

Abstract
In spite of preclinical efficacy and recent randomized, controlled studies with adenoviral vectors expressing herpes simplex virus-1 thymidine kinase (HSV1-TK) showing statistically significant increases in survival, most clinical trials using single therapies have failed to provide major therapeutic breakthroughs. Because glioma is a disease with dismal prognosis and rapid progression, it is an attractive target for gene therapy. Preclinical models using microscopic brain tumor models (e.g., ≤0.3 mm3) may not reflect the pathophysiology and progression of large human tumors. To overcome some of these limitations, we developed a syngeneic large brain tumor model. In this model, administration of single therapeutic modalities, either conditional cytotoxicity or immunostimulation, fail. However, when various immunostimulatory therapies were delivered in combination with conditional cytotoxicity (HSV1-TK), only the combined delivery of fms-like tyrosine kinase ligand (Flt3L) and HSV1-TK significantly prolonged the survival of large tumor-bearing animals (≥80%; P ≤ 0.005). When either macrophages or CD4+ cells were depleted before administration of viral therapy, TK + Flt3L therapy failed to prolong survival. Meanwhile, depletion of CD8+ cells or natural killer cells did not affect TK + Flt3L efficacy. Spinal cord of animals surviving 6 months after TK + Flt3L were evaluated for the presence of autoimmune lesions. Whereas macrophages were present within the corticospinal tract and low levels of T-cell infiltration were detected, these effects are not indicative of an overt autoimmune disorder. We propose that combined Flt3L and HSV1-TK adenoviral-mediated gene therapy may provide an effective antiglioma treatment with increased efficacy in clinical trials of glioma.
Introduction
Glioblastoma is the most common type of brain tumor in adults and accounts for 25% of all brain tumors diagnosed (1, 2). Unfortunately, glioblastoma is also an extremely malignant tumor with a mean survival of ~12 months postdiagnosis. Aggressive surgical resection can be combined with either chemotherapy or radiotherapy to reduce tumor burden (3); however, glioblastoma invariably recurs within 6 to 12 months. Consequently, novel therapies are urgently needed to improve the prognosis of glioblastoma patients (3).
Using a syngeneic (CNS-1 cells) intracranial glioma model in Lewis rats, we have shown that in spite of powerful antiglioma effects, RAdTK, which carries the herpes simplex virus-1 thymidine kinase (HSV1-TK) transgene combined with systemic ganciclovir, also elicits severe adverse effects in long-term survivors (4). Using syngeneic CNS-1 cells in Lewis rats, we previously evaluated adenoviral gene therapies in microscopic glioma models. Intratumoral injection of RAdTK or RAdFlt3L (an adenovirus expressing the human soluble Flt3L) were effective in causing tumor regression when used as single therapies in the microscopic brain tumor model (4, 5). Other experimental gene therapies include the expression of tumor suppressors, oncogene antagonists, angiogenesis inhibitors, immunostimulatory approaches, and more recently the use of replication-competent viral vectors and stem cells (6–9). These approaches have only been tested in microscopic brain tumor models (tumor size ≤0.3 mm3 at time of treatment) in preclinical studies; in the clinical setting, most experimental approaches have generally only provided marginally significant results (7, 9–11). In preclinical studies, therapies are usually administered when the tumor mass is very small and are efficient at inhibiting tumor progression. However, microscopic tumor models may not mimic the human disease state where well established, infiltrating tumor masses are encountered. Therefore, the experimental availability of a syngeneic large glioma tumor model in an immunocompetent animal is critical to assess the efficacy of adenoviral gene therapy approaches.
In the large model, RAdFlt3L, RAdCD40L, and RAdIL-12 were ineffective when used as single therapies. In contrast, coadministration of RAdFlt3L with RAdTK in this large model rescued 80% of animals. All other single or combined treatments tested did not significantly improve long-term survival of rats harboring large intracranial tumors. Immunosuppression using cyclosporine A reduced survival to levels attained by the treatment of RAdTK alone, suggesting a role for the immune system in the tumor regression process. Depletion of either macrophages or CD4+ cells before adenoviral treatment likewise reduced survival, whereas depletion of CD8+ or natural killer (NK) cells did not affect treatment efficacy. In summary, combined immunostimulatory and conditional cytotoxic gene therapy, using Flt3L in combination with TK, will provide a clinically relevant platform to further develop this experimental treatment for clinical trials for glioblastoma.
Materials and Methods
Adenoviral Vectors
RAdFlt3-L, expressing the human soluble Fms-like tyrosine kinase ligand; RAd35, expressing β-galactosidase; RAd128, expressing HSV1-TK; RAdCD40L, and RAdIL12: production and characterization
RAds used in this study are first-generation, replication-defective recombinant adenovirus type 5 vectors expressing the transgenes under the transcriptional control of the human cytomegalovirus intermediate early promoter within the E1 region. RAd35, an adenovirus encoding LacZ under the control of the hCMV promoter, was described by Wilkinson and Akrigg (12) and has been used previously by us (13); herein, it is called RAd β-gal. RAd128 has also been previously described and is indicated as RAd TK throughout (14). RAd0 carries no transgene and was used in our studies to balance total viral load between experimental conditions. RAds expressing CD40L and IL-12 have been described in detail elsewhere (15–17).
RAdFlt3L (RAdFlt3L) was generated by cloning the hsFlt3L cDNA (provided by Immunex, Seattle, WA) into the unique BamHI cloning site of the pAL119 shuttle vector. The shuttle vector was then cotransfected with the E1-deleted adenoviral vector plasmid PJM17 (Microbix Biosystems, Toronto, Ontario, Canada) into the HEK 293 cell line. The presence of the transgenes within the RAds was tested by restriction analysis of the viral DNA and by analysis of protein expression using immunocytochemistry (5). The methods for adenoviral generation, purification, characterization, large-scale production, and viral vector titration have been previously described (18, 19). Briefly, titrations were carried out in triplicate and in parallel for all viruses by end point dilution, cytopathic effect assay, with centrifugation of infected 96-well plates as described in detail by Nyberg-Hoffman and Aguilar-Cordova (20). All viral preparations were screened for the presence of replication competent adenovirus (21) and for lipopolysaccharide (LPS) contamination, using the Limulus amebocyte gel clot assay (Biowhittaker, Rockland, ME; ref. 22). Virus preparations used were free from replication-competent adenovirus and LPS contamination. All relevant adenoviral methods and quality control procedures are previously described in detail (18). Viruses were diluted in sterile saline for intracranial injection.
CNS-1 cells infection for Flt3L expression
CNS-1 cells (1 × 106) were mock infected or virally infected with RAdFlt3L, RAdTK, or RAdFlt3L + RAdTK at a multiplicity of infection (MOI) of 250 for 48 hours. Total viral load in single infections was balanced with RAd0. Supernatants were cleared of cellular debris and stored at −20°C until use in human Flt3L ELISA or β-galactosidase e nzymatic activity assay. Cell lysates were harvested by subjecting cells to three freeze-thaw cycles. Lysates were stored at −20°C until used in immunoblot, Flt3L ELISA, and β-galactosidase enzyme activity assays.
ELISA for hsFlt3L
ELISA for human Flt3L (R&D Systems, Minneapolis, MN) was conducted exactly as described by the manufacturer using cell lysates and supernatants collected from CNS-1 cells infected (MOI 250, 48 hours) above.
β-galactosidase enzymatic activity assay
β-galactosidase activity was measured b y conversion of o-nitrophenyl-β-d-galactopyranoside in 10 mmol/L MgCl2/0.45 mol/L 2-mercaptoethanol at 37°C. The enzymatic reaction was stopped with 1 mol/L Na2CO3. Standard curves were generated using bovine serum albumin (protein standard) or o-nitrophenol (nitrophenol standard). β-galactosidase enzyme activity was calculated using the following formula: enzymatic activity/min = [o-nitrophenol] / (time × [protein]).
Western blots
Thirty micrograms of cell lysate were electrophoreically separated on a 12% SDS-PAGE before transfer to polyvinylidene difluoride membranes (Amersham Biosciences, Piscataway, NJ). Membranes were blocked using 5% nonfat dry milk in PBST (0.1% Tween 20). Primary antibodies, generated in our laboratory, to β-galactosidase (1:2,000), TK (1:1,000), and hsFlt3L (1:1,000) were incubated overnight at 4°C. The anti-hsFlt3L antibody was generated by injection of an antigenic peptide (CETVFHRVSQDGLDL) into New Zealand white rabbits (New England Peptides, Gardner, MA). The antigenic peptide was coupled to keyhole limpet hemocyanin via the cysteine residue. Rabbits were prebled and immunized 14 and 28 days later with antibodies collected on days 3 5 and 42. Antibody specificity was verified by ELISA to the antigenic peptide and Western blotting to the purified Flt3L protein (results not shown). All primary antibodies were detected with goat anti-rabbit horseradish peroxidase secondary antibody (DAKO, Glostrup, Denmark). Secondary antibody visualization was detected with ECL (Amersham Biosciences).
