

Abstract
Precursors of cochlear and vestibular hair cells of the inner ear exit the cell cycle at midgestation. Hair cells are mitotically quiescent during late-embryonic differentiation stages and postnatally. We show here that the retinoblastoma gene Rb and the encoded protein pRb are expressed in differentiating and mature hair cells. In addition to Rb, the cyclin dependent kinase inhibitor (CKI) p21 is expressed in developing hair cells, suggesting that p21 is an upstream effector of pRb activity. p21 apparently cooperates with other CKIs, as p21-null mice exhibited an unaltered inner ear phenotype. By contrast, Rb inactivation led to aberrant hair cell proliferation, as analysed at birth in a loss-of-function/transgenic mouse model. Supernumerary hair cells expressed various cell typespecific differentiation markers, including components of stereocilia. The extent of alterations in stereociliary bundle morphology ranged from near-normal to severe disorganization. Apoptosis contributed to the mutant phenotype, but did not compensate for the production of supernumerary hair cells, resulting in hyperplastic sensory epithelia. The Rb-null-mediated proliferation led to a distinct pathological phenotype, including multinucleated and enlarged hair cells, and infiltration of hair cells into the mesenchyme. Our findings demonstrate that the pRb pathway is required for hair cell quiescence and that manipulation of the cell cycle machinery disrupts the coordinated development within the inner ear sensory epithelia.
Keywords: Inner ear, Cochlea, Vestibular organ, Hair cell, Proliferation, Differentiation, Apoptosis, Cell cycle, Mitosis, Polyploidy, Rb (Rb1), p21 (Cdkn1a), Mouse
Introduction
The auditory sensory epithelium of the cochlea, the organ of Corti, and the sensory epithelia of vestibular organs are composed of hair cells (HCs) and supporting cells. Inner ear HCs are mechanosensory cells, which convert mechanical force produced by sound waves and head movements into neural impulses. Hair cells and supporting cells have common precursors, as shown in the chick auditory organ (Fekete et al., 1998). In the mouse, these precursors permanently exit the cell cycle in midgestation (Ruben, 1967). In the presumptive organ of Corti, cell cycle exit is followed by the upregulation of the basic helix-loop-helix (bHLH) gene Math1 (Atoh1 – Mouse Genome Informatics), which initiates HC differentiation (Bermingham et al., 1999; Chen et al., 2002; Fritzsch et al., 2005). Differentiating and adult HCs are postmitotic and refractory to mitogens, a likely reason being the activity of negative cell cycle regulators.
A recent study by Chen et al. (Chen et al., 2003) showed that targeted disruption of the gene encoding a member of the Ink4 family of CKIs, p19 (Cdkn2d – Mouse Genome Informatics), leads to abnormal DNA synthesis in postnatal cochlear HCs. Bromodeoxyuridine incorporation was shown to occur at a low rate. Aberrant proliferation was accompanied by apoptosis and resulted in progressive hearing loss. In contrast to postnatal HCs, p19 inactivation did not affect the antiproliferative state of HCs during late-embryogenesis, although p19 was reported to be expressed in the embryonic organ of Corti (Chen et al., 2003). These data suggest that additional CKIs compensate for p19 deficiency in developing cochlear HCs. Cell cycle regulation downstream of CKIs has not been reported in the cochlea. Furthermore, the mechanisms underlying cell cycle arrest of vestibular HCs have not been explored.
A key regulator of the cell cycle is pRb, the protein product of the Rb (Rb1 – Mouse Genome Informatics) tumour suppressor gene (Weinberg, 1995). pRb is the prototypical member of the pocket protein family, which also comprises p107 and p130. Pocket proteins have both unique and overlapping functions in cell cycle control, in regulation of cell differentiation and survival, and in inhibition of oncogenic transformation (Classon and Harlow, 2002). pRb is a nuclear phosphoprotein. It binds members of the E2f transcription factor family during G1 and represses genes required for G1 to S-transition. In response to mitogens, pRb becomes phosphorylated (inactivated) by cyclin/cyclin dependent kinase (CDK) complexes, resulting in the release of bound E2fs, transcriptional de-repression and cell cycle progression. Mitogenic signals induce the cell cycle machinery at the level of cyclins and CDKs (Murray, 2004). CDK activation is regulated by various mechanisms, a particularly important one being the inhibition by CKIs (Vidal and Koff, 2000). Inhibition of CDK activity by CKIs maintains pRb in a hypophosphorylated (active) state. There are two families of CKIs, the Ink4 family (p15, p16, p18, p19) and the Cip/Kip family (p21, p27, p57).
Analyses of loss-of-function mutant mice have demonstrated the essential role of Rb as a repressor of cell cycle progression during embryogenesis. Rb knockouts die in midgestation, between embryonic day 13 (E13) and E14. In addition to ectopic cell cycles, development of the nervous system, skeletal muscles, lens and haematopoietic cells of the mutants is characterized by aberrant differentiation and extensive apoptosis (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1993; Morgenbesser et al., 1994; Zacksenhaus et al., 1996). Consistently, Rb is prominently expressed in these tissues (Jiang et al., 1997). Recent conditional mutagenesis and placental rescue indicate that the apoptotic phenotype of whole-embryo Rb knockouts is not caused by cell-autonomous mechanisms in all tissues: apoptosis in the brain largely occurs secondarily to other embryonic defects, whereas apoptosis in skeletal muscles, retina and lens appears to be a direct consequence of Rb inactivation (Ferguson et al., 2002; de Bruin et al., 2003; MacPherson et al., 2003; MacPherson et al., 2004; Wu et al., 2003; Chen et al., 2004; Zhang et al., 2004).
How Rb regulates cell differentiation is in general poorly understood. The role of Rb in differentiation appears to be more versatile than merely indirectly stimulating this process through the inhibition of cell cycle progression. In some cases, it has been possible to separate the effects of Rb on cell proliferation and differentiation (Sellers et al., 1998; Liu and Zacksenhaus, 2000; Takahashi et al., 2003; Zhang et al., 2004). In skeletal muscles, Rb transcriptionally upregulates genes involved in the late stages of differentiation through the bHLH gene myogenin (Gu et al., 1993; Novitch et al., 1996; Novitch et al., 1999). Similarly, adipocyte differentiation is induced by the positive effect of Rb on the transcriptional activity of CCAAT/enhancer-binding proteins (Chen et al., 1996).
We show here that pRb is expressed in inner ear HCs. The lethality of Rb knockouts at the stage when part of HCs have not yet started to differentiate precludes the use of these mutants in our studies. To genetically dissect the Rb pathway and unravel the requirement for pRb during HC development, we have analysed the inner ear sensory epithelia of mgRb:Rb−/− mutants, which are rescued to birth by a hypomorphic Rb transgene (Zacksenhaus et al., 1996). The transgene is expressed in the nervous system, but not in non-neuronal tissues (Jiang et al., 2001), including the inner ear HCs (this study). Our results suggest that pRb regulates HC quiescence and that, during development, p21 may act co-operatively with other CKI(s) as an upstream effector of pRb activity. Rb loss induced aberrant HC proliferation, but these cells also showed pathological features, including mitotic abnormalities and signs of apoptosis.
Materials and methods
Mice
The NMRI mouse strain was used for the analysis of pRb protein and Rb and p21 mRNA expression in the inner ear. Timed pregnancies were established by the detection of vaginal plug, taken the morning of plug observation as E0.5. Generation and genotyping of mgRb:Rb−/− mutant mice have been described previously (Zacksenhaus et al., 1996). p21−/− mice were obtained from Jackson Laboratory, originally described by Brugarolas et al. (Brugarolas et al., 1995). E2f1−/− and apoptosis protease-activating factor 1 (Apaf1)−/− mice have been described previously (Field et al., 1996; Yoshida et al., 1998). Their genotyping and generation of compound mgRb:Rb−/−:E2f1−/− and mgRb:Rb−/−:Apaf1−/− mutants have been described previously (Jiang et al., 2000; Guo et al., 2001).
Histology, immunohistochemistry and TUNEL staining
Whole heads of E12.5, E13.5, E14.5, E15.5 and E16.5 embryos, and dissected inner ears of E17.5 and E18.5 embryos and of postnatal day (PN) 2 and 6 pups were fixed overnight in 4% paraformaldehyde (PFA), embedded in paraffin and cut to 5-μm sections. Inner ears of 8-week-old mice were perilymphatically fixed with PFA, immersed in this fixative overnight, decalcified with 0.5 M EDTA and embedded in paraffin wax. Following antibodies were used: monoclonal Rb (BD Biosciences), polyclonal myosins VI and VIIa (Hasson et al., 1997; Pirvola et al., 2004), monoclonal calretinin and calbindin (Swant), polyclonal phospho-histone H3 (Ser10, Cell Signaling Technology) (Pirvola et al., 2004), polyclonal espin (Zheng et al., 2000), polyclonal p75 neurotrophin receptor (p75NTR) (Pirvola et al., 2002), monoclonal p27 (Neomarkers), and monoclonal (rabbit) cleaved caspase 3 (Cell Signaling Technology). Detection was carried out with the Vectastain Elite ABC kit or the Vectastain Mouse-On-Mouse kit and the diaminobenzidine substrate (Vector Laboratories). Methyl Green was used for counterstaining. For double-labelling experiments, PFA-fixed inner ears of E18.5 embryos were cryosectioned, and phospho-histone H3 and calretinin or calbindin were used as primary antibodies. Binding was visualized by fluorochrome-conjugated secondary antibodies (Alexa Fluor 488 and 568, Molecular Probes). In addition to cleaved caspase 3 immunostaining, TUNEL method-based Fluorescein In Situ Cell Death Detection Kit (Roche) was used to detect apoptotic cells.
