from T0VEGF in vitro and in vivo
Transduction of cultures of dissociated primary DRG neurons at a multiplicity of infection of 1 with vector T0VEGF (Figure 1) resulted in the release of 17–23 ng/ml of VEGF in supernatant collected at 24, 48, and 72 h after transduction (Figure 2a). Following subcutaneous inoculation of T0VEGF in the foot, viral genomes were detected in the ipsilateral DRG by polymerase chain reaction (PCR) using primers for the HSV UL27 gene (Figure 2b). Transduction of DRG in vivo by subcutaneous inoculation resulted in the production of substantial amounts of VEGF (52–55 pg/μg protein) that accumulated proximal to a ligature on the sciatic nerve (Figure 2c), indicating that VEGF was synthesized in the DRG with axonal transport in the sciatic nerve. The sciatic nerve of control animals contained 10–12 pg of VEGF per microgram of protein proximal to a ligature.
Figure 1.
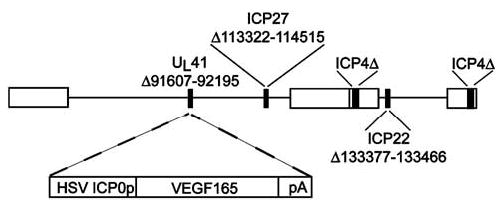
Schematic representation of the replication-incompetent HSV vector T0VEGF. VEGF expression is driven by the HSV ICP0 promoter, in a recombinant defective in ICP4, ICP22, and ICP27.
Figure 2.
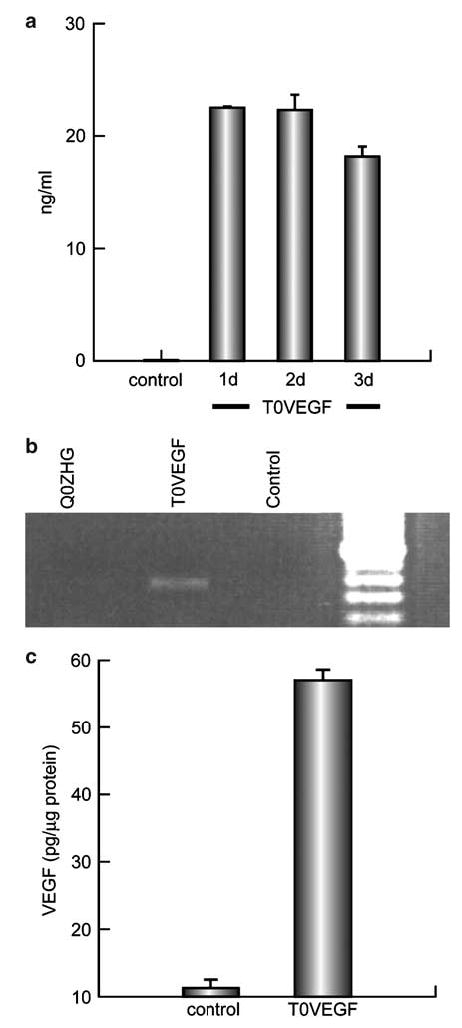
(a) Analysis of transgene (VEGF) production in vitro by ELISA. DRG cells infected with T0VEGF at a multiplicity of infection of 1 for 1 h released 17–23 ng/ml of VEGF in supernatant collected at 24, 48, and 72 h after transfection. (b) Detection of viral genomes. Viral genome was detected from the DRG of the animals, 7 days after footpad inoculation by PCR. (c) The amount of VEGF accumulating proximal to a ligature on sciatic nerve over 24 h was substantially greater in animals inoculated 7 days earlier with T0VEGF than in control animals.
Transduction of the DRG with T0VEGF preserved the sensory nerve amplitude in diabetic mice. Diabetes induced by STZ resulted in a sensory neuropathy manifested by a decrease in the foot sensory nerve amplitude (control 22.0±1.8 μV; STZ 12.5±1.3 μV; P<0.005). Transduction of dorsal root ganglia in vivo with T0VEGF by footpad inoculation protected against the decrease in foot sensory nerve amplitude (T0VEGF 31±1.6 μV and Q0ZHG 14.1±1.7 μV, respectively; P<0.005) measured 4 weeks after vector inoculation and 6 weeks after the induction of diabetes (Figure 3). We did not record sensory nerve conduction velocity because this parameter depends critically on measurement of the distance between the electrodes and is difficult to obtain accurately in mice. Motor nerve amplitude did not differ significantly between control and STZ diabetic mice, and was not affected by vector inoculation (data not shown).
Figure 3.
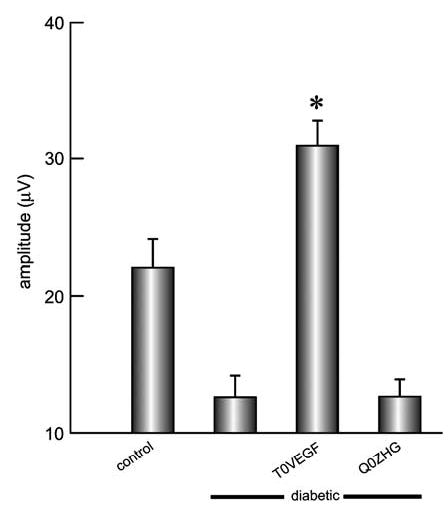
The amplitude of the evoked sensory response was reduced substantially in animals with diabetes, and this reduction was not affected by control vector inoculation. Inoculation of T0VEGF resulted in substantial preservation of the evoked sensory amplitude. Mean±s.e.m.; *P<0.005 compared to diabetic or diabetic inoculated with Q0ZHG; n = 8 animals per group.