Intracranial Glioma Models
Development of large brain tumor model
Animals treated on day 3 after CNS-1 cell implantation a re referred to as having a small tumor, whereas animals treated on day 1 0 after CNS-1 cell implantation are referred to as having a large tumor (see Fig. 2 for reference sizes). To develop a large glioma tumor model, male Lewis rats (220–250 g) were unilaterally injected into the striatum (+1 mm bregma, +3 mm lateral, and −4 mm from the dura) with 5 × 103 CNS-1 cells. Animals were sacrificed at 3, 6, and 10 days post tumor implantation (n = 10). Forty-millimeter-thick serial brain sections were cut using a vibratome. The section in which the tumor filled the largest area of the striatum was used for the calculation of tumor size. To determine the size of the tumors at each point, the following equation, which determines the volume of an ovoid, was used: 4 / 3 (πabc), where a is the shortest radius of cross-sectional face of the tumor, b is the largest radius of the cross-sectional face of the tumor, and c is the total thickness of the tumor in millimeter.
Figure 2.
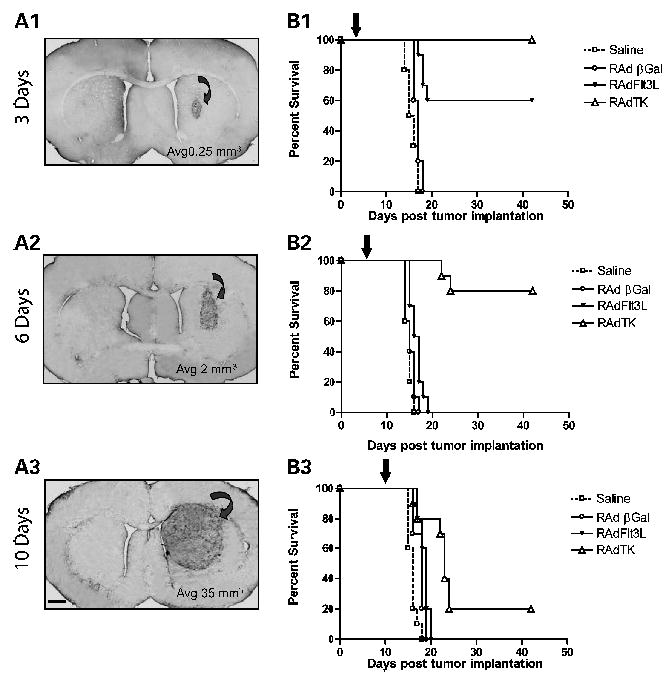
Single therapy with RAdTK improves survival in rats with microscopic brain tumors but is ineffective against large brain tumors. A, progressively increasing tumor sizes and volumes at 3 (A1), 6 (A2), and 10 (A3) days postimplantation of 5,000 CNS-1 cells into the brains of syngeneic Lewis rats. Tumor infiltration into the striatum was visualized using ED1, a marker of activated macrophages and microglial cells. Tumor volume was calculated to be 140 times greater at day 10 than at day 3. B, assessment of the survival efficiency of 8 × 107 plaque-forming units of either RAdTK (followed by 7 days of systemic GCV treatment) in rats bearing tumors of different sizes. Treatments were delivered to growing CNS-1 tumors either at 3 (B1), 6 (B2), or 10 (B3) days following tumor implantation (black arrow at the top of each of the graphs). Controls animals were injected with saline or RAdβgal. Notice the drop in effectiveness of RAdTK as it is injected into progressively larger tumors. Bar, for (A1) to (A3), shown in (A3), 150 μm.
To test survival of single therapies, doses of 8 × 107 infectious units (i.u.) of either RAdTK or RAdFlt3L were injected intratumorally 3, 6, and 10 days post CNS-1 cell implantation to determine the time point post tumor implantation a t which single therapies would fail (i.e., animals succumb within 30 days). O n the day after the injection of the virus, 25 mg/kg ganciclovir (Cytovene, Roche P roducts, Welwyn G arden City, United Kingdom) was injected i.p. twice daily for 7 days into all experimental animals. Control animals received saline or Rad β-gal. Animals were monitored daily and terminally perfused-fixed a t the first signs of moribund behavior. Brains were removed for histologic examination.
Gene therapy in the treatment of the large glioma tumor model
Groups of male Lewis rats (n = 10) were implanted with CNS-1 cells as above, and 10 days later viral vectors were injected intratumorally with a total viral load of 2 × 107 i.u. In injections of individual virus, 1 × 107 i.u. was balanced with an additional 1 × 107 i.u. RAd0, an adenoviral vector expressing no transgene. Beginning 24 hours after injection of the RAds, 25 mg/kg ganciclovir (Cytovene, Roche Products) was injected i.p. twice daily for 7 days. All animals including controls received ganciclovir. Animals were monitored daily and terminally perfuse-fixed at the first signs of moribund behavior.
Immune cell depletion in a large glioma model
Hybridoma cell lines for CD8+ cell depletion [OX-8, European Collection of Animal Cell Cultures (ECACC), Porton Village, United Kingdom] and CD4+ cell depletion (OX-34, ECACC), NK cell depletion (NK 3.2.3, provided by William Chambers, University of Pittsburgh Medical Center, Pittsburgh, PA) were produced by Bioexpress Cell Culture Services (West Lebanon, NH). Depletion of macrophages was conducted with liposome encapsulated clodronate (Cl2MDP; ref. 23). Clodronate was a gift of Roche Diagnostics GmbH (Mannheim, Germany). It was then encapsulated in liposomes as previously described (23). One milliliter of suspensions of liposomes containing 6 mg clodronate was injected per 100 mg animal weight. Groups of Lewis rats (n = 6 ) were unilaterally injected with 5,000 CNS-1 cells into striatum. Nine days later, 1 mg of OX-8, OX-34, or normal mouse IgG; 0.5 mg of NK 3.2.3, or 2 mL of clodronate or PBS-filled lipsomes were injected i.p. into tumor-bearing Lewis rats. Animals surviving 2 weeks after initial immune cell depletion were injected i.p. one additional time. On day 10 after CNS-1 implantation, animals received intratumoral injection of either saline or RAdTK + RAdFlt3L. GCV was administered twice daily for 7 days. Animals were sacrificed by terminal perfusion at the first signs of moribund behavior. To ensure that depleting antibodies were thoroughly and accurately depleting their respective immune cell types, naïve animals were injected as above (n = 4). Spleens were collected 7 days later and analyzed by flow cytometry.
Splenocytes were harvested and RBC lysed with ACK. Cells were counted and labeled with antibodies for analysis by flow cytometry. Cells (1 × 106) were resuspended in 1 mL of cell surface staining buffer (0.1 mol/L PBS, without Ca2+, Mg2+, with 1% fetal bovine serum, 0.1% sodium azide). Cells were washed and resuspended in 100 μL cell surface staining buffer containing the combinations of antibodies described below before incubation in the dark for 30 minutes at 4°C. Samples were washed in cell surface staining buffer and analyzed by flow cytometry. CD4 and CD8 cells were detected by labeling with CD3-FITC, CD4-PE-Cy5, CD8-PE. NK cells were detected by labeling with CD3-FITC and CD161a-PE. Macrophages were detected by labeling with CD45-PE, CD4-PE-cy5, CD11b-FITC (all antibodies from BD PharMingen, San Jose, CA).
Cyclosporine A treatment
In immunosuppression experiments, animals were immunosuppressed beginning 7 days after CNS-1 cell implantation. Animals received 10 mg/kg of cyclosporine A (Neoral, Sandoz)/300 μL olive oil given p.o. by gavage twice daily until the termination of the experiment. Control animals were given 300 μL olive oil. Whole blood cyclosporine A concentrations were measured using a monoclonal RIA kit, following the instructions provided by the manufacturer (Incstar, Stillwater, MN).
All animal studies were conducted in accordance with the institutional guidelines and oversight of the Department of Comparative Medicine, Cedars-Sinai Medical Center, Los Angeles, CA.