Semi-thin sections
Inner ears of mgRb:Rb−/− mice and control littermates were dissected at E18.5 and fixed overnight in 2.5% glutaraldehyde, postfixed in 1% osmium tetroxide and embedded in Epon. Sections (0.5 μm) were cut in transverse (midmodiolar) plane and stained with 2% Toluidine Blue.
In situ hybridization
In situ hybridization was performed with 35S-labelled riboprobes on PFA-fixed paraffin wax sections according to the protocol by Wilkinson and Green (Wilkinson and Green, 1991). Rb, p107 (Rbl1 – Mouse Genome Informatics), p130 (Rbl2 – Mouse Genome Informatics), p21 (Cdkn1a – Mouse Genome Informatics), Math1, fibroblast growth factor 8 (Fgf8), Fgf10, Brn3c (Pou4f3 – Mouse Genome Informatics) and brain-derived neurotrophic factor (Bdnf) cDNAs were used.
Results
pRb expression in the inner ear sensory epithelia
We first studied pRb expression in normal inner ears by immunohistochemistry (Fig. 1A-H). We confirmed at all stages that pRb expression pattern corresponded to that of Rb mRNA, as analysed by in situ hybridization. Weak and diffuse pRb expression was initially, at E12.5, detected in the presumptive vestibular sensory patches. The expression was upregulated in the early-differentiating vestibular HCs at E13.5 (Fig. 1A). In the vestibular sensory epithelia (saccular and utricular maculae, and ampullary cristae) of late embryonic and early postnatal mice, pRb was detected in the nuclei of the entire HC population (Fig. 1A-D). By contrast, adult vestibular organs showed expression only in a subpopulation of HCs (Fig. 1E). From E12.5 onwards, pRb was weakly expressed in the caudal wall of the cochlear duct, which contains sensory precursor cells (data not shown). In the developing organ of Corti, pRb expression was upregulated at E15.5, at the early stage of morphological differentiation of cochlear HCs. This upregulation was confined to HCs and followed a base-to-apex gradient, corresponding to the differentiation gradient of the auditory organ. Both inner and outer HCs showed pRb-staining during late embryogenesis and early postnatal period (Fig. 1F,G), while during adulthood staining was detected only in inner HCs (Fig. 1H). Supporting cells adjacent to cochlear and vestibular HCs did not show detectable expression (Fig. 1A-H). Importantly, by in situ hybridization, we did not detect expression of the related genes, p107 and p130, in developing HCs (data not shown), suggesting that functional redundancy among pocket proteins is unlikely in these cells.
Fig. 1.
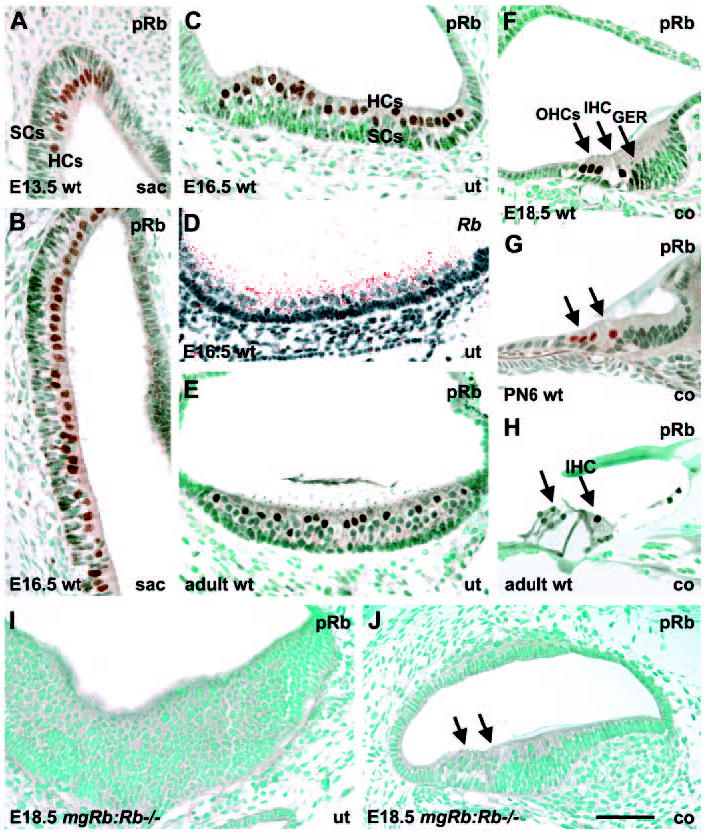
pRb expression in hair cells of the inner ear. pRb immunostaining; Rb in situ hybridization. (A,B) pRb staining in the early-and later-differentiating HCs of the embryonic saccule. (C,D) pRb and Rb expressions in HCs of the late-embryonic utricle. (E) A subpopulation of HCs is pRb-positive in the adult utricle. (F,G) pRb expression in inner and outer HCs of the cochlea at birth and early postnatal life. In addition to HCs, the greater epithelial ridge shows weak expression at birth. (H) In the mature organ of Corti, pRb expression is detected only in inner HCs. Supporting cells are negative at all stages. (I,J) pRb is not expressed in the hyperplastic sensory epithelium of utricle and cochlea of mgRb:Rb−/− mutants at E18.5. Abbreviations: wt, wild type; sac, saccule; ut, utricle; co, cochlea; IHC, inner hair cell; OHCs, outer hair cells; GER, greater epithelial ridge; HCs, hair cells; SCs, supporting cells. Arrows in G,H,J indicate HCs. Scale bar: 70 μm.
The dynamic expression of pRb and the lack of detectable levels of p107 and p130 in differentiating HCs prompted us to test the in vivo consequences of Rb loss in the inner ear. To circumvent midgestational lethality of mice homozygous for a null mutation in Rb, we used mgRb:Rb−/− mice, which are rescued to birth by transgenic expression of a wild-type Rb minigene (consisting of a genomic fragment spanning 1.3 kb of the mouse Rb promoter plus the first exon and intron fused to exons 2 to 27 of the mouse Rb cDNA) (Zacksenhaus et al., 1996). This genetic manipulation leads to suppression of the neurogenic phenotype seen in Rb knockouts and reveals a role for pRb in skeletal myogenesis. A severe lens defect and incomplete erythropoiesis have also been noted in these mutant fetuses (Zacksenhaus et al., 1996; Liu and Zacksenhaus, 2000; Jiang et al., 2000). Consistent with this, the minigene, fused to lacZ reporter gene, directs expression exclusively to the nervous system (Jiang et al., 2001). In line with the results obtained in other non-neuronal tissues of mgRb:Rb−/− embryos, we did not detect Rb or pRb expression in their HCs (Fig. 1I,J). Hence, these cells can be viewed as null for Rb.
Hyperplasia of the developing auditory sensory epithelium in the absence of Rb
Global inner ear morphology of mgRb:Rb−/− mutants at E17.5 (n=8 ears) and E18.5 (n=15 ears) was comparable with controls, but distinct differences were seen in the sensory epithelia (Figs 2 and 3). Only minor variations were observed between individual mutants, both at the level of vestibular and auditory sensory epithelia. The normal organ of Corti shows one inner HC and three outer HCs in a midmodiolar, radially sectioned cochlear duct. This was apparent from histology and from the positive staining with myosin VI, a cytoplasmic marker (Fig. 2A). One layer of supporting cells is normally located below HCs on the basilar membrane. The organ of Corti of mgRb:Rb−/− mutants was hyperplastic because of excessive HC formation (Fig. 2A′). Hyperplasticity was most prominent in the basal half of the cochlea, where cellular differentiation is more advanced than in the apical half. When analysing transverse sections from the upper basal turn of E18.5 cochleas, myosin VI-positive area showed sixfold increase in the numbers of HC nuclei (n=6 mutant cochleas, mean±s.d. 23.2±4.3, contrasting with controls showing four HC nuclei in the same view). We first asked whether supernumerary HCs were generated at the expense of supporting cells. We used the CKI p27 (Fig. 2B,B′) and p75NTR (Fig. 2C,C′) as markers for differentiating supporting cells, the former one being expressed in the nuclei of several supporting cell populations, including pillar, Deiters’, Hensen’s and Claudius’ cells, and the latter in the cytoplasm of pillar and Hensen’s cells. p75NTR (Ngfr – Mouse Genome Informatics) was also expressed in the cochlear ganglion neurons. At E17.5 and E18.5, supporting cell numbers were not decreased in the mutants, rather, their numbers were increased, but not to the extent of HC overproduction. Based on the analysis of transverse sections from the upper basal turn of E18.5 cochleas, a threefold increase in the numbers of p27- positive Deiters’ plus pillar cells was found below HCs (n=6 mutant cochleas, mean±s.d. 14.5±3.1, contrasting with controls with five of these cells in a corresponding view).
Fig. 2.