Diabetic animals showed reduced thermal sensation, manifested by an increased latency to withdraw (control 19.18±0.7 s; diabetic 31.75±0.9 s; P<0.001). Animals inoculated with T0VEGF showed preservation of pain sensation (latency 20.52±0.1 s) while control vector-inoculated animals were indistinguishable from the diabetic untreated cohort (Figure 4).
Figure 4.
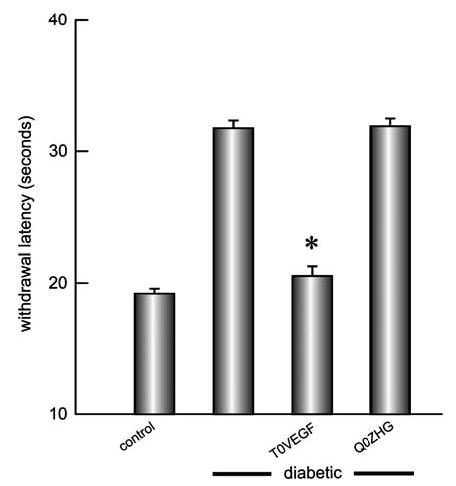
Thermal pain sensation was evaluated with a hotplate. Diabetic animals showed sensory nerve impairment manifested by an increased latency to withdraw. Diabetic mice inoculated with T0VEGF showed near-normal thermal pain sensation. Mean±s.e.m.; * P<0.005 compared to diabetic or diabetic inoculated with Q0ZHG; n = 8 animals per group.
Autonomic innervation of sweat glands in the paw was evaluated by measuring the number of sweat droplets formed over 10 min after injection of pilocarpine. Reduction in the number of sweat droplets in diabetic mice reflecting autonomic neuropathy (control 82.6±4.6 droplets; STZ 20.3±1.2 droplets; P<0.0001) was prevented in animals inoculated with T0VEGF (52±7.2 droplets; Q0ZHG 20.6±2.4 droplets; P<0.005) (Figure 5).
Figure 5.
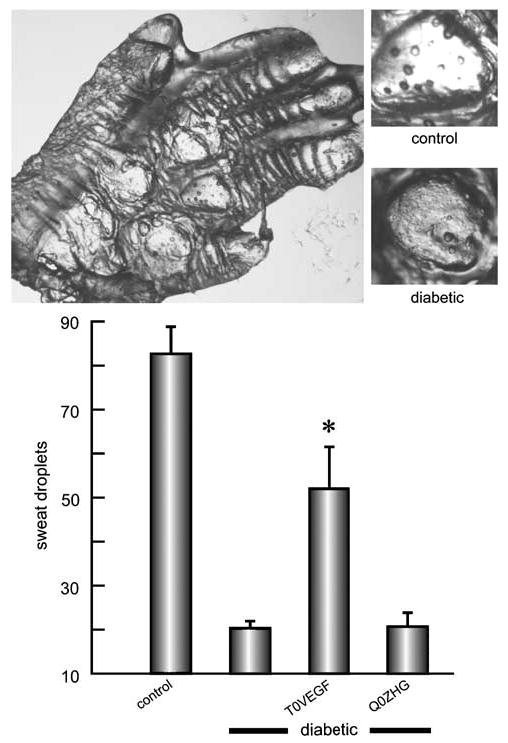
Sudomotor function after pilocarpine injection was evaluated by silicone imprint. The number of sweat droplets per sweat gland was substantially reduced in diabetic compared to control animals (illustrated top, first and second bars in graph). Diabetic animals inoculated with T0VEGF (third bar) showed substantial preservation of sweating compared to diabetic alone or Q0ZHG-inoculated diabetic mice. Mean±s.e.m.; * P<0.005 compared to diabetic or diabetic inoculated with Q0ZHG; n = 5 animals per group.
At the conclusion of electrophysiologic and behavioral measurements, the animals were killed and morphological and immunohistochemical assessments were performed in the DRG, sciatic nerve, and dorsal horn of spinal cord. The extension of damage to nerve fibers was visualized by immunocytochemistry for PGP 9.5. In the footpads of control animals, nerve fibers were evenly distributed, whereas in the diabetic or diabetic animals inoculated with control vector, few nerve fibers were seen, and those were not evenly distributed through the entire footpad (Figure 6). Innervation of the skin, quantitated as the proportional area of the skin and subcutaneous tissue occupied by nerve branches, was substantially reduced in diabetic animals (control 0.057±0.004; diabetic 0.006±0.001; P<0.005). The animals inoculated with T0VEGF showed substantial preservation of skin innervation (proportional area 0.035±0.002), while animals inoculated with Q0ZHG were indistinguishable from diabetic animals (proportional area 0.007±0.001; P<0.005, T0VEGF compared to Q0ZHG).
Figure 6.