Brain Immunohistochemistry
Forty-micrometer-thick coronal sections were cut through the striatum using a vibratome. Free-floating immunohistochemistry was done to detect inflammatory and immune cell markers. Endogenous peroxidase was inactivated with 0.3% hydrogen peroxide, and sections were blocked with 10% horse serum (Life Technologies, Carlsbad, CA) before incubating overnight with primary antibody. Antibodies ED1 (1:1,000), CD8 (1:1,000), CD45R (1:100), CD161a (1:500), MHC I (1:1,000), and MHC II (1:1,000) were obtained from Serotec (Raleigh, NC). Vimentin (1:1,000; Sigma, St. Louis, MO) was used to label CNS-1 cells. Secondary antibodies were biotinylated antimouse (1:1,000; The Jackson Laboratory, Bar Harbor, ME) were detected using the Vectastain Elite ABC horseradish peroxidase method (Vector Laboratories, Burlingame, CA). After developing with diaminobenzidine and glucose oxidase, sections were mounted on gelatinized glass slides and dehydrated through graded ethanol solutions. Tissues were analyzed and photographed using a Zeiss Axioplan microscope.
Spinal Cord Immunocytochemistry
Animals were perfused with 4% paraformaldehyde in 0.1 mol/L phosphate buffer. The spinal cord was dissected and 12 to 14 blocks from all levels of the cord were embedded in paraffin. Five-micrometer sections were stained with H&E, Luxol fast blue myelin stain, and Bielschowsy’s silver impregnation for axons. Adjacent serial sections were s ubjected to immunocytochemistry for T-cells (W3/13; Seralab, Loughborough, United Kingdom) and macrophages (ED1; Serotec). Binding of primary antibodies was visualized using biotinylated anti-mouse immunoglobulin (Amersham, Piscataway, NJ) and avidin peroxidase (Sigma). Peroxidase reaction was developed with diaminobenzidine (Fluka, St. Louis, MO).
Statistical Analysis
In vivo experiments were conducted twice to thrice with an n = 6 or n = 1 0 a s detailed above. Survival data were analyzed by Kaplan-Meier estimator analysis and compared using the log-rank tests. Analyses were done using SPSS (SPSS, Inc., Chicago, IL) and PRISM software (GraphPad Software, Inc., San Diego, CA).
Results
Coexpression of hsFlt3L and HSV1-TK within RAd-Infected CNS-1 Cells
First-generation, replication-deficient adenoviral vectors Radβgal, RAdTK, and RAdFlt3L (Fig. 1A) were used to show that coinfection with two RAds would result in the efficient expression of both proteins. CNS-1 cells were infected in vitro with either RAdTK or RAdFlt3L alone and in combination, using MOI 250. In all experiments herein, total viral particles were balanced with RAd0, which expresses no transgene such that all cultures received identical levels of infectious units. Immunoblot analysis showed that β-galactosidase was only present in the cell extracts of cells infected with RAdβgal, TK in cell extracts from cells infected with RAdTK and coinfected with RAdTK plus RAdFlt3L, and Flt3L was only detected in cell extracts from cells infected with RAdFlt3L and coinfected with RAdTK plus RAdFlt3L (Fig. 1B). Whereas β-galactosidase and TK are intracellular enzymes, Flt3L is a secreted protein. To insure Flt3L was secreted efficiently, w e tested for the presence of Flt3L in supernatants from infected and coinfected CNS-1 cells using a human Flt3L ELISA. Only supernatants of RAdFlt3L-infected and RAdTK plus RAdFlt3L–coinfected CNS-1 cells showed secretion of Flt3L protein (2,640.5 and 2,537.8 pg/mL, respectively; Fig. 1C). Intracellular expression of Flt3L was similarly restricted (641.5 and 320.8 pg/mL total protein; Fig. 1D). Whereas β-galactosidase enzymatic activity was detected in lysates of cells infected with RAdβgal, no β-galactosidase activity was detected in supernatants from RAdβgal-infected CNS-1 cells, consistent with expectations for an intracellular protein (Fig. 1C). This also indicated that no cellular lysis as a result of RAd infection occurred. These results indicate that coinfection of CNS-1 glioma cells with two different RAds in vitro elicits the efficient biosynthesis of both transgenes (i.e., TK and Flt3L) within the coinfected cells. Expression of one transgene did not block expression of the other. However, upon coinfection, both transgenes were expressed at lower levels. TK a nd Flt3L expression is d riven by t he same hCMV promoter. Thus, competition for the transcriptional and translational machinery in coinfected cells could result in lower expression of each individual transgene in vitro in the coinfected (RAdTK + RAdFlt3L) CNS-1 cells.
Figure 1.
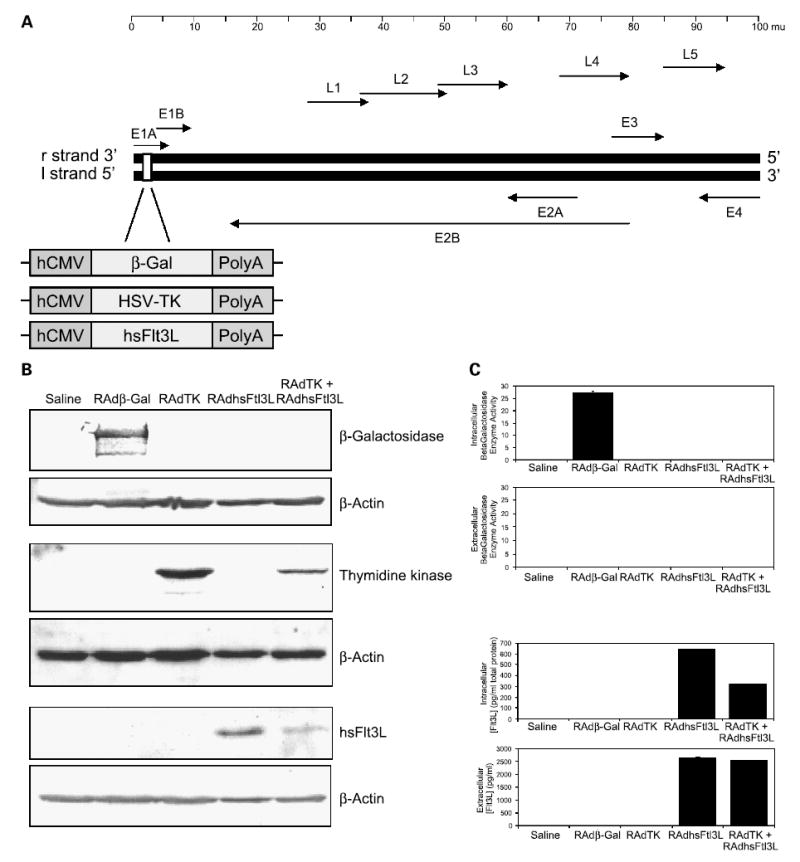
Generation and functional characterization of RAdFlt3L. A, a schematic diagram depicting the organization of the adenovirus type 5 genome (Ad5). Expression cassettes with Escherichia coliLacZ gene (β-Gal), HSV1-TK cDNA, or human soluble Flt3L cDNA (Flt3L) under the control of the hCMV promoter were created. These expression vectors were subsequently inserted into the E1 region of the adenovirus genome as shown, generating the recombinant adenoviral vectors RAdβgal, RAdTK, and RAdFlt3L. B, Western blot to detect RAd-generated protein from of CNS-1 cells supernatants and lysates that were mock infected, infected with RAdβgal, RAdTK, RAdFlt3L, or RAdTK + RAdFlt3L. C, β-galactosidase enzymatic activity assay from cell lysates and supernatants infected with viruses described in (B). Only intracellular protein extracts from RAdβgal-infected CNS-1 cells displayed significant β-galactosidase activity when compared with control samples (27.25 μg o-nitrophenol/μg protein/min). No secreted β-galactosidase activity was evident in any samples. D, ELISA to detect intracellular and secreted Flt3L expressed from cells infected with the viruses described in (B). Intracellular human Flt3L expression was only observed in RAdFlt3L (641.5 pg/mg total protein) and RAdTK + RAdFlt3L (320.8 pg/mg total protein)–infected CNS-1 cells. In addition, secreted human Flt3L was only detected in the media of RAdFlt3L and RAdTK + RAdFlt3L–infected CNS-1 cells at concentrations of 2,640.5 and 2,537.8 pg/mL, respectively.
Development of a Large Syngeneic Glioma Model where Single Gene Therapies Fail
In previous work, we had implemented successful gene therapy strategies using a microscopic syngeneic CNS-1 glioma modeL ( 4, 5). To d evelop a large model of g lioma, implanted 5,000 CNS-1 cells and injected RAds 3 , 6 , and 10 days later (i.e., at times of progressively larger tumor mass). The tumor volume was found to increase 140-fold between days 3 and 10, growing from 0.25 to 35 mm3 (Fig. 2A1-A3). We sought to determine whether tumor volume significantly affects survival when treated with recombinant adenoviral vectors using RAdTK as our positive therapeutic control treatment. Animals were treated with RAdTK, RAdβgal, or saline 3, 6, and 10 postimplantation of CNS-1 cells. In agreement with previous studies undertaken by our group and others (4, 5, 24–28), RAdTK treatment was 100% effective when delivered 3 days post tumor implantation, 80% effective when injected 6 days post tumor implantation, and only 20% effective when given 10 days post tumor implantation (Fig. 2B). Our results highlight the importance of tumor size on the efficacy of various adenoviral-mediated glioma therapies. Furthermore, this model reproduces more closely the human disease condition where tumor size at the time of treatment determines the treatment outcome.