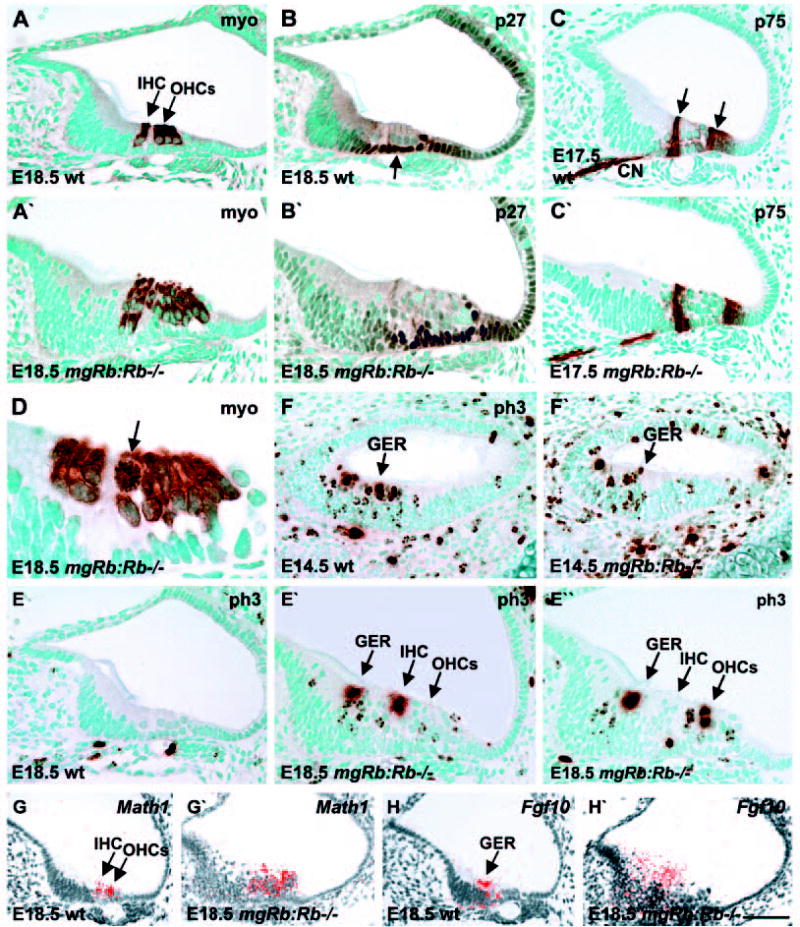
Proliferation and differentiation in the cochleas of mgRb:Rb–/– embryos. Myosin VI-and phospho-histone H3 immunostaining; Math1 and Fgf10 in situ hybridization. (A,A′) At birth, myosin VI is expressed in inner and outer HCs of control cochleas and also in supernumerary HCs of mutants. (B-C′) At late-embryogenesis, p27- and p75NTR-positive supporting cell populations (arrows) are seen both in controls and mutants. (D) A myosin VI-stained mitotic HC (arrow) in a mutant cochlea at birth. (E,E′) At birth, phospho-histone H3 staining shows ectopic mitoses in the greater epithelial ridge and the region of inner and outer HCs in mutant cochleas. These are not present in controls. (E″) Most of these mitotic HCs with strongly stained condensed chromatin are located near the lumen. (F,F′) At E14.5, phospho-histone H3- stained cells are seen in the caudal wall of the cochlea of both mutants and controls. (G,G′) At birth, Math1 is expressed in cochlear HCs of controls and also in supernumerary HCs of mutants. (H,H′) At birth, Fgf10 is expressed in the greater epithelial ridge of control cochleas and in the thickened ridge of mutants. Abbreviations: wt, wild type; ph3, phospho-histone H3; myo, myosin VI; GER, greater epithelial ridge; IHC, inner hair cell; OHCs, outer hair cells; CN, cochlear nerve. Scale bar: 70 μm for A-C′,E-F′); 30 μm for D; 80 μm for G-H′).
Fig. 3.
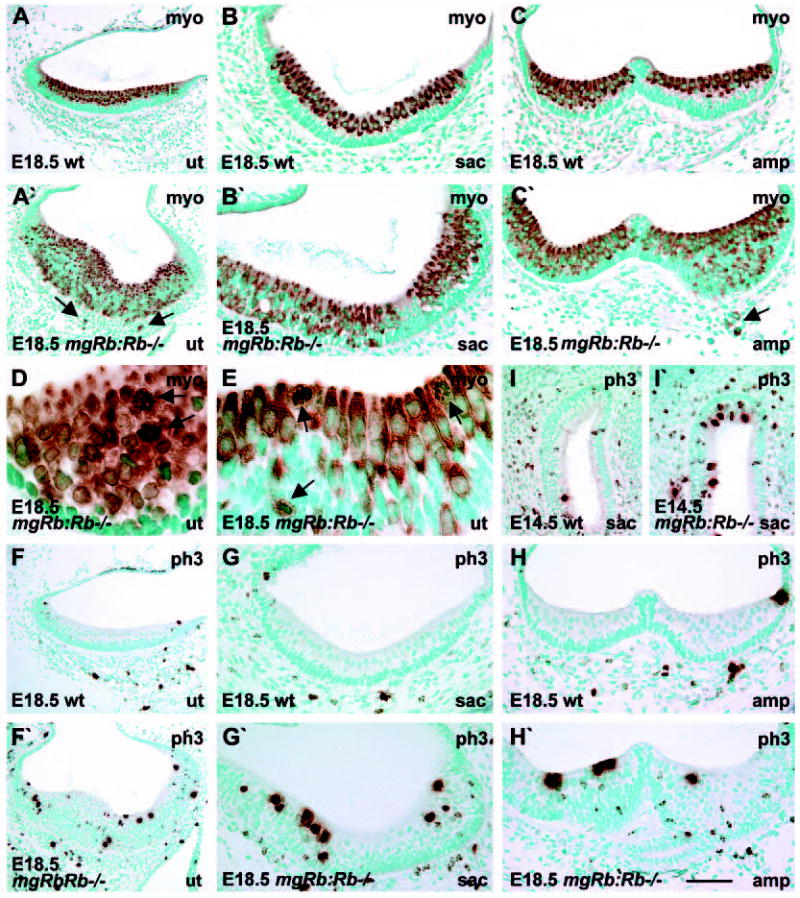
Mitosis and differentiation of vestibular hair cells of mgRb:Rb−/− embryos. Myosin VI- and phospho-histone H3- immunostaining. (A,A′) When compared with controls at birth, utricular sensory epithelia of mutants are hyperplastic because of an excess of myosinVI-positive HCs, some of which have penetrated into the mesenchyme (arrows). (B,B′) In contrast to the saccular sensory epithelium of controls in which HCs occupy the lumenal layer, HCs are distributed throughout the epithelium in mutants. (C,C′) In contrast to controls, ampullary sensory epithelia of mutants are hyperplastic because of overproduction of myosin VI-positive HCs, some of which invade the mesenchyme (arrow). (D) An oblique section through the utricular sensory epithelium of a mutant shows mitotic figures (arrows) in myosin VI-stained HCs. (E) A cross-section through the utricle shows that mitotic HCs (arrows) have rounded shape and occupy both lumenal and deeper epithelial layers. (F-H′) Phospho-histone H3- stained utricular (F,F′), saccular (G,G′) and ampullary (H,H′) sensory epithelia at birth show higher numbers of mitotic cells in mutants when compared with controls. (I,I′) At E14.5, early-differentiating saccular HCs of mutants undergo mitoses, in contrast to controls. Differentiating HCs occupy the lumenal layer and have a large nucleus. Abbreviations: wt, wild type; myo, myosin VI; ph3, phospho-histone H3. Scale bar: 120 μm for A,A′,F,F′); 70 μm for B-C′,G-I′; 30 μm for D,E.
Differentiating cochlear hair cells proliferate in the absence of Rb
A possible reason for the presence of supernumerary cochlear HCs in mgRb:Rb−/− mutants could be aberrant cell cycling. Perturbations in the activity of negative cell cycle regulators often lead to ectopic DNA synthesis, but manipulated cells can arrest at G2 and do not enter M phase (Novitch et al., 1996; Lipinski et al., 2001). To study the cell cycle status in the organ of Corti at E17.5 and E18.5, we used an M- and late G2-phase marker, phospho-histone H3. Although mitotic cells were not present in the control organ of Corti, prominent induction of mitoses was seen in the auditory sensory epithelium of mgRb:Rb−/− mice (Fig. 2E-E″). The condensed chromatin of mitotic cells reacted strongly with the phospho-histone H3 antibody, while the G2-phase cells showed patchy staining of heterochromatin. Rb inactivation led to proliferation of differentiating HCs, as confirmed by the presence of mitotic figures in myosin VI-positive HCs (Fig. 2D). This was further confirmed by co-expression of phospho-histone H3 and calbindin, another marker for cochlear HCs (data not shown). Both inner (Fig. 2E′) and outer (Fig. 2E″) HCs were cycling in the mutants. Mitotic HCs showed a rounded morphology, characteristic of dividing cells (Fig. 2D). Most of them were found at the lumenal surface, but some also in deeper epithelial layers. Mitoses were not detected in the supporting cell layer. In addition to the organ of Corti, the greater epithelial ridge of the mutants showed prominent increase in thickness and high numbers of mitotic nuclei (Fig. 2E-E″), these results being in accordance with the observations of weak Rb/pRb expression in this ridge in normal animals (Fig. 1F). In conclusion, in contrast to controls where cells of the organ of Corti and almost all cells of the greater epithelial ridge are postmitotic at E18.5, Rb inactivation leads to ectopic cell cycles. Strikingly, differentiating HCs undergo mitoses in the Rb-deficient organ of Corti.