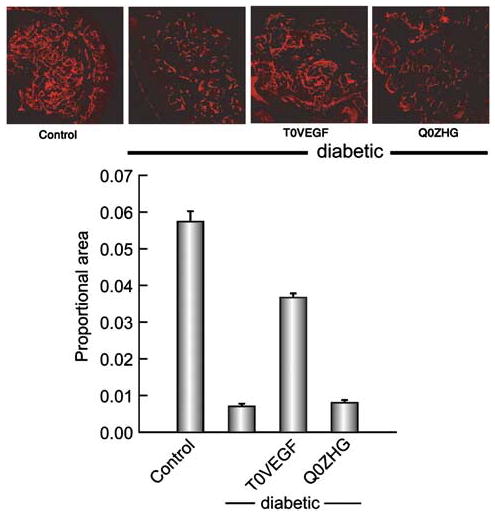
Nerve fibers in the footpad visualized by PGP 9.5 immunoreactivity. The nerve area, quantitated as proportional area occupied by the nerve in the footpad, was substantially reduced in diabetic animals but preserved in diabetic animals inoculated with T0VEGF. Mean±s.e.m.; * P<0.005 compared to diabetic or diabetic animals inoculated with Q0ZHG; n = 5 animals per group.
Diabetic neuropathy is characterized by reductions in neuropeptide content in sensory ganglia and sensory nerve terminals in the dorsal spinal cord. The amount of CGRP immunoreactivity in the dorsal horn was reduced in diabetic animals (control 5.137±1.1 mm2; diabetic 2.138±1.4 mm2; P<0.0001). Animals inoculated with T0VEGF showed substantial preservation of CGRP-immunoreactivity (5.591±3.3 mm2), whereas animals inoculated with Q0ZHG were indistinguishable from diabetic animals (2.95±2.9 mm2; P<0.0001; T0VEGF compared to Q0ZHG) (Figure 7).
Figure 7.
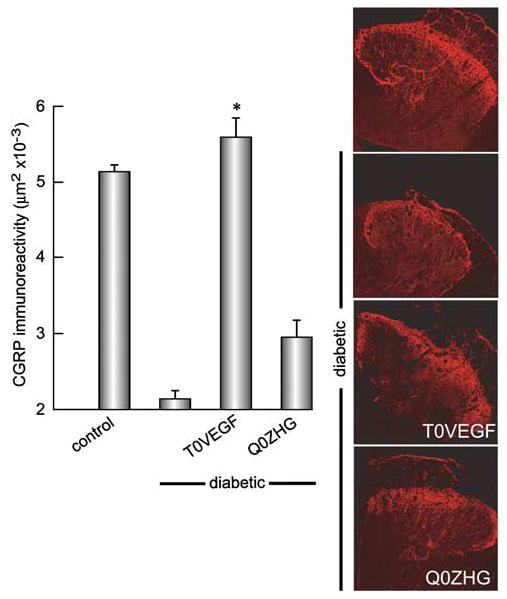
CGRP-immunoreactivity in dorsal horn was reduced in diabetic animals, but the area of immunoreactive terminals was substantially preserved in animals inoculated with T0VEGF compared to Q0ZHG. Mean±s.e.m.; * P<0.0001 compared to diabetic or diabetic inoculated with Q0ZHG; n = 5 animals per group.
Levels of SP immunoreactivity were also reduced in the spinal cord of diabetic animals compared to control (control 4.102±2.3 mm2; diabetic 1.996±3.5 mm2; P<0.0001). Animals with T0VEGF showed preservation of SP-immunoreactivity (T0VEGF 4.312±2.3 mm2; Q0ZHG 1.873±5.0 mm2; P<0.0001) (Figure 8).
Figure 8.
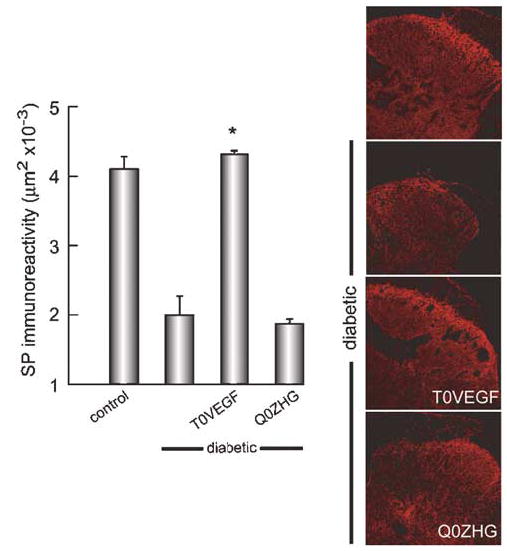
SP immunoreactivity was reduced in diabetic animals compared to control (right); diabetic animals inoculated with T0VEGF showed substantial preservation of SP-IR (P<0.0001) compared to diabetic or diabetic inoculated with Q0ZHG. Mean±s.e.m.; * P<0.0001 compared to diabetic or diabetic inoculated with Q0ZHG; n = 5 animals per group.
In diabetic mice, the number and size of blood vessels in nerve were reduced (Figure 9). Diabetic animals transduced with T0VEGF showed a number of vessels per nerve similar to those of nondiabetic control mice (18.6±0.8 versus 21.0±1.1 vessels per cross-section in nondiabetic mice versus T0VEGF-treated mice, respectively; P = NS). In diabetic animals transduced with T0VEGF, the average area of the vessels in nerve was increased compared with that of the diabetic untreated animals (7.76±0.3 μm2 in T0VEGF versus 5.740±0.07 μm2; P<0.01). The number of vessels and the area of vessel distribution in the three groups are shown in the histogram (Figure 9).