Combined Treatment with RAdFlt3L + RAdTK Significantly Improves the Long-term Survival of Rats with Large Brain Tumors
In view of the results shown in Fig. 2, we sought to develop a combined gene therapy strategy aimed at promoting long-term survival and tumor regression in animals bearing large intracranial gliomas. Dendritic cells are absent from the naïve brain, but are detected within the brain under conditions of inflammation (29–32). The lack of dendritic cells from the brain, together with the high levels of immunosuppressive transforming growth factor-β expressed by glioblastomas (33), could help explain the difficulties in stimulating antiglioma immune responses. We hypothesized that in the large model, codelivery of RAdTK with a powerful immune stimulant could provide the necessary signals to trigger an effective antitumor immune response. We tested codelivery with three potential candidates to stimulate immune responses (i.e., Flt3L, CD40L, and IL-12). Codelivery of the conditional cytotoxic gene therapy, TK with Flt3L, would increase the number of dendritic cells (34), whereas CD40L would activate dendritic cells (35). IL-12 would stimulate the immune responsiveness of various immune cell types (36, 37). We hypothesized that TK-mediated killing of tumor cells would provide antigenic tumor epitopes to Flt3L-stimulated dendritic cells in situ that could eventually stimulate an antitumor immune response. This in turn would result in tumor elimination and prolonged survival.
Our results show that the combined treatment with RAdTK + RAdFlt3L rescued a highly significant percentage of animals from tumor-induced death (Fig. 3A). Even as late as 6 months after tumor implantation, >70% of animals remained alive, compared with animals in all control groups (i.e., treated with saline, RAd0, RAdFlt3L, RAdIL-12, or RAdCD40L), which were all dead by day 25 post tumor implantation (Fig. 3A-C). RAdTK alone only protected 20% of animals, and this protection was not increased by the addition of RAdIL12 (Fig. 3C). Although treatment with RAdTK plus RAdCD40L rescued 40% of the animals, this value failed to reach statistical significance compared with animals treated with TK alone (Fig. 3B).
Figure 3.
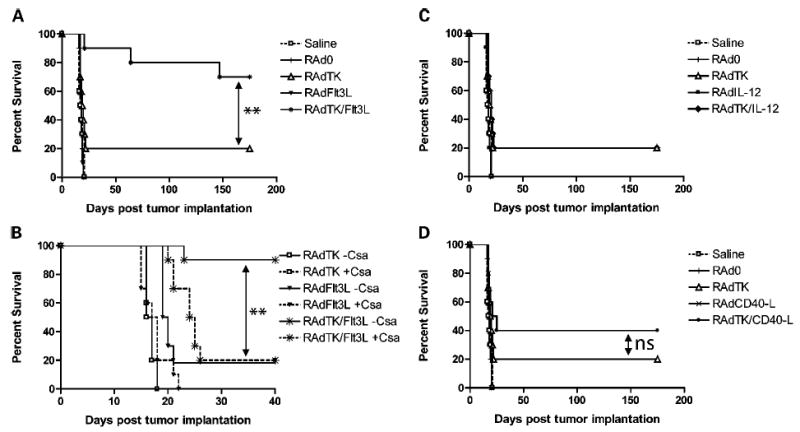
Combined treatment with RAdTK and RAdFlt3L significantly enhances survival in a large brain tumor model and cyclosporine A eliminates the efficiency of combined treatment. A, rats with large tumors were treated 10 days after CNS-1 cell implantation with either saline (□), or 1 × 107 i.u. RAd0 (|), RAdTK (▵), RAdFlt3L (▾), or RAd TK/Flt3L (*). Whereas neither saline, RAd0, or RadFlt3L alter survival with all animals dying ~25 days after tumor implantation, RAd TK prolonged survival in 20% of animals. RAdTK + Flt3L prolonged survival in 70% of animals. B, rats with large tumors were treated 10 days after CNS-1 cell implantation with either saline (□) or 1 × 107 i.u. RAd0 (|), RAdTK (▵), RAdCD40L (×), or RAd TK/RAdCD40L (•). Treatment with RAdTK resulted in 20% survival, whereas combined treatment with RAdCD40L resulted in 40% survival. C, rats with large tumors were treated 10 days after CNS-1 cell implantation with either saline (□) or 1 × 107 i.u. RAd0 (|), RAdTK (▵), RAdIL-12 (▪), or RAd TK + RAdIL-12 (♦). Treatment with either RAdTK or RAdTK + RAdIL-12 resulted in 20% survival. Statistical analysis for (A) to (C) was done as indicated in Materials and Methods. Survival of animals treated with RAdTK + RAdFlt3L was statistically significantly increased compared with animals treated with either RAdTK or RAdTK + RAdIL-12 (**, P < 0.005); although the survival of animals treated with RAdTK + RAdCD40L seemed increased compared with animals treated with either RAdTK or RAdTK + RAdIL-12, this difference was not statistically significant. No statistically significant differences were found between RAdTK and RAdTK/RAdIL-12, or between RAd0, RAdFlt3L, RAdIL-12, or RAdCD40L treatments. D, survival of animals treated with the immunosuppressant cyclosporine A (solid lines are without cyclosporine A and dashed lines are with cyclosporine A in each treatment condition). Animals were immunosuppressed with cyclosporine A beginning day 7 after tumor implantation and continuing daily throughout the experiment. Animals treated with RAdTK + Flt3L displayed increased survival only when not treated with cyclosporine A. Treatment of animals injected in the brain with RAdTK + Flt3L with cyclosporine A reduced the efficiency of the combined treatment to that of RAdTK alone. Treatment of animals with RAdTK and cyclosporine A reduced the efficiency of RAdTK further, potentially suggesting that the effectiveness of RAdTK is due to immunostimulation. Animals treated with RAdhsFlt3L, with or without cyclosporine A, had no effect on animal survival. Statistical analysis was done as described in Materials and Methods. Animals treated with RAdTK + Flt3L − cyclosporine A showed statistically significant increased survival compared with those injected with RAdTK + RAdFlt3L + cyclosporine A, or those treated only with RAdTK, indicated by arrows (**, P < 0.005). None of the other survival curves were statistically significantly different.
Immunosuppression blocks RAdTK + Flt3L–Mediated tumor regression
To determine if combined gene therapy with RAdTK + RAdFlt3L was acting through an immune-mediated mechanism, we repeated the survival experiment but a group of animals was treated with the powerful immunosuppressant, cyclosporine A, twice daily (38). We detected circulating levels of cyclosporine A >3,000 ng/mL in all experimental animals treated with cyclosporine A, whereas cyclosporine A levels were not detectable in vehicle-treated control animals (results not shown). Cyclosporine A treatment did not have a significant effect on the life span of any of the control animals, but completely abolished the increased survival provided by TK combined with Flt3L. At 40 days post tumor implantation and 30 days post gene therapy, survival of animals treated with both TK and Flt3L and cyclosporine A was reduced to the level of survival of animals treated with TK alone (Fig. 3D). Thus, the beneficial therapeutic effects of TK + Flt3L in the large glioma model was completely abolished by immunosuppression by cyclosporine A (Fig. 3D).
Brain Neuropathology of Long-term Survivors after Gene Therapy with RAdTK + RAdFlt3L
Rats that survived long-term after RAdTK + RAdFlt3L treatment were subjected to detailed neuropathologic analysis to assess the potential existence of inflammatory and neuropathologic side effects. All animals displayed enlarged ipsilateral ventricles indicative of striatial tissue loss as well as scar tissue at the site of tumor implantation and RAd injection (Fig. 4A, full brain image). High levels of ED1, MHC I, and MHC II staining were observed in the ipsilateral hemisphere near the injection site (Fig. 4A). CNS-1 cells express high levels of the intermediate filament vimentin. Whereas vimentin staining consistent with tumor cells was not detected (Fig. 4A), vimentin staining was up-regulated on the ipsilateral hemisphere consistent with reactive gliosis. Low but above background levels of CD8-, CD45R-, and CD161a-expressing cells were also detected near the site of tumor location and virus injection (Fig. 4B).
Figure 4.