To determine the developmental time window of aberrant mitoses in the cochleas of mgRb:Rb−/− mutants, we next focused on earlier developmental stages. The bHLH gene Math1 is one of the earliest markers for the HC lineage, being first expressed in the cochlea at E14.5 (Chen et al., 2002). This is followed by the induction of myosin VI at E15.5. At E13.5 (n=4 ears of mutants and controls each) and E14.5 (n=4 ears of mutants and controls each), the caudal wall of the cochlear duct, which houses precursor cells, showed comparable structure in mgRb:Rb−/− mutants and controls. At these stages, there was a slight increase in the numbers of phospho-histone 3-positive cells at the site of future organ of Corti in the mutants, but the increase was not statistically significant (Fig. 2F,F′, data not shown). By contrast, the adjacent mesenchyme of the mutants showed a clear increase in the amount of dividing cells, this being in line with the knowledge that Rb is expressed in the embryonic mesenchyme (Leezer et al., 2002). In the organ of Corti of the mutants, increased mitotic activity and overproduction of myosin VI-positive HCs became clear at E15.5 (n=8 ears of mutants and controls each), at the stage when pRb expression ensues in cochlear HCs of normal animals.
Available data show that in addition to a role as a repressor of cell cycle progression, depending on the cell context, Rb positively regulates differentiation by inducing expression of cell type-specific genes (Chen et al., 1996; Novitch et al., 1996; Novitch et al., 1999). To find out the possible role of Rb on cochlear HC differentiation, we studied the expression of a panel of early (Math1, myosins VI and VIIa, calbindin, Fgf8) and late (Brn3c) differentiation markers in mgRb:Rb−/− mutants at E17.5 and E18.5. Math1 (Fig. 2G,G′) and the other markers (data not shown) were expressed in Rb-null cochlear HCs, similar to controls, although consistent with the expansion of HCs the signal covered a correspondingly wider zone. In normal animals, Fgf8 is expressed in inner HCs, but not in outer HCs (Pirvola et al., 2002). Likewise, in the mutants, Fgf8 expression was restricted to supernumerary inner HCs, indicating that cochlear HCs were subtyped despite their overproduction (data not shown). Thus, despite the altered cytoarchitecture of the organ of Corti, cochlear HCs of the mutants showed a normal profile of molecules involved in differentiation and maturation.
In addition, the greater epithelial ridge, the epithelial domain located medially to the organ of Corti, was hyperplastic in mgRb:Rb−/− mutants at birth (Fig. 2). Earlier studies have shown that Fgf10 is expressed in the greater epithelial ridge (Pirvola et al., 2000; Pauley et al., 2003) (Fig. 2H) and this structure has been suggested to contain sensory precursor-like cells (Zheng and Gao, 2000; Woods et al., 2004). At birth, Fgf10 was expressed throughout the abnormally thick greater epithelial ridge of the mutants, suggesting that ectopic cells in this region have characteristics of sensory precursor cells (Fig. 2H,H′).
Differentiating vestibular hair cells mitose in the absence of Rb
In addition to cochlear HCs, pRb was expressed in vestibular HCs (Fig. 1). We therefore studied the consequences of Rb loss in the vestibular sensory epithelia of mgRb:Rb−/− mice at E17.5 (n=8 ears) and E18.5 (n=15 ears). Prominent abnormalities were found (Fig. 3). Similar to the organ of Corti, Rb inactivation led to increase in the thickness of the vestibular sensory epithelia and to overproduction of myosin VI-positive HCs (Fig. 3A-C′). In normal sensory epithelia, HCs are situated lumenally and supporting cells basally. In the mutants, HCs were intermixed with supporting cells in the middle layers and the basal part of the epithelium was filled with supernumerary HCs. Moreover, some myosin VI-stained HCs were dislocated through the basal lamina into the mesenchyme (Fig. 3A′,C′). Mitotic figures were found in rounded, myosin VI-positive cells, demonstrating divisions of differentiating vestibular HCs (Fig. 3D,E), similar to Rb-null cochlear HCs (Fig. 2D). Most mitotic vestibular HCs were located at the lumenal surface, but some were seen at deeper epithelial levels (Fig. 3E) and in the mesenchyme. Accordingly, high numbers of phospho-histone H3-positive cells were observed in the vestibular sensory epithelia of the mutants, the majority of them at the lumenal surface, but some in deeper layers. By contrast, only a few dividing cells were found in control specimens (Fig. 3F-H′). Double-labelling experiments showed co-expression of phospho-histone H3 and calretinin, a marker for vestibular HCs, these data confirming that vestibular HCs of mgRb:Rb−/− mice undergo mitosis (data not shown). Similar to the situation at birth, at E14.5, high numbers of dividing HCs were found in the early-differentiating vestibular sensory epithelia of the mutants, in contrast to controls (n=6 ears of mutants and controls each) (Fig. 3I,I′).
Owing to differences in the timing of onset of differentiation, vestibular HCs at birth represent a more mature HC status when compared with cochlear HCs. Therefore, we also focused on the development of HC stereocilia in the vestibular organs. Math1, myosins VI and VIIa, calretinin, Fgf8, Bdnf and Brn3c were expressed in vestibular HCs of the mutants, similar to controls (data not shown). The fact that the neurotrophic factor Bdnf was expressed in supernumerary HCs suggests that these cells can attract neuronal endings for the establishment of synaptic contacts. Together, molecular differentiation of Rb-deficient vestibular HCs appears well advanced, similar to Rb-null auditory HCs. However, HCs of the mutants showed abnormalities in the stereociliary bundle development, as evidenced by espin staining (Fig. 4A-H). Controls showed espin expression exclusively in stereocilia (Fig. 4A,E,G). In the mutants, espin-staining showed abnormal stereociliary bundle morphologies, particularly in utricles (Fig. 4B-F), but near-normal bundles were also seen, often in ampullae (Fig. 4G,H). Interestingly, supernumerary HCs in the deeper epithelial layers and in the mesenchyme showed espin-positive, disorganized cilia-like protusions and staining along cell membrane, indicating that the apicobasal polarity of these cells was lost (Fig. 4B-F).
Fig. 4.
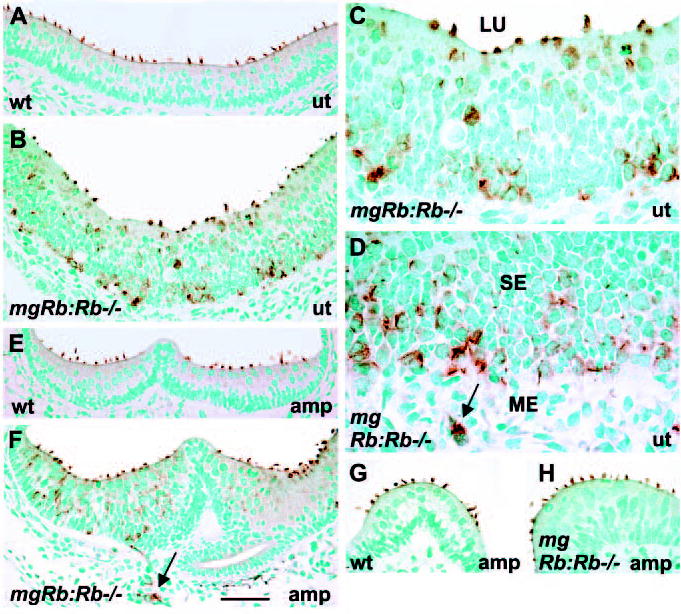
Stereociliary bundles of vestibular hair cells of mgRb:Rb−/− mutants at birth, as shown by espin-immunostaining. (A) Espin is localized to HC stereocilia of control utricules. (B) In mutants, espin-positive HCs are situated throughout the thickened utricular sensory epithelium. Several HCs at the surface show abnormal bundles. (C,D) High magnification views show HCs with aberrant stereocilia in the utricle and in the adjacent mesenchyme (arrow) of mutants. (E,F) In contrast to controls, mutants show espin-stained HCs at different levels of the ampullary sensory epithelium. HCs in the mesenchyme are also espin positive (arrow). (G,H) In addition to the distinct bundle abnormalities, stereociliary bundle morphologies comparable with controls are seen in some HCs of mutants, especially in ampullae. Abbreviations: wt, wild type; ut; utricle; amp, ampulla; SE, sensory epithelium; ME, mesenchyme; LU, lumen. Scale bar: 70 μm for A-F; 60 μm for G,H; 30 μm for C,D.