Figure 9.

Histogram of the number of blood vessels in control, diabetic, and diabetic T0VEGF-inoculated animals, by vessel cross-sectional area. The number of vessels in the diabetic animals was reduced compared to controls, but it was preserved in the vector-treated group (11.3±3.1 versus 21.0±1.1 vessels per cross-section in diabetic mice versus T0VEGF-treated mice, respectively; P<0.05).
Discussion
The results of our study indicate that transduction of DRG neurons by subcutaneous inoculation of an HSV vector expressing VEGF can prevent the progression of diabetic neuropathy in a mouse model. Although VEGF is best known for its effects on blood vessels,17–19 the peptide has trophic effects on neurons and Schwann cells in vitro20 and in vivo,21 producing neuroprotective effects in models of focal cerebral ischemia, Parkinson’s disease, and a transgenic mouse model of amyotrophic lateral sclerosis.22–24
The role of VEGF in diabetic neuropathy is complex. VEGF levels are increased in Schwann cells of rats with diabetic neuropathy,25,26 perhaps in response to nerve hypoxia. Systemic expression of VEGF achieved by the intramuscular gene transfer of naked DNA encoding VEGF has been shown to restore large- and small-fiber peripheral nerve function in a rat model of diabetic neuropathy and a rabbit model of alloxan-induced diabetic neuropathy.26
In a human trial,27 VEGF165, a plasmid coding, was injected into the muscle of patients with leg ischemia (six of whom were diabetic), resulting in significant clinical improvement in neuropathic symptoms, nerve conduction measures, and vibration threshold. Although a human trial of VEGF gene transfer by injection of naked plasmid into muscle for the treatment of ischemic neuropathy has been proposed, there is concern that systemic delivery of the VEGF peptide may result in aberrant angiogenesis or acceleration of tumor growth.15,16
HSV-mediated gene transfer to the DRG provides for local release of neurotrophic substances that may act in an autocrine or paracrine manner to protect peripheral sensory neurons from axonal degeneration. We have previously demonstrated the effect of HSV-mediated gene transfer in models of neuropathy induced by overdose of pyridoxine in the rat, STZ diabetes in the mouse, and neuropathy induced by the chemotherapeutic drug cisplatin in the rat.4,6,7 Our study extends those observations to demonstrate that the local release of VEGF achieved by transduction of DRG neurons with an HSV-based vector can prevent the progression of diabetic neuropathy in the mouse, and suggests an alternative approach to the use of VEGF in the treatment of this disease.
The single VEGF gene produces several isoforms by alternative splicing of mRNA.28 In the mouse, four different isoforms have been identified with molecular mass of 115, 120, 164, and 188 kDa.29 In humans, five isoforms of VEGF with molecular mass of 121, 145, 165, 189, and 206 kDa have been identified.30,31 VEGF isoforms are highly conserved across species,32–34 and human VEGF165 is homologous to mouse VEGF164.
In our study, we used the human VEGF165 gene, because the biological properties of that isoform correspond most closely to the array of effects mediated by the full set of splice isoforms,35 except for effects on the reproductive system produced exclusively by VEGF145.31,36 The effects of VEGF are mediated by high-affinity binding to two phosphotyrosine kinase receptors; fetal liver kinase (flk-1 or VEGFR-2) and fms-like tyrosine (flt-1 or VEGFR-1).37,38 The different isoforms of VEGF vary in their affinity for their cognate receptors.39 Activation of flk-1 receptor by VEGF results in a mitogenic endothelial cell response, while the activation of flt-1 by VEGF does not induce cell proliferation.40 VEGF stimulation of Schwann cells in vitro leads to the phosphorylation of both the flt-1 and flk-1 receptors, but hypoxic culture conditions increase the expression of flt-1 but not flk-1.14 Neurons in both cultured superior cervical ganglia and DRG contain the flk-1 receptor and show an increased mRNA expression and immunoreactivity for flk-1 after 48 h in culture. In superior cervical ganglia but not in DRG, double immunostaining for flk-1 and VEGF reveals coexpression in many neurons, implying that VEGF may exert both autocrine and paracrine actions.41
Endothelial cells also contain a second VEGF165 responsive receptor, coded by the neuropilin-1 or neuropilin-2 genes, which has a short intracellular domain. Neuropilin-1 is an important regulator of blood vessel development42 and has also been isolated in developing neurons, where it functions as a component of the receptor complex for VEGF as well as for the class 3 semaphorins.43,44 The neuropilin-1 receptor is present in 60% of nerve cell bodies in DRG in culture, and 80% of those cells are also flk-1 positive.41 We cannot determine from the studies performed whether the VEGF effect we observed in diabetic neuropathy requires neuropilin-1 or not, but in agreement with previous reports,26 we found that VEGF treatment resulted in preservation of peripheral nerve microvasculature in the diabetic animals. The vector genomes are restricted to the DRG neurons, and the precise site from which VEGF was released to achieve the effect on nerve vasculature is not established by the studies performed. It would appear most likely that the peptide is released from the axons of transduced neurons within the nerve.