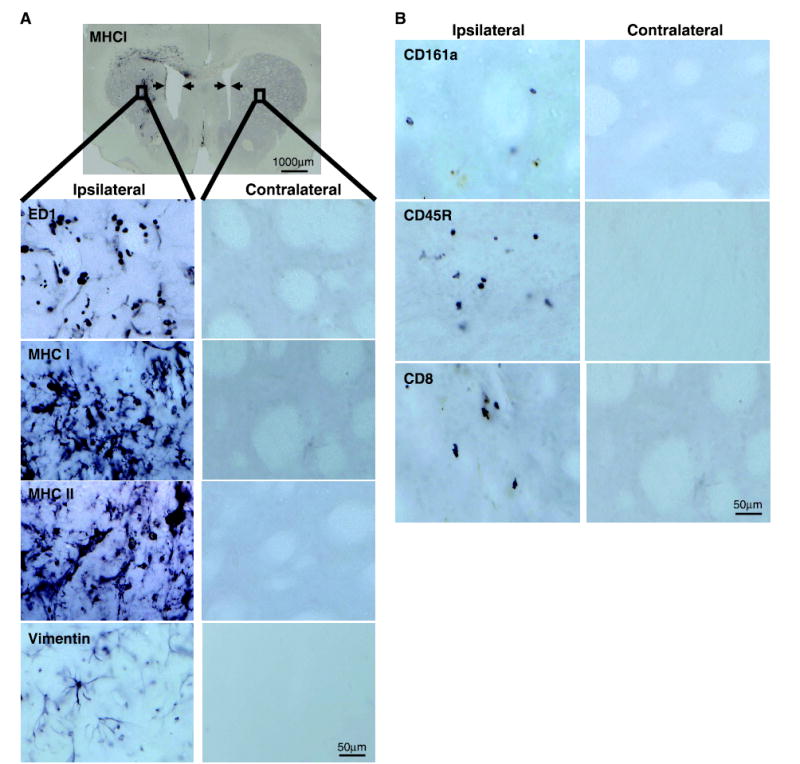
Inflammatory responses observed in the CNS of rats in which RAdTK + RAdFlt3L resulted in tumor regression. Male Lewis rats bearing large tumors were injected with RAdTK+RAdFlt3L. Animals still surviving at 2 months were sacrificed and brain sections were stained. A, whereas no residual tumor was observed, animals show enlarged ventricles (indicated by arrows in MHC I staining of full brain) and immune marker up-regulation. Ipsilateral and contralateral sections of striatum are shown. Evidence of immune cell infiltration at and around the injection site is observed in increased levels of ED1-, MHC I–, MHC II–, and vimentin-positive cells. B, CD161a-, CD45R-, and CD8-positive cells were observed near the injection site at low levels. Low-magnification images were taken at ×1; bar, 1,000 μm. Higher-magnification images were taken at ×40 near the injection site as indicated by a black box in the low-magnification field; bars, 50 μm.
Depletion of Macrophages or CD4+ Cells but not CD8+ or Natural Killer Cells Eliminates Therapeutic Efficacy of RAdTK + RAdFlt3L Treatment
To evaluate the extent and specificity of the depleting antibody treatments, naïve, non–tumor-bearing animals were injected once with 1 mg OX-8, OX-34, or normal mouse IgG; 0.5 mg NK3.2.3 or 2 mL clodronate or PBS-filled liposomes. Seven days later, spleens were harvested and evaluated for cell depletion. Each treatment responded as expected, specifically eliminating its target immune cell type (Fig. 5B) either NK (3.2 × 108 ± 1.2 × 108 control, 9.3 × 106 ± 2.7 × 106 NK 3.2.3 depleted), macrophages (7.8 × 108 ± 2.3 × 108control, 2.87 ×108±1.4 ×108clodronate depleted), CD8 (1.6 ×109 109 ± 6 × 108 control, 4.1 × 107 ± 3.3 × 107 OX-8 depleted), or CD4 (6.9 × 108 ±2.4 × 108 control, 4.5 × 107 ±4.5 × 107 OX-34 depleted).
Figure 5.
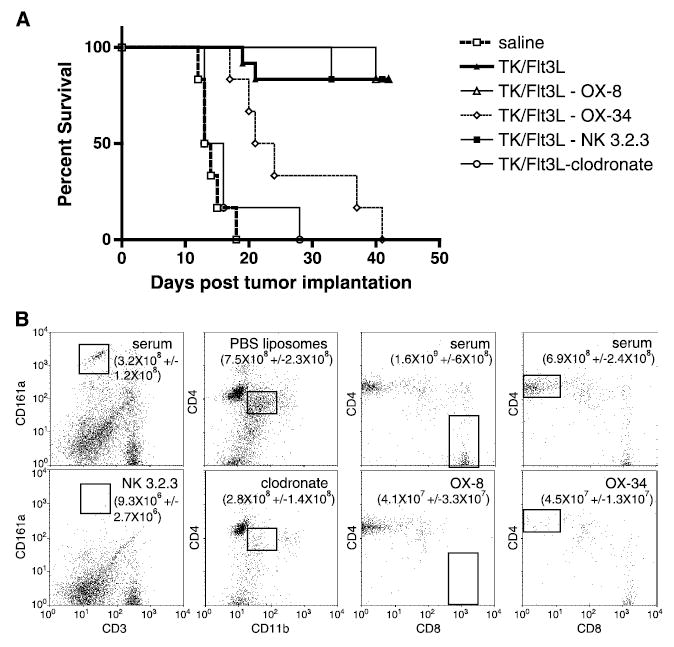
Depletion of specific subsets of immune cell alters therapeutic efficacy of RAdTK + Flt3L in macrophage and CD4+ cell–depleted animals. A, all animals were injected with 5,000 CNS-1 glioma cells. Nine days later, specific subsets of immune cells were depleted by i.p. injection of 1 mg OX-8, OX-34 or normal mouse serum, 0.5 mg of NK3.2.3, or 2 mL clodronate or PBS-filled liposomes (n = 6). Twenty-four hours following immune cell depletion, saline or RAdTK + Flt3L was intratumorally injected. A, Kaplan-Meier survival curve of all depleted animals (serum and PBS liposome, ▴; OX-8, ▵; OX-34, ⋄; NK3.2.3, ▪; and clodronate, ○) and control groups (saline treated, □). B, depletion efficiency and specificity was evaluated by flow cytometry in splenocytes from immune cell depleted or control animals seven days after depletion (n = 4). All samples were gated on lymphocyte populations by FSC and SSC. NK cells were identified as CD3, CD161a+. Macrophages were identified after gating CD45+ lymphocytes as CD4+, CD11b intermediate populations. CD4 cells were identified as CD4+, CD8−, and CD8 cells were identified as CD4−, CD8+ after gating respective populations for CD3+ lymphocytes. Numbers represent total number of a given cell population per spleen ± SE (n = 4).
To deplete cells in large tumor-bearing animals, the latter were injected: OX-8 (to deplete CD8+ cells), OX-34 (to deplete CD4+ cells), NK 3.2.3 (to deplete NK cells), or clodronate (clodronate-filled liposomes; to deplete macrophages). Control animals were injected with normal mouse immunoglobulin or PBS-filled liposomes. Twenty-four hours after depletion, animals were intratumorally injected with either saline or RAdTK + RAdFlt3L. Whereas animals treated only with saline succumbed to brain tumors 13 to 19 days following CNS-1 cell implantation, 70% of RAdTK + RAdFlt3L + normal mouse immunoglobulin or PBS-filled liposomes–treated animals survived 35 days after implantation of CNS-1 cells (Fig. 5). Animals treated with RAdTK + RAdFlt3L and depleted of macrophages succumbed to brain tumors as rapidly as saline-treated animals. CD4+ cell–depleted animals survived longer than macrophage-depleted or saline-treated animals but did not survive long-term. Most CD8+ and NK cell–depleted animals survived as well as control mouse immunoglobulin or PBS liposome–treated animals. Comparison of all survival curves by log rank analysis (GraphPad Prism software) indicates a statistically significant difference for both macrophage and CD4+-depleted animals (P ≤ 0.0001).
Brain and Spinal Cord Autoimmunity Were Not Found in Animals Treated with RAdFlt3L + RAdTK
A possible concern with an immunologic therapy against brain tumors is that the delivery of powerful proinflammatory/immunostimulatory genes might induce systemic anti–central nervous system (CNS) immune responses. Experimental allergic encephalomyelitis (EAE), for example, primarily targets the spinal cord. To determine the presence of such putative adverse side effects, spinal cords of RAdTK + RAdFlt3L–treated animals that had survived for up to 6 months (Fig. 6) were analyzed to determine the presence and distribution of inflammatory infiltrates (T cells and macrophages) in the meninges and within the spinal cords. Inflammatory infiltrates of T cells across all groups were very low (10–30 cells/section), but higher than normal control animals (0–2 T cells per section). In one animal (injected with TK + IL12), we found a few typical perivascular inflammatory infiltrates, similar in quantity to those found in very mild subclinical EAE (this animal had the largest forebrain tumor present; data not shown). No animal displayed overt symptoms of EAE. The distribution of macrophages throughout the spinal cord, and especially throughout the corticospinal tract, indicates a lesion in the pyramidal tract, most likely a lesion to the axon tracts of the internal capsule due to the tumor growth or even a small lesion to the axon fibers of the internal capsule. Even a direct or indirect (compression) lesion to the capsula interna, where the pyramidal tract is located in the forebrain, will produce visible lesions within the corticospinal tract in the spinal cord. This spinal cord inflammation is compatible with ongoing axon tract degeneration, but no evidence of autoimmune spinal cord inflammation and/or oligodendrocyte attack was detected.