Supernumerary hair cell production is partially compensated by apoptosis
In addition to the regulation of cell cycle progression and differentiation, several studies have shown that Rb inactivation is associated with apoptosis. Because ectopic proliferation in the absence of Rb is rapidly balanced by massive apoptosis in many tissues, there are no obvious hyperplastic lesions (Zacksenhaus et al., 1996; Guo et al., 2001; Chen et al., 2004; MacPherson et al., 2004, Zhang et al., 2004). In contrast to other tissues, cleaved caspase 3- and TUNEL staining revealed apoptotic profiles only in part of sections through the mutant inner ear sensory epithelia at E17.5 and E18.5 (n=6 mutant ears at each stage). Myosin VI staining of nearby sections localized apoptosis to the regions of supernumerary cochlear (Fig. 5A-C) and vestibular HCs (Fig. 5D-F). Control sensory epithelia (n=6 ears at E18.5) showed very few apoptotic profiles (data not shown).
Fig. 5.
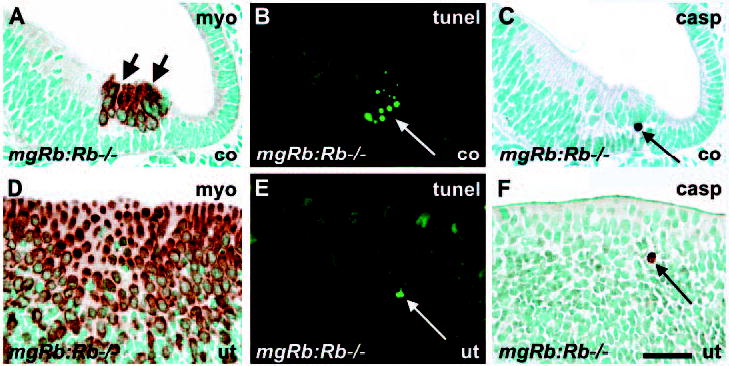
Apoptosis in the inner ear of mgRb:Rb−/− mutants at E18.5. (A-C) In a mutant cochlea, the myosin VI-stained region of supernumerary HCs, which comprises mitotic figures (A, short arrows), shows TUNEL- (B) and cleaved caspase 3- (C) positive cells. Views are from adjacent sections. Compare with normal morphology in Fig. 2A. (D-F) TUNEL- and caspase 3-positive cells are present in the myosin VI-stained region of a hyperplastic utricular sensory epithelium. Compare with normal morphology in Fig. 3A. Long arrows indicate apoptotic profiles.
Abbreviations: co, cochlea; ut, utricle; myo, myosin VI; casp, cleaved caspase 3. Scale bar: 60 μm.
Rb loss can induce cell-autonomous apoptosis in certain tissues through the E2f1/p53/Apaf1 pathway (Morgenbesser et al., 1994; Macleod et al., 1996; Tsai et al., 1998; Jiang et al., 2000; Guo et al., 2001). To further evaluate the contribution of apoptosis to the mutant phenotype, we studied the compound mgRb:Rb−/−:Apaf1−/− mutants (n=5 ears) and mgRb:Rb−/−:E2f1−/− mutants (n=5 ears) at E18.5. The expectation was that Rb lossinduced apoptosis might be rescued to some extent by the concomitant Apaf1 or E2f1 inactivation and, thus, that the compound mutants might exhibit an increase in the amount of supernumerary HCs. The analysis revealed that, first, the morphology of the inner ear sensory epithelia of Apaf1 and E2f1 single null mutants was indistinguishable from controls and, second, the extent of HC overproduction in both types of compound mutants was comparable with mgRb:Rb−/− mutants (data not shown). Thus, apoptosis seems to be part of the altered phenotype, but does not prevent the distinct hyperplasia of Rb-deficient inner ear sensory epithelia at birth. The fact that we did not find deregulated cell cycle activity in these epithelia of E2f1-null mutants suggests that other E2f family members, which can bind pRb (E2f2, E2f3), regulate the expression of genes required for the G1- to S-phase progression either alone or redundantly with E2f1.
Rb loss induces hair cell multinucleation
To further study the developmental status of supernumerary HCs of mgRb:Rb−/− mutants at birth, we analysed 0.5 μm Toluidine Bluestained plastic sections. Intermixture of HCs and supporting cells within the hyperplastic vestibular sensory epithelia was readily observed in the absence of Rb (Fig. 6A-H). In contrast to controls (Fig. 6A,G), the mutant sensory epithelia contained large numbers of HCs with a single, bizarre-shaped nucleus or with two (occasionally three or four) nuclei (Fig. 6B-E,H). Most of these HCs showed decondensed DNA. The occurrence of multinucleated HCs suggests that nuclear divisions had occurred without cytokinesis. Despite these distinct nuclear abnormalities, many of the lumenally located HCs showed near-normal stereociliary bundles (Fig. 6B,E,H). Many multinucleated HCs seemed to have a giant size, extending from the lumenal surface to the deeper epithelial strata (Fig. 6B,C,E). In addition to HCs with aberrant nuclear morphologies, small and rounded cells with mitotic figures in a single nucleus were seen, most of them at the epithelial surface (Fig. 6C-E,H). Many of these mitotic cells were identified as HCs, based on their immature stereociliary bundles and, in paraffin sections, on myosin VI expression (Fig. 3E). Thus, the hyperplastic phenotype of the inner ear sensory epithelia of mgRb:Rb−/− mutants seems to be caused by increase both in numbers and size of HCs. The relationship between the two types of Rb-null HCs is unclear, although in some cases HCs undergoing mitosis appeared to be derived from multinucleated HCs (data not shown). Based on observations made in some semi-thin sections, we cannot exclude the possibility that some supporting cells normally located basally in the sensory epithelia translocate to the lumenal surface and divide there (the mitotic cell marked with arrowhead in Fig. 6C). Consistent with an increase in apoptosis, as revealed by TUNEL- and cleaved caspase 3 staining (Fig. 5), semi-thin sections revealed scattered apoptotic profiles in the inner ear sensory epithelia of the mutants (Fig. 6D,F). The cytoplasm of some mitotic HCs exhibited signs of degeneration and, in a some cases, appeared apoptotic with condensed and fragmented nuclei (Fig. 6E,F). The organ of Corti of the mutants (Fig. 6I,J) showed similar pathological features to the vestibular sensory epithelia.
Fig. 6.
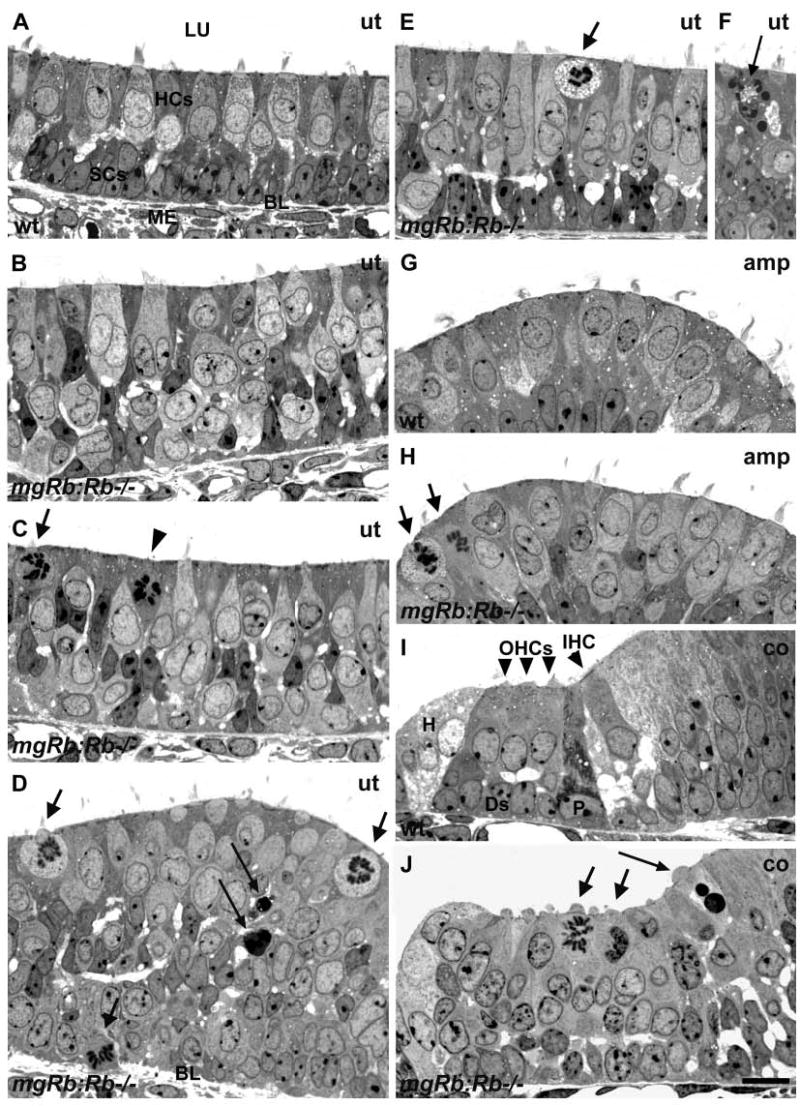
Mitosis, apoptosis and multinucleation of hair cells of mgRb:Rb−/− mutants at E18.5, as shown in semi-thin sections. (A) Normal utricular sensory epithelium shows HCs at the lumenal surface and underlying supporting cells. (B) A mutant utricle shows disorganized cell layering and HCs with an atypical nucleus or with two or more nuclei. Some of these HCs show near-normal stereociliary bundles. Some HCs seem to extend from the surface deep into the epithelium. (C) Mitotic cells with rounded morphology and nascent stereocilia (arrow) at the lumenal surface are intermixed with HCs with aberrant nuclear morphologies. A putative supporting cell that has translocated to the surface for mitosis is marked by an arrowhead. (D) Mitotic HCs (short arrows) are found mostly at the lumenal surface, but also in deeper layers, such as in this view where a cell undergoing mitosis is penetrating through the basal lamina. Scattered apoptotic profiles are also found (long arrows). (E,F) A mitotic cell (short arrow) showing signs of degeneration in the cytoplasm. Mitotic cells occasionally showed apoptotic figures (long arrow). (G,H) In contrast to controls, the ampullary sensory epithelia of mutants contain mitotic cells (arrows). In addition, several HCs with abnormal nuclei are found and many of them have stereociliary bundles with a morphology comparable with controls. (I,J) A transverse view through the organ of Corti of a normal cochlea shows four HCs and the underlying supporting cells. In the region of organ of Corti of mutants, high numbers of nuclei are seen, some of them having mitotic figures (short arrows) and a few apoptotic profiles (long arrow). Abbreviations: wt, wild type; ut, utricle; amp, ampulla; co, cochlea; LU, lumen, BL, basal lamina; ME, mesenchyme; HCs, hair cells; SCs, supporting cells; IHC, inner hair cell; OHCs, outer hair cells; P, pillar cell; Ds, Deiters’ cell, H, Hensen’s cell. Scale bar: 15 μm for A-J.