The results presented in our study demonstrate preservation of sensory nerves in diabetic animals by electrophysiologic and behavioral measures. Neuropep-tide expression in the fibers of the peripheral nerve was also preserved by this treatment. There is no evidence to suggest that VEGF directly affects neuropeptide gene expression, but rather, it is more likely that the effect of VEGF on preserving nerve function results in the preservation of the neuropeptide phenotype. The effect of activation of flk-1 on neuropeptide expression has not previously been demonstrated. Sondell et al41 reported that small DRG neurons coexpress flk-1 and CGRP. Most flk-1-positive neurons in the DRG were also positive for neuropilin-1. Specific flk-1 inhibitor blocks VEGF-induced axonal outgrowth, suggesting in our study that the action of VEGF165 may be mediated through flk-1 receptor.
Diabetic neuropathy commonly affects the longest nerve fibers first, so that quantification of axon number demonstrates the loss of cutaneous nerve fibers in diabetic patients.45,46 In STZ-diabetic mice, there is also a significant loss in cutaneous innervation in the hind limb. We found a decrease in total cutaneous innervation in the footpad of mice by PGP 9.5 immunostaining, which was prevented by transgene expression of VEGF in the DRG, suggesting that VEGF acts as a neurotrophic factor and plays an important role in peripheral nerve regeneration. Similar effects have been demonstrated previously by intrathecal treatment with the glial-derived neurotrophic factor and with neurturin.47
Sudomotor dysfunction is a common feature of diabetic autonomic neuropathy, and correlates with a reduction in neuropeptide concentration and sweat gland activity.48 In our study, VEGF expression also preserved autonomic function, measured by pilocarpine-induced sweating. We believe that this most likely results from retrograde transport of the vector from the skin to the autonomic ganglia, although this has not been established conclusively. An alternative possibility is that VEGF released from sensory nerve terminals in the skin interacts with receptors on the autonomic fibers peripherally.
Despite substantial evidence in animal models demonstrating that neurotrophic factors are beneficial in the treatment or prevention of peripheral neuropathy, the treatment of human neuropathies has been limited, in part because of the short life of these peptide factors and the side effects engendered by systemic administration. Systemic administration of VEGF may enhance the growth of tumors, but restricted local expression on nerve achieved by HSV-mediated gene transfer to DRG through footpad inoculation may avoid that potential complication. The neuroprotective effects of VEGF in diabetic neuropathy offer an important alternative to the use of traditional neurotrophic factors by gene transfer in the treatment of this significant clinical disease.
Materials and methods
Vector construction
The VEGF-expressing vector T0VEGF was constructed by cotransfection of PacI linearized plasmid p41:0VEGF containing human VEGF165 coding sequence and the HSV ICP0 IE promoter with T0ZHG,49 an HSV recombinant defective for the HSV IE functions ICP4, ICP22, and ICP27 (Figure 1). The composition of the recombinant vector was confirmed by Southern blot, and the virus was purified by three rounds of limiting dilution.
Analysis of VEGF production in vitro
DRG from 17-day-old rat embryos were dissociated with 0.25%. trypsin, 1 mM ethylenediaminetetraacetic acid for 30 min at 37°C with constant shaking and plated on poly-d-lysine-coated coverslips at 105 cells per well in a 24-well plate, in 500 μl of defined neurobasal media containing B27, Glutamax I, Albumax II, and penicillin/streptomycin (Gibco BRL, Div. of Invitrogen Corp., Carlsbad, CA, USA), supplemented with 100 ng/ml of 7.0S nerve growth factor per milliliter (Sigma-Aldrich Co., St Louis, MO, USA). Cells were infected with T0VEGF at a multiplicity of infection of 1 for 1 h, after which the medium was changed and collected after 24, 48, and 72 h of infection. The amount of VEGF released from the transduced cells was determined using a commercial VEGF ELISA kit (R&D Systems, Inc., Minneapolis, MN, USA).
Animal studies
Experiments were conducted on young adult male mice weighing 25–30 g, and were in compliance with approved institutional animal care and use protocols. A total of 32 mice were rendered diabetic by injection of STZ (100 mg/kg twice in 48 h). The blood glucose of the diabetic mice was greater than 300 mg/dl at 2 weeks and remained elevated throughout 6 weeks after STZ administration. Subsets of eight mice received 10 μl containing 1 × 107 PFU of HSV-based vector expressing VEGF (T0VEGF), a control vector expressing GFP under the control of the HSV ICP0 promoter and lacZ under the control of the human cytomegalovirus IEp (vector Q0ZHG),7 or vehicle alone subcutaneously into both hind feet. Eight animals did not receive STZ and served as normal controls. All analyses were carried out by an observer blinded to the treatment group.
Analysis of VEGF production in vivo
Male Sprague–Dawley rats (because the in vitro studies were carried out in primary neurons from fetal rats) weighing 250 g were inoculated subcutaneously in the plantar surface of one hind foot with 30-μl containing 3 × 107 PFU T0VEGF. At 5 days after inoculation, the animals were anesthetized and the sciatic nerve ligated with 4-0 silk sutures. After 24 h, the animals were killed and the DRG, sciatic nerve, dorsal roots, and the dorsal quadrant of spinal cord attached to those roots were collected fresh. Tissue was homogenized in buffer.50 The amount of VEGF in the sample was determined using a commercial ELISA kit (R&D Systems, Inc., Minneapolis, MN, USA).