Figure 6.
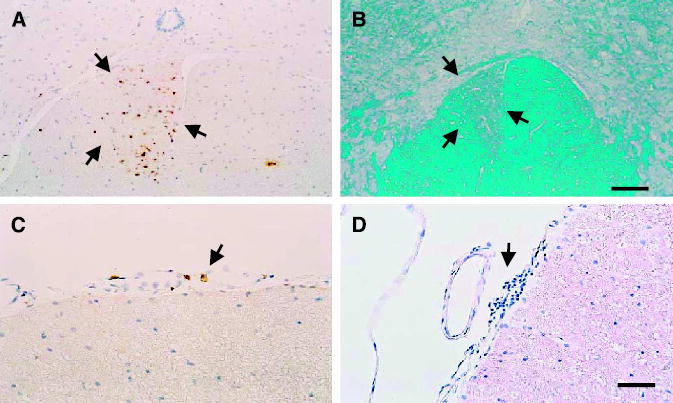
Spinal cord neuropathology of animals surviving a large brain tumor for 6 months. Representative animals are illustrated herein. A and C, animals treated with RAdTK + RAdFlt3L and that survived tumor treatment for 6 months; A, distribution of macrophages (stained with ED1) in the corticospinal tract (outlined by black arrows), compatible with forebrain lesions to the pyramidal tract as caused by the growing tumor, and (C) indicates the distribution of T cells in the meninges of the same group of animals. Although the values of T cells in the meninges of these animals is extremely low, it is nevertheless higher than in animals not receiving any treatment. B, spinal cord of an animal treated with RAdTK and RAdCD40L stained with the Kluwer histochemical stain for myelin. Notice the unilateral area of demyelination in the spinal cord (outlined by black arrows), compatible with a lesion to the descending spinocortical tract, most possibly due to forebrain lesions to the pyramidal tract. D, largest infiltration of the meninges with T cells of any of the examined animals, and that was seen in an animal treated with RAdFlt3L and RAdIL12 (this animal displayed the largest tumor growth and brain inflammation at the time of sacrifice; data not shown). Bar, for all panels, shown in (D), 80 μm.
Discussion
Recombinant adenovirus vectors have shown promise in preclinical, and most recently in randomized, controlled clinical studies (9–11, 24, 39, 40). Complete tumor elimination has not been as forthcoming in clinical studies (9, 10, 24, 39, 40). This highlights shortcomings in the brain tumor models currently used to evaluate the clinical efficacy of adenoviral therapies. To address this issue, we have developed a large syngeneic brain tumor model in immunocompetent rats. This model more faithfully predicts the outcome of gene therapies observed in a clinical setting.
Vaccine immunotherapy for cancer has recently shown great promise with the discovery that dendritic cells play a pivotal role in antigen presentation. The use of dendritic cell–based vaccines is currently ongoing in about 20 phase II and at least 1 phase III clinical trial in the United States, many of which are showing promising results (41–48). In addition, early studies have indicated that the toxicity of dendritic cell vaccination is mild and limited to local reactions at the site of injection (49). As an alternative to dendritic cell vaccination, we sought to attract dendritic cells to the tumor in situ, and then provide antigen epitopes by inducing tumor cell death.
To function effectively as afferent antigen-presenting cells capable of antigen presentation to naïve T cells, dendritic cell precursors must first differentiate into dendritic cells. Flt3L has been reported to promote this differentiation and expansion of dendritic cell precursors in vivo (50–54). Both resting and activated dendritic cells can present antigen to naïve T lymphocytes. However, if resting dendritic cells present antigen to naïve T lymphocytes, the T lymphocytes usually die prematurely in the absence of costimulatory molecules. This could lead to peripheral tolerance to that particular antigen (55). Tumors often masquerade as healthy tissues and cause little or no dendritic cell activation or inflammatory immune responses. Although absent from the naïve brain, infiltration of dendritic cells has been observed under conditions that cause inflammation (29–32, 56). We utilized recombinant adenoviruses with the ability to modulate dendritic cell differentiation (RAdFlt3L), dendritic cell activation (RAdCD40L), or a powerful cytokine that activates TH1 responses (RAdIL-12). Interleukin-12 is a powerful stimulator of cell-mediated antitumor immune responses and has shown promising results in various clinical trials (57, 58). Because RAdTK would be cytotoxic to tumor cells, we hypothesized that tumor killing would provide antigenic tumor epitopes to dendritic cells in situ when coadministered with RAdFlt3L or RAdCD40L. Eventually, this could stimulate an antitumor immune response, tumor reduction or elimination, and increased survival. Coadministration of RAdFlt3L + RAdTK was found to greatly increase the survival of rats in our large brain tumor model (≥70%, P ≤ 0.005). Also, increased survival was noted when we used RAdCD40L in combination with RAdTK, but this did not achieve statistical significance. RAdIL-12 did not increase survival in animals bearing large tumors when codelivered with RAdTK. Cyclosporine A completely negated the improved survival with RAdFlt3L + RAdTK; this supports the putative role played by the immune system in this antitumor response.
We observed elevated reactive gliosis, macrophage/microglial, MHC I, and MHC II staining around the site of injection in long-term survivors injected RAdTK + RAdFlt3L. In addition, CD8, CD45R, and CD161a expression was also observed in these animals, demonstrating that the combination therapy used was capable of inducing significant inflammatory responses at the site of injection. The inhibitory effect of cyclosporine A strongly suggests the presence of an immunocomponent mediating the effect of the combined gene therapies on tumor regression. However, due to the complex mechanism of action of this powerful immunosuppressant, individual lymphocyte subtypes mediating glioma elimination cannot be identified. To advance toward the mechanism of action of our combined gene therapy approach, we treated tumors with the combined gene therapies in the presence of depleting antibodies or clodronate-filled liposomes that would selectively eliminate individual populations of lymphocytes and macrophages. These experiments show that presence of CD4+ T cells and macrophages are necessary for the combined gene therapy with Flt3L and TK to be effective, but that CD8+ T cells and NK cells are dispensable for glioma eradication.
A number of cells in the brain can express MHC II, and thus could present tumor antigens to CD4+ T cells. Activated macrophages and microglia, as well as endothelial cells in the brain, express MHC II, and we have tested that the CNS-1 cells are also able to express MHC II, although at much lower levels than MHC I. Thus, a number of cells within the brain could present tumor antigens to the CD4+ T cells, which upon entry into the brain environment could release IFNγ, as well as other cytokines to stimulate the activity, and especially the phagocytic capacity of recruited macrophages. The killing of tumor cells with TK and ganciclovir would increase local inflammation and allow tumor cells to express signals to the macrophages, eventually leading to their demise. Although CD4+ T cells could be thought of having other roles, e.g., as regulatory T cells, the fact that CD8+ T cells are not necessary for tumor rejection makes this unlikely. Also, in our model, CD4+ T cells could themselves be cytotoxic to the tumors, either through the release of IFNγ or tumor necrosis factor α, or through interactions between FasL and Fas. Although the role of CD4+ T cells in tumor elimination has not be clearly elucidated, similar CD4+ T cell–centered tumor elimination mechanisms have been recently reported in models of myeloma (59), lymphoma (60), and cervical cancer (61).
A concern of all vaccine immunotherapies is the development of autoimmune diseases in response to the vaccine. Although adverse autoimmune reactions have not yet been observed in clinical studies, the use of adenovirus to stimulate an immune response against tumor-specific antigens differs from these therapies. Lewis rats are susceptible to the induction of EAE and thus provide a powerful model to assess long-term inflammatory or autoimmune side effects. Although we observed elevated T-lymphocyte infiltration in the meninges and within the spinal cord of animals treated with RAdFlt3L compared with control animals, the levels of infilt ration were very low and no animal displayed clinical symptoms of EAE.
In conclusion, we have developed a large preclinical model for glioma that reflects more accurately the clinical situation when administering recombinant adenoviral therapies. We have shown that RAdFlt3L strongly promotes survival in rats bearing large brain tumors when coadministered with RAdTK. Further, this therapy does not elicit an appreciable autoinflammatory or autoimmune response in animals. Our data suggests that RAdFlt3L + RAdTK may be a useful adjuvant for increasing the numbers of dendritic cells and enhancing long-term survival and tumor regression in glioblastoma and warrants further development for its implementation in clinical trials.