p21 is expressed in differentiating hair cells, but the null mutants have an unaltered inner ear phenotype
In our efforts to understand cell cycle regulation of HCs, we found that, in addition to Rb, the CKI p21 is expressed in developing vestibular and cochlear HCs (Fig. 7A-C). p21 expression was induced in vestibular HCs at E12.5 and in cochlear HCs at E14.5 (data not shown). By PN6, this expression was downregulated in vestibular HCs, while weak expression was still seen in cochlear HCs. Hair cells of the adult inner ear were devoid of p21 (data not shown). Thus, in contrast to Rb, p21 expression in HCs is restricted to developmental stages. In addition to HCs, p21 was strongly expressed in some other structures of the developing inner ear: spiral limbus and stria vascularis of the cochlear duct; nonsensory epithelium of vestibular organs; endolymphatic duct; and the neurons of the cochlear and vestibular ganglia (Fig. 7A,C).
Fig. 7.
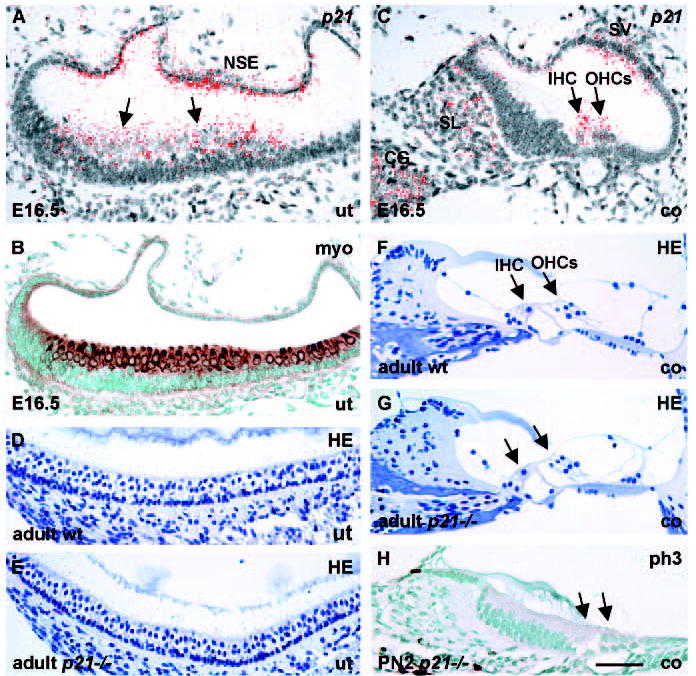
p21 expression in the developing inner ear and the phenotype of p21 knockout mice. p21 in situ hybridization; myosin VI and phospho-histone H3 immunostaining; Haematoxylin staining. (A,B) As shown in adjacent sections, p21 and myosin VI are expressed in utricular HCs (arrows) at E16.5. p21 is also found in the utricular non-sensory epithelium. (C) In the cochlea at E16.5, p21 is expressed in HCs and in stria vascularis, spiral limbus and cochlear ganglion neurons. (D,E) Histology of the utricular sensory epithelium of adult p21−/− mice is indistinguishable from controls, as shown by Haematoxylin staining. (F,G) Histology of the organ of Corti of adult p21 knockouts is comparable with controls. (H) Mitoses are not induced in the cochleas of p21-null mutants, as shown at P2 by phospho-histone H3-staining (compare with control in Fig. 2E). Arrows in G,H indicate HCs. Abbreviations: wt, wild type; ut, utricle; co, cochlea; HE, Haematoxylin; SL, spiral limbus; CG, cochlear ganglion; SV, stria vascularis; IHC, inner hair cell; OHCs, outer hair cells; NSE, non-sensory epithelium; myo, myosin VI; ph3, phospho-histone H3. Scale bar: 70 μm for A-C,H; 80 μm for D-G.
To address the role of p21 in the inner ear, we analysed the consequences of p21 inactivation (Fig. 7D-H). Earlier studies have shown that p21 knockout embryos develop into phenotypically normal adults (Deng et al., 1995), most likely owing to functional compensation between the CKI family members. However, previous studies have not specifically analysed p21 null inner ears. At E16.5, E18.5, P2 and 2 months postnatally, we did not find morphological abnormalities in these mutant inner ears. Specifically, the cytoarchitecture of the vestibular and auditory sensory epithelia appeared normal (Fig. 7D-G) and there was no induction of proliferation (Fig. 7H). Taken together, the intriguing co-expression of p21 and Rb in developing HCs suggests that p21 is an upstream regulator of pRb activity and that both cell cycle repressors account for the postmitotic state of developing HCs. The fact that p21 knockouts exhibit a normal inner ear phenotype suggests redundancy between p21 and another CKI(s).
Discussion
The present study shows that the Rb tumour suppressor gene and its encoded protein, pRb, are expressed in cochlear and vestibular HCs, both during the stages of differentiation and during adulthood. We have dissected the requirement for Rb during HC development in mgRb:Rb−/− mutant mice, in which HCs are Rb negative. A main finding is the aberrant cycling of differentiating HCs. Differentiating and mature HCs are normally characterized by their permanent withdrawal from the cell cycle. Analysis of mgRb:Rb−/− mutants at birth, together with the expression data, suggest that pRb is required in a cellautonomous manner for keeping differentiating HCs in a postmitotic state. As pRb expression was also detected in postnatal HCs, it is likely that its role as a negative cell cycle regulator is maintained during adulthood.
In several tissues, precursor cells permanently withdraw from the cell cycle before the onset of differentiation. This has also been shown to be the case in the embryonic cochlea (Chen et al., 2002). Although we could not see a clear effect of Rbinactivation on the size of the precursor cell pool at E13.5 and E14.5, other observations suggest that pRb regulates the timing of their terminal mitoses. First, at these stages, there was a slight increase in the amount of phospho-histone 3-positive cells at the site of presumptive organ of Corti in mgRb:Rb−/− mice. Second, the greater epithelial ridge, which harbours precursor-like cells that can develop into HCs (Zhang et al., 2000; Woods et al., 2004), contained aberrantly cycling cells in the late-embryonic mutants. Third, in the cochleas of E18.5 mutants, the supporting cell population was to some extent expanded. As mitoses were not detected in the supporting cell layer of mutant cochleas, elevated numbers of precursor cells might contribute to this expansion. Based on our studies on the expression of members of the pocket protein family in the midgestational inner ear (U.P. and J.M., unpublished), the role of pRb on precursor cells appears to be to some extent redundant with other pocket proteins.
The present data show that HCs differentiate despite abnormal cell cycling, suggesting that HC proliferation and differentiation are uncoupled processes. Supernumerary HCs expressed a wide panel of markers characteristic for differentiating (and mature) HCs. During skeletal myogenesis, Rb stimulates the expression of differentiation markers by influencing Myod1 activity (Gu et al., 1993; Novitch et al., 1996; Novitch et al., 1999; Zacksenhaus et al., 1996). A similar mechanism might occur in the developing inner ear, but as the interacting genes of the equivalent bHLH gene (Math1) in HCs have not been characterized, this possibility remains to be explored. Rb-null HCs at birth showed molecular evidence of differentiation, including expression of integral molecular constituents of stereocilia. However, supernumerary HCs situated in the deeper epithelial strata showed distinctly abnormal stereociliary bundles. At the lumenal surface, where stereociliary development normally occurs, large numbers of nascent bundles were seen. In many cases, this immaturity could be linked with HCs undergoing mitosis. Thus, the aberrant stereociliary development seems to be a consequence of deregulated cell cycles. The fact that stereociliary bundles of some of the supernumerary HCs appeared histologically almost comparable with controls implies they are functional in terms of mechanotransduction (Sage et al., 2005).