PCR for the detection of viral genome
Rats were inoculated with either T0VEGF or Q0ZHG subcutaneously in the foot using the same protocol. At 7 days after the inoculation, the animals were killed and total DNA was isolated from fresh DRG using QIA-Amp DNA mini-kit (QIAGEN, Inc., Valencia, CA, USA). PCR was performed for 35 cycles (94°C for 45 s, 60°C for 45 s, and 72°C for 45 s) using primers for the HSV UL27 flanking region (UL27-F: CGG TGG TTC GTC GTA TG; UL27-R: GGC GTC ACC GTC TTT GTT G). The PCR products were separated by 1% agarose gel and visualized with ethidium bromide.
Electrophysiologic measurement
Nerve conduction recordings were performed on the right hind foot using a Nicolet Viking II (Nicolet Biomedical, Madison, WI, USA). Mice were anesthetized with ketamine/xylazine (80/10 mg/kg i.p.), the hind limbs were secured at an angle of 30–45° relative to the long axis of the body, and motor nerve conduction velocity and amplitude in the sciatic nerve was determined. The recording electrode was inserted into the gastrocnemius muscle and the stimulating electrode pair were placed at the sciatic notch or the knee; the reference electrode was inserted subcutaneously into the fifth digit of the hind limb. A ground electrode was inserted into the tail, and subcutaneous temperature was maintained at 36–37°C. Both latency and baseline-to-peak amplitudes were determined. For foot sensory nerve recordings, the electrode placed in the sciatic notch was used as the recording electrode, with the stimulating electrode placed at the ankle and the reference electrode placed at the first digit. The statistical significance of the difference between groups was determined by analysis of variance using the Bonferroni adjustment for multiple post hoc analyses (Systat 9.0 software, SPSS, Inc., Chicago, IL, USA).
Sudomotor test
Autonomic function, reflecting innervation of sweat glands in the paw, was evaluated by measuring the number of sweat droplets induced by injection of pilocarpine (Sigma-Aldrich Co., St Louis, MO, USA) using the silicone imprint technique.51 The animals were injected with 50 μl of 0.75% bupivacaine (Sigma-Aldrich Co., St Louis, MO, USA) at the ankle, after which sweating was stimulated by subcutaneous injection of pilocarpine nitrate (0.3 mg/kg). At 10 min after the injection, silicone (Silaplast and Silasoft, Detax GmbH & Co., Ettlingen, Germany) was spread over the plantar surface of the hind paw and left in place for 10 min. The amount of sweating was determined by counting the number of sweat droplet impressions made in the silicone mold.
Hotplate test
Small fiber (thermal pain) sensory function was assessed by hotplate test. Animals were placed on a metal plate heated to 47°C, and the temperature increased at 2°C per minute. The withdrawal latency from the plate was measured in seconds.
Immunocytochemistry
Mouse hearts were perfused with 0.9% NaCl followed by Zamboni’s fixative.6 The spinal cord, DRG, and footpads were removed, post-fixed with Zamboni’s fixative for 2 h, then cryo-protected with 30% sucrose in phosphate-buffered saline (PBS). In all, 10-μm cryostat sections were collected on gelatin-coated slides and fixed with 2% paraformaldehyde for 15 min, washed with PBS, and incubated with blocking solution (PBS with 1% NGS and 0.3% Triton X-100) for 1 h, then washed once. The DRG and spinal cord were incubated with the primary antibody, either rabbit anti-calcitonin gene-related peptide (CGRP) and/or anti-substance P (SP), 1:1000 dilution (Peninsula Laboratories, Inc., San Carlos, CA, USA), for 2 h at room temperature. The footpads were incubated with anti-protein gene product (PGP) 9.5, 1:1000 dilution (Chemicon International, Inc., Temecula, CA, USA) overnight at 4°C, and washed 3 times. After incubation in the secondary fluorescent antibody, Alexa Fluor 594 goat anti-rabbit IgG, 1:500 dilution (Molecular Probes, Eugene, OR, USA), for 2 h at room temperature, the specimens were washed 3 times and mounted in water-based Fluoromount G (Electron Microscopy Sciences, Fort Washington, PA, USA).
Morphometric analysis
Digitized images of immunostained sections were captured with an Olympus FluoView BX61 laser-scanning microscope, and analyzed using the MCID computer-based image analysis program (Imaging Research, Inc., St Catharines, Ontario, Canada). The area occupied by immunostained central sensory nerve terminals in the dorsal horn of the spinal cord, or immunostained peripheral nerve terminals in the subcutaneous tissue of the footpad, was determined as a percentage of the total area. The statistical significance of the difference between groups was determined by analysis of variance using the Bonferonni adjustment for multiple post hoc analyses (Systat 9.0 software, SPSS, Inc., Chicago, IL, USA). To assess the number of blood vessels in nerve, nerves obtained at the time of killing were postfixed in 2% glutaraldehyde, osmicated and embedded in epoxy resin (tEPON812, Tousimis Research Corp., Rockville, MD, USA), cut into 0.5 μm sections, mounted on slides and stained with 1% toluidine blue. Digitized images were obtained using a Nikon E1000 microscope, and the number and area of blood vessels determined using the MetaMorph computer-based image analysis system (Universal Imaging Corporation, Downingtown, PA, USA). Three cross-sections of each animal were randomly selected from each specimen, vessels per cross-section were counted, and area of each vessel was measured by an investigator blinded to the treatment group. Data are expressed as mean±s.e.m. (n = 5 per study group). All of the analyses were performed by an observer blinded to the treatment condition.