Acknowledgments
Grant support: NIH/National Institute of Neurological Disorders and Stroke grant 1R01 NS44556.01; National Institute of Diabetes and Digestive and Kidney Diseases grant 1 R O3 TW006273-01 (M.G. Castro); NIH/National Institute of Neurological Disorders and Stroke grants 1 RO1 NS 42893.01, U54 NS045309-01, and 1R21 NS047298-01, and Bram and Elaine Goldsmith Chair in Gene Therapeutics (P.R. Lowenstein); The Linda Tallen and David Paul Kane Annual Fellowship (M.G. Castro and P.R. Lowenstein); and the Board of Governors at Cedars Sinai Medical Center.
We thank S. Melmed, R . Katzman, and D. Meyer for their academic leadership and superb administrative and organizational support.
Footnotes
Note: S. Ali and G. King contributed equally to this work. S. Ali is currently in The Paterson Institute for Cancer Research, Manchester, United Kingdom.
References
- 1.Counsell CE, Grant R. Incidence studies of primary and secondary intracranial tumors: a systematic review of their methodology and results. J Neurooncol. 1998;37:241–50. doi: 10.1023/a:1005861024679. [DOI] [PubMed] [Google Scholar]
- 2.Pobereskin LH, Chadduck JB. Incidence of brain tumours in two English counties: a population based study. J Neurol Neurosurg Psychiatry. 2000;69:464–71. doi: 10.1136/jnnp.69.4.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castro MG, Cowen R, Williamson IK, et al. Current and future strategies for the treatment of malignant brain tumors. Pharmacol Ther. 2003;98:71–108. doi: 10.1016/s0163-7258(03)00014-7. [DOI] [PubMed] [Google Scholar]
- 4.Dewey RA, Morrissey G, Cowsill CM, et al. Chronic brain inflammation and persistent herpes simplex virus 1 thymidine kinase expression in survivors of syngeneic glioma treated by adenovirus-mediated gene therapy: implications for clinical trials. Nat Med. 1999;5:1256–63. doi: 10.1038/15207. [DOI] [PubMed] [Google Scholar]
- 5.Ali S, Curtin JF, Zirger JM, et al. Inflammatory and anti-glioma effects of an adenovirus expressing human soluble Fms-like tyrosine kinase 3 ligand (hsFlt3L): treatment with hsFlt3L inhibits intracranial glioma progression. Mol Ther. 2004;10:1071–84. doi: 10.1016/j.ymthe.2004.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fulci G, Chiocca EA. Oncolytic viruses for the therapy of brain tumors and other solid malignancies: a review. Front Biosci. 2003;8:e346–60. doi: 10.2741/976. [DOI] [PubMed] [Google Scholar]
- 7.Klatzmann D, Valery CA, Bensimon G, et al. A phase I/II study of herpes simplex virus type 1 thymidine kinase ‘‘suicide’’ gene therapy for recurrent glioblastoma. Study Group on Gene Therapy for Glioblastoma. Hum Gene Ther. 1998;9:2595–604. doi: 10.1089/hum.1998.9.17-2595. [DOI] [PubMed] [Google Scholar]
- 8.Li S, Tokuyama T, Yamamoto J, Koide M, Yokota N, Namba H. Bystander effect-mediated gene therapy of gliomas using genetically engineered neural stem cells. Cancer Gene Ther. 2005;12:600–7. doi: 10.1038/sj.cgt.7700826. [DOI] [PubMed] [Google Scholar]
- 9.Sandmair AM, Loimas S, Puranen P, et al. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Hum Gene Ther. 2000;11:2197–205. doi: 10.1089/104303400750035726. [DOI] [PubMed] [Google Scholar]
- 10.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–74. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 11.Rampling R, Cruickshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859–66. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 12.Wilkinson GW, Akrigg A. Constitutive and enhanced expression from the CMV major IE promoter in a defective adenovirus vector. Nucleic Acids Res. 1992;20:2233–9. doi: 10.1093/nar/20.9.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas CE, Schiedner G, Kochanek S, Castro MG, Lowenstein PR. Peripheral infection with adenovirus causes unexpected long-term brain inflammation in animals injected intracranially with first-generation, but not with high-capacity, adenovirus vectors: toward realistic long-term neurological gene therapy for chronic diseases. Proc Natl Acad Sci U S A. 2000;97:7482–7. doi: 10.1073/pnas.120474397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas WS, Neal-Beliveau BS, Joyce JN. There is a limited critical period for dopamine’s e ffects on D1 receptor expression in the developing rat neostriatum. Brain Res Dev Brain Res. 1998;111:99–106. doi: 10.1016/s0165-3806(98)00126-6. [DOI] [PubMed] [Google Scholar]
- 15.Godiska R, Chantry D, Raport CJ, et al. Human macrophage-derived chemokine (MDC), a novel chemoattractant for monocytes, monocyte-derived dendritic cells, and natural killer cells. J Exp Med. 1997;185:1595–604. doi: 10.1084/jem.185.9.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lasarte JJ, Corrales FJ, Casares N, et al. Different doses of adenoviral vector expressing IL-12 enhance or depress the immune response to a coadministered antigen: the role of nitric oxide. J Immunol. 1999;162:5270–7. [PubMed] [Google Scholar]
- 17.Sun Y, Peng D, Lecanda J, et al. In vivo gene transfer of CD40 ligand into colon cancer cells induces local production of cytokines and chemokines, tumor eradication and protective antitumor immunity. Gene Ther. 2000;7:1467–6. doi: 10.1038/sj.gt.3301264. [DOI] [PubMed] [Google Scholar]
- 18.Cowsill C, Southgate TD, Morrissey G, et al. Central nervous system t oxicity of two adenoviral vectors encoding variants of the herpes simplex virus type 1 thymidine kinase: reduced cytotoxicity of a truncated HSV1-TK. Gene Ther. 2000;7:679–85. doi: 10.1038/sj.gt.3301147. [DOI] [PubMed] [Google Scholar]
- 19.Shering AF, Bain D, Stewart K, et al. Cell type-specific expression in brain cell cultures from a short human cytomegalovirus major immediate early promoter depends o n whether it is inserted into herpesvirus o adenovirus vectors. J Gen Virol. 1997;78:445–59. doi: 10.1099/0022-1317-78-2-445. [DOI] [PubMed] [Google Scholar]
- 20.Nyberg-Hoffman C, Aguilar-Cordova E. Instability o f adenoviral vectors during transport and its implication for clinical studies. Nat Med. 1999;5:955–7. doi: 10.1038/11400. [DOI] [PubMed] [Google Scholar]
- 21.Dion LD, Fang J, Garver RI., Jr Supernatant rescue assay vs. polymerase chain reaction for detection of wild type adenovirus-contaminating recombinant adenovirus stocks. J Virol Methods. 1996;56:99–107. doi: 10.1016/0166-0934(95)01973-1. [DOI] [PubMed] [Google Scholar]
- 22.Cotten M, Baker A, Saltik M, Wagner E, Buschle M. Lipopolysaccharide is a frequent contaminant of plasmid DNA preparations and can be toxic to primary human cells in the presence of adenovirus. Gene Ther. 1994;1:239–46. [PubMed] [Google Scholar]
- 23.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 24.Izquierdo M, Martin V, de Felipe P, et al. Human malignant brain tumor response to herpes simplex thymidine kinase ( HSVtk)/ganciclovir gene therapy. Gene Ther. 1996;3:491–5. [PubMed] [Google Scholar]
- 25.Jia WW, McDermott M, Goldie J, Cynader M, Tan J, Tufaro F. Selective destruction of gliomas in immunocompetent rats by thymidine kinase-defective herpes simplex virus type 1. J Natl Cancer Inst. 1994;86:1209–15. doi: 10.1093/jnci/86.16.1209. [DOI] [PubMed] [Google Scholar]
- 26.Moriuchi S, Wolfe D, Tamura M, et al. Double suicide gene therapy using a replication defective herpes simplex virus vector reveals reciprocal interference in a malignant glioma model. Gene Ther. 2002;9:584–91. doi: 10.1038/sj.gt.3301693. [DOI] [PubMed] [Google Scholar]
- 27.Nanda D, Vogels R, Havenga M, Avezaat CJ, Bout A, Smitt PS. Treatment of malignant gliomas with a replicating adenoviral vector expressing herpes simplex virus-thymidine kinase. Cancer Res. 2001;61:8743–50. [PubMed] [Google Scholar]
- 28.Tyynela K, Sandmair AM, Turunen M, et al. Adenovirus-mediated herpes simplex virus thymidine kinase gene therapy in BT4C rat glioma model. Cancer Gene Ther. 2002;9:917–24. doi: 10.1038/sj.cgt.7700515. [DOI] [PubMed] [Google Scholar]