Using a phospho-histone H3 antibody, we were able to determine the kinetics of dividing HCs. Phospho-histone H3 is an M-phase marker, based on the fact that histone H3 Ser10 phosphorylation coincides with chromosome condensation of mitotic cells. Initiation of this phosphorylation is associated with heterochromatin of late G2 phase, and, thus, this antibody is not completely M-phase specific (Hendzel et al., 1997; Van Hooser et al., 1998). The present results demonstrate, in controls and mutants, the difference between the strong staining of condensed chromatin of mitotic cells and the ‘patchy’ staining of heterochromatin of G2-phase cells. In the inner ear sensory epithelia of normal embryos, at the stages before HC differentiation, mitotic nuclei were invariably located to the lumenal surface. The majority of late G2-phase (this study) and S-phase cells (bromodeoxyuridine-positive) (e.g. Pirvola et al., 2002) are situated deeper in the sensory epithelia. This kind of relationship between nuclear position and the phase of cell cycle, termed as interkinetic nuclear migration, has been found in several types of developing epithelia, including the traumatized inner ear sensory epithelia of birds (Bhave et al., 1995). Also in mgRb:Rb−/− mutants, most mitotic HCs were located to the epithelial surface. However, HCs undergoing mitosis were also seen deeper in the epithelia, indicating that translocation of nucleus or cell body to the surface is not a prerequisite for mitosis.
Our results suggest that postmitotic HCs retain the potential for cell cycle entry, but the pRb pathway acts as a guard to prohibit proliferation. The activity of pRb is probably kept in check by post-translational modifications, such as phosphorylation. Mitogens activate CDKs through upregulation of cyclins. A primary mechanism inhibiting CDK activity is the binding of CKIs to these kinases. CKIs efficiently inhibit CDKs even in the presence of cyclins (Olson et al., 2000). Indirect evidence for the importance of CDK regulation by CKIs in HCs comes from the fact that HCs do not proliferate in response to serum or mitogenic growth factors (Ryan, 2003).
Our data show that the CKI p21 is expressed in the differentiating cochlear and vestibular HCs, and that the expression is induced at the initiation of HC differentiation. In the auditory sensory epithelium, p21 expression was initiated at E14.5, at the stage when Math1 expression has been first detected (Chen et al., 2002). It is possible that p21 induction in HCs is regulated by Math1, in analogy to the positive role of bHLH proteins such as Myod1 and myogenin in skeletal myogenesis (Halevy et al., 1995; Guo et al., 1995). Thereafter, p21 together with other CKI(s) (see below) might have an active role in keeping pRb in a hypophosphorylated form. Thus, negative regulation at the level of both pRb and CKIs seems to be responsible for the maintenance of HC quiescence.
We did not find phenotypic alterations or aberrant mitoses in the inner ears of developing or adult p21−/− mice (Fig. 7). Interestingly, in addition to p21, another CKI, p19, has been shown to be expressed in the late-embryonic organ of Corti, but its inactivation does not result in developmental abnormalities (Chen et al., 2003). Thus, functional redundancy may exist between p21 and p19 in developing cochlear HCs. In addition, developing vestibular HCs express p21, but do not show phenotypic changes following targeted gene disruption, most probably owing to functional compensation. The identity of the CKI that may cooperate with p21 in vestibular HCs remains to be identified, as p19 expression and the consequences of p19 inactivation have not been reported in vestibular organs.
In contrast to developmental stages, we did not detect p21 expression in adult HCs. Consistent with these observations are the data that cochlear HCs of p19-null mice show aberrant S-phase entry only during postnatal life (Chen et al., 2003). Interestingly, in the mature cochlea, p19 inactivation was shown to have a stronger effect on inner HCs when compared with outer HCs (Chen et al., 2003), suggesting differences in the regulation of postmitotic state between the two cochlear HC subtypes. There might be differences in the expression of p19 and/or other CKIs in these cells (postnatal expression of p19 was not shown by Chen et al.) or cell cycle regulation downstream of CKIs might be different. The latter possibility is supported by the present data showing detectable expression of pRb in inner, but not outer, HCs of the adult cochlea. Interestingly, also in adult vestibular organs, pRb and Rb were expressed in a subset of HCs. Although non-labelled HCs may express pRb levels that are below the detection limit of the methods used, it remains to be determined whether this non-homogenous expression is linked to the morphological classification into type I and type II vestibular HCs (Wersäll and Bagger-Sjöbäck, 1974).
Existing data provide evidence for the sensitization of cells to apoptosis because of Rb loss-induced unscheduled proliferation at ectopic sites, although there is also evidence that pRb harbours inherent anti-apoptotic functions (Chau and Wang, 2003). We observed low-level apoptosis in the inner ear sensory epithelia of mgRb:Rb−/− mice at birth. The extent of this death did not compensate for the prominent hyperplasia of these epithelia, in marked contrast to several other Rb-deficient tissues showing massive cell-autonomous apoptosis in conjunction with ectopic proliferation (Zacksenhaus et al., 1996; Guo et al., 2001; Chen et al., 2004; MacPherson et al., 2004; Zhang et al., 2004). Thus, at least during development, the extent of apoptosis associated with Rb loss appears to be context dependent. The present data raise the issue of the fate of Rb-null HCs. Intriguingly, Rb loss induced HC cycling, but a large part of supernumerary HCs had defects in the completion of the cell cycle, with apparent failures in cytokinesis. The formation of multinucleated HCs, most of which appeared binucleated, implies for polyploidy. Based on the knowledge that polyploidy induced by manipulation of the cell cycle machinery often triggers cell death (Storchova and Pellman, 2004), the rate of apoptosis of supernumerary HCs may accelerate during postnatal life, an issue that can not be studied in the mgRb:Rb−/− mice because of their lethality at birth. The data that supernumerary HCs of adult p19 knockout mice, which are generated postnatally rather than during embryogenesis, apoptose (Chen et al., 2003) support the possibility that Rbdeficient HCs may ultimately be lost. However, the aberrant proliferation, polyploidy and infiltration of Rb-null HCs into the mesenchyme may lead to neoplastic transformation.
In conclusion, the present work reveals unexpected plasticity of differentiating HCs. We show that the normally quiescent HCs proliferate in response to Rb loss and that these divisions are to a large extent tolerated, as analysed at birth. Continued Rb and pRb expression in mature HCs speaks for the role of this tumour suppressor as a guard against mitoses during adulthood as well. Our results point to the importance of upstream effectors of the CKI family in modulating pRb activity. While our work was under review, Sage et al. showed, in agreement with our results, that Rb inactivation induces HC proliferation (Sage et al., 2005). Similar to our data, they also showed that stereociliary bundles of Rb-null HCs were disoriented, but they also demonstrated that supernumerary HCs can function as mechanoelectric transducers. Our findings on the pathology of Rb-null HCs, including apoptosis, polyploidy and disorganization within and outside the inner ear sensory epithelia, were not reported by Sage et al. but suggest that HC quiescence is essential for the maintenance of coordinated development of these epithelia. Our results suggest that forced HC proliferation may induce tumour cell-like and death-prone phenotypes. These data are likely to have implications in the design of future therapies to induce HC re-growth.
Acknowledgments
We thank Juha Partanen for comments on the manuscript and Maria von Numers for technical assistance. We are grateful to J. Bartles (espin), M. Chao (p75NTR), T. Hasson (myosins) for providing antibodies; and B. Vogelstein (p21), J. Johnson (Math1), M. Xiang (Brn3c), C. MacArthur (Fgf8) and B. Hogan (Fgf10) for providing in situ hybridization probes. This work was supported by the Academy of Finland, Sigrid Jusélius Foundation and the European Commisssion FP6 Integrated Project EuroHear (U.P.), and the Canadian Institute for Health Research (E.Z.). E.Z. holds a CRS/CIHR New Investigator Scholarship.