Acknowledgments
This work was supported by grants from the National Institutes of Health, the Department of Veterans Affairs, and the Juvenile Diabetes Research Foundation to Drs Mata and Fink. We acknowledge the excellent technical assistance of Xiaoping Hu, Vikram Thakur, Mingdi Zhang, and Veljko Puskovic. We extend our special thanks to Dr James Goss.
Dr. Mata's work was supported by R01 NS43247 (M Mata, PI), Gene Transfer for Spinal Root Trauma.
References
- 1.Tomlinson DR, Fernyhough P, Diemel LT. Role of neurotrophins in diabetic neuropathy and treatment with nerve growth factors. Diabetes. 1997;46 (Suppl 2):S43–S49. doi: 10.2337/diab.46.2.s43. [DOI] [PubMed] [Google Scholar]
- 2.Ishii DN. Implication of insulin-like grwoth factors in the pathogenesis of diabetic neuropathy. Brain Res. 1995;20:47–67. doi: 10.1016/0165-0173(94)00005-a. [DOI] [PubMed] [Google Scholar]
- 3.Apfel SC. Nerve growth factor for the treatment of diabetic neuropathy: what went wrong, what went right, and what does the future hold? Int Rev Neurobiol. 2002;50:393–413. doi: 10.1016/s0074-7742(02)50083-0. [DOI] [PubMed] [Google Scholar]
- 4.Goss JR. Herpes simplex-mediated gene transfer of nerve growth factor protects against peripheral neruropathy in streptozotocin-induced diabetes in the mouse. Diabetes. 2002;51:2227–2232. doi: 10.2337/diabetes.51.7.2227. [DOI] [PubMed] [Google Scholar]
- 5.Glorioso JC, Fink DJ. Herpes vector-mediated gene transfer in treatment of diseases of the nervous system. Annu Rev Microbiol. 2004;58:253–271. doi: 10.1146/annurev.micro.58.030603.123709. [DOI] [PubMed] [Google Scholar]
- 6.Chattopadhyay M. Protective effect of herpes simplex virus-mediated neurotrophin gene transfer in cisplatin neuropathy. Brain. 2004;127:929–939. doi: 10.1093/brain/awh103. [DOI] [PubMed] [Google Scholar]
- 7.Chattopadhyay M. In vivo gene therapy of pyridoxine-induced neuropathy by HSV-mediated gene transfer of neurotrophin-3. Ann Neurol. 2002;51:19–27. doi: 10.1002/ana.10061. [DOI] [PubMed] [Google Scholar]
- 8.Chattopadhyay M. Protective effect of HSV-mediated gene transfer of nerve growth factor in pyridoxine neuropathy demonstrates functional activity of trkA receptors in large sensory neurons of adult animals. Eur J Neurosci. 2003;17:732–740. doi: 10.1046/j.1460-9568.2003.02500.x. [DOI] [PubMed] [Google Scholar]
- 9.Breier G, Albrecht U, Sterrer S, Risau W. Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development. 1992;114:521–532. doi: 10.1242/dev.114.2.521. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi T, Abe K, Itoyama Y. Reduction of ischemic damage by application of vascular endothelial growth factor in rat brain after transient ischemia. J Cereb Blood Flow Metab. 1998;18:887–895. doi: 10.1097/00004647-199808000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Zhang ZG. VEGF enhances angiogenesis and promotes blood–brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell B, Chuhran C, Lefer AM. Vascular endothelial growth factor attenuates trauma-induced injury in rats. Br J Pharmacol. 2000;129:71–76. doi: 10.1038/sj.bjp.0703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Widenfalk J. Vascular endothelial growth factor improves functional outcome and decreases secondary degeneration in experimental spinal cord contusion injury. Neuroscience. 2003;120:951–960. doi: 10.1016/s0306-4522(03)00399-3. [DOI] [PubMed] [Google Scholar]
- 14.Schratzberger P. Favorable effect of VEGF gene transfer on ischemic peripheral neuropathy. Nat Med. 2000;6:405–413. doi: 10.1038/74664. [DOI] [PubMed] [Google Scholar]
- 15.Antonetti DA. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. 1999;274:23463–23467. doi: 10.1074/jbc.274.33.23463. [DOI] [PubMed] [Google Scholar]
- 16.Castro-Rivera E, Ran S, Thorpe P, Minna JD. Semaphorin 3B (SEMA3B) induces apoptosis in lung and breast cancer, whereas VEGF165 antagonizes this effect. Proc Natl Acad Sci USA. 2004;101:11432–11437. doi: 10.1073/pnas.0403969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carmeliet P. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 18.Breier G, Risau W. The role of vascular endothelial growth factor in blood vessel formation. Trends Cell Biol. 1996;6:454–456. doi: 10.1016/0962-8924(96)84935-x. [DOI] [PubMed] [Google Scholar]
- 19.Couffinhal T. Mouse model of angiogenesis. Am J Pathol. 1998;152:1667–1679. [PMC free article] [PubMed] [Google Scholar]
- 20.Sondell M, Lundborg G, Kanje M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J Neurosci. 1999;19:5731–5740. doi: 10.1523/JNEUROSCI.19-14-05731.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hobson MI, Green CJ, Terenghi G. VEGF enhances intraneural angiogenesis and improves nerve regeneration after axotomy. J Anat. 2000;197 (Part 4):591–605. doi: 10.1046/j.1469-7580.2000.19740591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111:1843–1851. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yasuhara T. Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson’s disease. Eur J Neurosci. 2004;19:1494–1504. doi: 10.1111/j.1460-9568.2004.03254.x. [DOI] [PubMed] [Google Scholar]