- 29.Fischer HG, Bonifas U, Reichmann G. Phenotype and functions of brain d endritic cells emerging d chronic i nfection of mice with Toxoplasma gondii. J Immunol. 2000;164:4826–34. doi: 10.4049/jimmunol.164.9.4826. [DOI] [PubMed] [Google Scholar]
- 30.Fischer HG, Reichmann G. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol. 2001;166:2717–26. doi: 10.4049/jimmunol.166.4.2717. [DOI] [PubMed] [Google Scholar]
- 31.McMenamin PG. Distribution and phenotype o f dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in whole-mount preparations. J Comp Neurol. 1999;405:553–62. [PubMed] [Google Scholar]
- 32.Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am J P athol. 2000;157:1991–2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bodmer S, Strommer K, Frei K, et al. Immunosuppression and transforming growth factor-β in glioblastoma. Preferential production of transforming growth factor-β 2. J Immunol. 1989;143:3222–9. [PubMed] [Google Scholar]
- 34.Banchereau J, Schuler-Thurner B, Palucka AK, Schuler G. Dendritic cells as vectors for therapy. Cell. 2001;106:271–4. doi: 10.1016/s0092-8674(01)00448-2. [DOI] [PubMed] [Google Scholar]
- 35.Zou GM, Tam YK. Cytokines in the generation and maturation of dendritic cells: recent advances. Eur Cytokine Netw. 2002;13:186–99. [PubMed] [Google Scholar]
- 36.Banchereau J, Paczesny S, Blanco P, et al. Dendritic cells: controllers of the immune system and a new promise for immunotherapy. Ann N Y Acad Sci. 2003;987:180–7. doi: 10.1111/j.1749-6632.2003.tb06047.x. [DOI] [PubMed] [Google Scholar]
- 37.Puccetti P, Belladonna ML, Grohmann U. Effects of IL-12 and IL-23 o n antigen-presenting cells at the interface between innate and adaptive immunity. Crit Rev Immunol. 2002;22:373–90. [PubMed] [Google Scholar]
- 38.Guillot C, Coathalem H, Chetritt J, et al. Lethal hepatitis after gene transfer of IL-4 in the liver is independent of immune responses and dependent on apoptosis of hepatocytes: a rodent model of IL-4-induced hepatitis. J Immunol. 2001;166:5225–35. doi: 10.4049/jimmunol.166.8.5225. [DOI] [PubMed] [Google Scholar]
- 39.Ram Z, Culver KW, Oshiro EM, et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat Med. 1997;3:1354–61. doi: 10.1038/nm1297-1354. [DOI] [PubMed] [Google Scholar]
- 40.Trask TW, Trask RP, Aguilar-Cordova E, et al. Phase I study of adenoviral delivery o f the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol Ther. 2000;1:195–203. doi: 10.1006/mthe.2000.0030. [DOI] [PubMed] [Google Scholar]
- 41.Gitlitz BJ, Belldegrun AS, Zisman A, et al. A pilot trial of tumor lysate-loaded dendritic cells for the treatment of metastatic renal c ell carcinoma. J Immunother. 2003;26:412–9. doi: 10.1097/00002371-200309000-00004. [DOI] [PubMed] [Google Scholar]
- 42.Hernando JJ, Park TW, Kubler K, Offergeld R, Schlebusch H, Bauknecht T. Vaccination with autologous tumour antigen-pulsed dendritic cells in advanced gynaecological malignancies: clinical and immunological evaluation of a phase I trial. Cancer Immunol Immunother. 2002;51:45–52. doi: 10.1007/s00262-001-0255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackensen A, Herbst B, Chen JL, et al. Phase I study in melanoma patients of a vaccine with peptide-pulsed dendritic cells generated in vitro from CD34(+) hematopoietic progenitor cells. Int J Cancer. 2000;86:385–92. doi: 10.1002/(sici)1097-0215(20000501)86:3<385::aid-ijc13>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 44.Marten A, Renoth S, Heinicke T, et al. Allogeneic dendritic cells fused with tumor cells: preclinical results and outcome of a clinical phase I/II trial in patients with metastatic renal cell carcinoma. Hum Gene Ther. 2003;14:483–94. doi: 10.1089/104303403321467243. [DOI] [PubMed] [Google Scholar]
- 45.O’Rourke MG, Johnson M, Lanagan C, et al. Durable complete clinical responses in a phase I/II trial using an autologous melanoma cell/dendritic cell vaccine. Cancer Immunol Immunother. 2003;52:387–95. doi: 10.1007/s00262-003-0375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ridgway D. The first 1000 dendritic cell vaccinees. Cancer Invest. 2003;21:873–86. doi: 10.1081/cnv-120025091. [DOI] [PubMed] [Google Scholar]
- 47.Slingluff CL, Jr, Petroni GR, Yamshchikov GV, et al. Clinical and immunologic results of a randomized phase II trial of vaccination using four melanoma peptides either administered in granulocyte-macrophage colony-stimulating factor in adjuvant or pulsed on dendritic cells. J Clin Oncol. 2003;21:4016–26. doi: 10.1200/JCO.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 48.Yamanaka R, Abe T, Yajima N, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer. 2003;89:1172–9. doi: 10.1038/sj.bjc.6601268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Svane IM, Soot ML, Buus S, Johnsen HE. Clinical application of dendritic cells in cancer vaccination therapy. APMIS. 2003;111:818–34. doi: 10.1034/j.1600-0463.2003.11107813.x. [DOI] [PubMed] [Google Scholar]
- 50.Maraskovsky E, Brasel K, Teepe M, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med. 1996;184:1953–62. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masten BJ, Olson GK, Kusewitt DF, Lipscomb MF. Flt3 ligand preferentially increases t he number of functionally active myeloid dendritic cells in the lungs of mice. J Immunol. 2004;172:4077–83. doi: 10.4049/jimmunol.172.7.4077. [DOI] [PubMed] [Google Scholar]
- 52.McKenna HJ, Stocking KL, Miller RE, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95:3489–97. [PubMed] [Google Scholar]
- 53.Parajuli P, Mosley RL, Pisarev V, et al. Flt3 ligand and granulocyte-macrophage colony-stimulating factor preferentially expand and stimulate different dendritic and T-cell subsets. Exp Hematol. 2001;29:1185–93. doi: 10.1016/s0301-472x(01)00722-6. [DOI] [PubMed] [Google Scholar]
- 54.Shurin GV, Chatta GS, Tourkova IL, Zorina TD, Esche C, Shurin MR. Regulation of dendritic cell expansion in aged athymic nude mice by FLT3 ligand. Exp Gerontol. 2004;39:339–48. doi: 10.1016/j.exger.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Spiotto MT, Fu YX, Schreiber H. Tumor immunity meets autoimmunity: antigen levels and dendritic cell maturation. Curr Opin Immunol. 2003;15:725–30. doi: 10.1016/j.coi.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 56.Serot JM, Foliguet B, Bene MC, Faure GC. Ultrastructural and immunohistological evidence for dendritic-like cells within human choroid plexus epithelium. Neuroreport. 1997;8:1995–8. doi: 10.1097/00001756-199705260-00039. [DOI] [PubMed] [Google Scholar]
- 57.Jenks S. After initial setback, IL-12 regaining popularity. J Natl Cancer Inst. 1996;88:576–7. doi: 10.1093/jnci/88.9.576. [DOI] [PubMed] [Google Scholar]
- 58.Mazzolini G, Qian C, Xie X, et al. Regression of colon cancer and induction of antitumor immunity by intratumoral injection of adenovirus expressing interleukin-12. Cancer Gene Ther. 1999;6:514–22. doi: 10.1038/sj.cgt.7700072. [DOI] [PubMed] [Google Scholar]
- 59.Corthay A, Skovseth DK, Lundin KU, et al. Primary antitumor immune response mediated by CD4+ T cells. Immunity. 2005;22:371–83. doi: 10.1016/j.immuni.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 60.Lundin KU, Screpanti V, Omholt H, et al. CD4+ T cells kill Id+ B-lymphoma cells: FasLigand-Fas interaction is dominant in vitro but is redundant in vivo. Cancer Immunol Immunother. 2004;53:1135–45. doi: 10.1007/s00262-004-0538-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daniel D, Chiu C, Giraudo E, et al. CD4+ T cell-mediated antigen-specific immunotherapy in a mouse model of cervical cancer. Cancer Res. 2005;65:2018–25. doi: 10.1158/0008-5472.CAN-04-3444. [DOI] [PubMed] [Google Scholar]