References
- Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, Zoghbi HY. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–1841. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- Bhave SA, Stone JS, Rubel EW, Coltrera MD. Cell cycle progression in gentamicin-damaged avian cochleas. J Neurosci. 1995;16:4618–4628. doi: 10.1523/JNEUROSCI.15-06-04618.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003;3:130–138. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- Chen PL, Riley DJ, Chen Y, Lee WH. Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev. 1996;10:2794–2804. doi: 10.1101/gad.10.21.2794. [DOI] [PubMed] [Google Scholar]
- Chen P, Johnson JE, Zoghbi HY, Segil N. The role of Math1 in inner ear development: Uncoupling the establishment of the sensory primordium from hair cell fate determination. Development. 2002;129:2495–2505. doi: 10.1242/dev.129.10.2495. [DOI] [PubMed] [Google Scholar]
- Chen P, Zindy F, Abdala C, Liu F, Li X, Roussel MF, Segil N. Progressive hearing loss in mice lacking the cyclin-dependent kinase inhibitor Ink4d. Nat Cell Biol. 2003;5:422–426. doi: 10.1038/ncb976. [DOI] [PubMed] [Google Scholar]
- Chen D, Livne-bar I, Vanderluit JL, Slack RS, Agochiya M, Bremner R. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell. 2004;5:539–551. doi: 10.1016/j.ccr.2004.05.025. [DOI] [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2:910–917. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- de Bruin A. Wu, L. Saavedra, H. I. Wilson, P. Yang, Y. Rosol, T. J. Weinstein, M. Robinson, M. L, Leone, G Rb function in extraembryonic lineages suppresses apoptosis in the CNS of Rb-deficient mice. Proc Natl Acad Sci USA. 2003;100:6546–6551. doi: 10.1073/pnas.1031853100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- Fekete DM, Muthukumar S, Karagogeos D. Hair cells and supporting cells share a common progenitor in the avian inner ear. J Neurosci. 1998;18:7811–7821. doi: 10.1523/JNEUROSCI.18-19-07811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK, Slack RS. Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J. 2002;21:3337–3346. doi: 10.1093/emboj/cdf338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG, Jr, Livingston DM, Orkin SH, Greenberg ME. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- Fritzsch, B., Matei, V. A., Nichols, D. H., Bermingham, N., Jones, K., Beisel, K. W. and Wang, V. Y. (2005). Atoh1 null mutants show directed afferent fiber growth to undifferentiated ear sensory epithelia followed by incomplete fiber retention. Dev. Dyn (in press). [DOI] [PMC free article] [PubMed]
- Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- Guo K, Wang J, Andres V, Smith RC, Walsh K. MyoDinduced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol. 1995;15:3823–3829. doi: 10.1128/mcb.15.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Yikang S, Yoshida H, Mak TW, Zacksenhaus E. Inactivation of the retinoblastoma tumor suppressor induces apoptosis protease-activating factor-1 dependent and independent apoptotic pathways during embryogenesis. Cancer Res. 2001;61:8395–8400. [PubMed] [Google Scholar]
- Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- Hasson T, Gillespie PG, Garcia JA, MacDonald RB, Zhao Y, Yee AG, Mooseker MS, Corey DP. Unconventional myosins in inner-ear sensory epithelia. J Cell Biol. 1997;137:1287–1307. doi: 10.1083/jcb.137.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Zacksenhaus E, Gallie BL, Phillips RA. The retinoblastoma gene family is differentially expressed during embryogenesis. Oncogene. 1997;14:1789–1797. doi: 10.1038/sj.onc.1201014. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Liang P, Leng R, Guo Z, Liu Y, Liu X, Bubnic S, Keating A, Murray D, Goss P, Zacksenhaus E. E2F1 and p53 are dispensable, whereas p21(Waf1/Cip1) cooperates with Rb to restrict endoreduplication and apoptosis during skeletal myogenesis. Dev Biol. 2000;227:8–41. doi: 10.1006/dbio.2000.9892. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Guo Z, Saad FA, Ellis J, Zacksenhaus E. Retinoblastoma gene promoter directs transgene expression exclusively to the nervous system. J Biol Chem. 2001;276:593–600. doi: 10.1074/jbc.M005474200. [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Leezer JL, Hackmiller RC, Greene RM, Pisano MM. Expression of the retinoblastoma family of tumor suppressors during murine embryonic orofacial development. Orthod Craniofacial Res. 2002;6:32–47. doi: 10.1046/j.1439-0280.2003.2c035.x. [DOI] [PubMed] [Google Scholar]
- Lipinski MM, Macleod KF, Williams BO, Mullaney TL, Crowley D, Jacks T. Cell-autonomous and non-cell-autonomous functions of the Rb tumor suppressor in developing central nervous system. EMBO J. 2001;20:3402–3413. doi: 10.1093/emboj/20.13.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zacksenhaus E. E2F1 mediates ectopic proliferation and stage-specific p53-dependent apoptosis but not aberrant differentiation in the ocular lens of Rb deficient fetuses. Oncogene. 2000;19:6065–6073. doi: 10.1038/sj.onc.1203996. [DOI] [PubMed] [Google Scholar]
- Macleod KF, Hu Y, Jacks T. Loss of Rb activates both p53- dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Crowley D, Trumpp A, Bronson RT, Jacks T. Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol Cell Biol. 2003;3:1044–1053. doi: 10.1128/MCB.23.3.1044-1053.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Kim T, Ho D, McLaughlin ME, Jacks T. Cell type-specific effects of Rb deletion in the murine retina. Genes Dev. 2004;18:1681–1694. doi: 10.1101/gad.1203304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenbesser SD, Williams BO, Jacks T, DePinho RA. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature. 1994;371:72–74. doi: 10.1038/371072a0. [DOI] [PubMed] [Google Scholar]
- Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116:221–234. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- Novitch BG, Mulligan GJ, Jacks T, Lassar AB. Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J Cell Biol. 1996;135:441–456. doi: 10.1083/jcb.135.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch BG, Spicer DB, Kim PS, Cheung WL, Lassar AB. pRb is required for MEF2-dependent gene expression as well as cellcycle arrest during skeletal muscle differentiation. Curr Biol. 1999;9:449–459. doi: 10.1016/s0960-9822(99)80210-3. [DOI] [PubMed] [Google Scholar]
- Olson NE, Kozlowski J, Reidy MA. Proliferation of intimal smooth muscle cells. Attenuation of basic fibroblast growth factor 2-stimulated proliferation is associated with increased expression of cell cycle inhibitors. J Biol Chem. 2000;275:11270–11277. doi: 10.1074/jbc.275.15.11270. [DOI] [PubMed] [Google Scholar]
- Pauley S, Beisel K, Wright T, Pirvola U, Ornitz D, Fritzsch B. Expression and function of FGF10 in mammalian inner ear development. Dev Dyn. 2003;227:203–215. doi: 10.1002/dvdy.10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Spencer-Dene B, Xing-Qun L, Kettunen P, Thesleff I, Fritzsch B, Dickson C, Ylikoski J. FGF/FGFR-2(IIIb) signaling is essential for inner ear morphogenesis. J Neurosci. 2000;20:6125–6134. doi: 10.1523/JNEUROSCI.20-16-06125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hebert JM, McConnell SK, Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Zhang X, Mantela J, Ornitz DM, Ylikoski J. Fgf9 signaling regulates inner ear morphogenesis through epithelialmesenchymal interactions. Dev Biol. 2004;273:350–360. doi: 10.1016/j.ydbio.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Ryan AF. The cell cycle and the development and regeneration of hair cells. Curr Top Dev Biol. 2003;57:449–466. doi: 10.1016/s0070-2153(03)57014-4. [DOI] [PubMed] [Google Scholar]
- Ruben RJ. Development of the inner ear of the mouse: a radioautographic study of terminal mitoses. Acta Otolaryngol Suppl. 1967;220:1–44. [PubMed] [Google Scholar]
- Sage C, Huang M, Karimi K, Gutierrez G, Vollrath MA, Zhang DS, Garcia-Anoveros J, Hinds PW, Corwin JT, Corey DP, Chen ZY. Proliferation of functional hair cells in vivo in the absence of the retinoblastoma protein. Science. 2005;307:1114–1118. doi: 10.1126/science.1106642. [DOI] [PubMed] [Google Scholar]
- Sellers WR, Novitch BG, Miyake S, Heith A, Otterson GA, Kaye FJ, Lassar AB, Kaelin WG., Jr Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 1998;12:95–106. doi: 10.1101/gad.12.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nature Rev. 2004;5:45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- Takahashi C, Bronson RT, Socolovsky M, Contreras B, Lee KY, Jacks T, Noda M, Kucherlapati R, Ewen ME. Rb and Nras function together to control differentiation in the mouse. Mol Cell Biol. 2003;23:5256–5268. doi: 10.1128/MCB.23.15.5256-5268.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell. 1998;3:293–304. doi: 10.1016/s1097-2765(00)80274-9. [DOI] [PubMed] [Google Scholar]
- Van Hooser A, Goodrich DW, Allis CD, Brinkley BR, Mancini MA. Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. J Cell Sci. 1998;111:3497–3506. doi: 10.1242/jcs.111.23.3497. [DOI] [PubMed] [Google Scholar]
- Vidal A, Koff A. Cell-cycle inhibitors: three families united by a common cause. Gene. 2000;247:1–15. doi: 10.1016/s0378-1119(00)00092-5. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Wersäll, J. and Bagger-Sjöbäck, D. (1974). Morphology of the vestibular sense organ. In: Handbook of Sensory Physiology. Vestibular System. Basic Mechanisms (ed. H. H. Kornhuber), pp. 123–170. New York: Springer.
- Wilkinson, D. G and Green, J. (1991). In situ hybridization and the threedimensional construction of serial sections. In: Postimplantation Mammalian Embryos (ed.. A. J. Copp and D. L. Cockroft), pp. 155–171. Oxford, UK: IRL Press.
- Woods C, Montcouquiol M, Kelley MW. Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat Neurosci. 2004;7:1310–1318. doi: 10.1038/nn1349. [DOI] [PubMed] [Google Scholar]
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003;421:942–947. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 1996;10:3051–3064. doi: 10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]
- Zhang J, Gray J, Wu L, Leone G, Rowan S, Cepko CL, Zhu X, Craft CM, Dyer MA. Rb regulates proliferation and rod photoreceptor development in the mouse retina. Nat Genet. 2004;36:351–360. doi: 10.1038/ng1318. [DOI] [PubMed] [Google Scholar]
- Zheng JL, Gao WO. Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat Neurosci. 2000;3:580–586. doi: 10.1038/75753. [DOI] [PubMed] [Google Scholar]
- Zheng L, Sekerkova G, Vranich K, Tilney LG, Mugnaini E, Bartles JR. The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell. 2000;102:377–385. doi: 10.1016/s0092-8674(00)00042-8. [DOI] [PMC free article] [PubMed] [Google Scholar]