- 24.Azzouz M. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- 25.Samii A, Unger J, Lange W. Vascular endothelial growth factor expression in peripheral nerves and dorsal root ganglia in diabetic neuropathy in rats. Neurosci Lett. 1999;262:159–162. doi: 10.1016/s0304-3940(99)00064-6. [DOI] [PubMed] [Google Scholar]
- 26.Schratzberger P. Reversal of experimental diabetic neuropathy by VEGF gene transfer. J Clin Invest. 2001;107:1083–1092. doi: 10.1172/JCI12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simovic D. Improvement in chronic ischemic neuropathy after intramuscular phVEGF165 gene transfer in patients with critical limb ischemia. Arch Neurol. 2001;58:761–768. doi: 10.1001/archneur.58.5.761. [DOI] [PubMed] [Google Scholar]
- 28.Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–865. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- 29.Sugihara T, Wadhwa R, Kaul SC, Mitsui Y. A novel alternatively spliced form of murine vascular endothelial growth factor, VEGF 115. J Biol Chem. 1998;273:3033–3038. doi: 10.1074/jbc.273.5.3033. [DOI] [PubMed] [Google Scholar]
- 30.Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 31.Poltorak Z. VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix. J Biol Chem. 1997;272:7151–7158. doi: 10.1074/jbc.272.11.7151. [DOI] [PubMed] [Google Scholar]
- 32.Tischer E. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991;266:11947–11954. [PubMed] [Google Scholar]
- 33.Shima DT. The mouse gene for vascular endothelial growth factor. Genomic structure, definition of the transcriptional unit, and characterization of transcriptional and post-transcriptional regulatory sequences. J Biol Chem. 1996;271:3877–3883. doi: 10.1074/jbc.271.7.3877. [DOI] [PubMed] [Google Scholar]
- 34.Ng YS. Differential expression of VEGF isoforms in mouse during development and in the adult. Dev Dyn. 2001;220:112–121. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1093>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 35.Houck KA. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992;267:26031–26037. [PubMed] [Google Scholar]
- 36.Charnock-Jones DS. Identification and localization of alternately spliced mRNAs for vascular endothelial growth factor in human uterus and estrogen regulation in endometrial carcinoma cell lines. Biol Reprod. 1993;48:1120–1128. doi: 10.1095/biolreprod48.5.1120. [DOI] [PubMed] [Google Scholar]
- 37.de Vries C. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255:989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 38.Quinn TP. Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc Natl Acad Sci USA. 1993;90:7533–7537. doi: 10.1073/pnas.90.16.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keyt BA. The carboxyl-terminal domain (111–165) of vascular endothelial growth factor is critical for its mitogenic potency. J Biol Chem. 1996;271:7788–7795. doi: 10.1074/jbc.271.13.7788. [DOI] [PubMed] [Google Scholar]
- 40.Waltenberger J. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 41.Sondell M, Sundler F, Kanje M. Vascular endothelial growth factor is a neurotrophic factor which stimulates axonal outgrowth through the flk-1 receptor. Eur J Neurosci. 2000;12:4243–4254. doi: 10.1046/j.0953-816x.2000.01326.x. [DOI] [PubMed] [Google Scholar]
- 42.Soker S. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 43.Ishihama H. Colocalization of neuropilin-1 and Flk-1 in retinal neovascularization in a mouse model of retinopathy. Invest Ophthalmol Vis Sci. 2001;42:1172–1178. [PubMed] [Google Scholar]
- 44.Kawasaki T. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126:4895–4902. doi: 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- 45.Hirai A. Evaluation of diabetic neuropathy through the quantitation of cutaneous nerves. J Neurol Sci. 2000;172:55–62. doi: 10.1016/s0022-510x(99)00290-7. [DOI] [PubMed] [Google Scholar]
- 46.Kennedy WR, Wendelschafer-Crabb G, Johnson T. Quantitation of epidermal nerves in diabetic neuropathy. Neurology. 1996;47:1042–1048. doi: 10.1212/wnl.47.4.1042. [DOI] [PubMed] [Google Scholar]
- 47.Christianson JA, Riekhof JT, Wright DE. Restorative effects of neurotrophin treatment on diabetes-induced cutaneous axon loss in mice. Exp Neurol. 2003;179:188–199. doi: 10.1016/s0014-4886(02)00017-1. [DOI] [PubMed] [Google Scholar]
- 48.Navarro X, Verdu E, Wendelschafer-Crabb G, Kennedy WR. Immunohistochemical study of skin reinnervation by regenerative axons. J Comp Neurol. 1997;380:164–174. doi: 10.1002/(sici)1096-9861(19970407)380:2<164::aid-cne2>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 49.Krisky DM. Deletion of multiple immediate-early genes from herpes simplex virus reduces cytotoxicity and permits long-term gene expression in neurons. Gene Therapy. 1998;5:1593–1603. doi: 10.1038/sj.gt.3300766. [DOI] [PubMed] [Google Scholar]
- 50.Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001;103:529–539. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- 51.Kennedy WR, Navarro X. Sympathetic sudomotor function in diabetic neuropathy. Arch Neurol. 1989;46:1182–1186. doi: 10.1001/archneur.1989.00520470036023. [DOI] [PubMed] [Google Scholar]