

Abstract
The CTRΔe13 splice variant of the rabbit calcitonin receptor, which lacks the 14 amino acids of the seventh transmembrane domain (TMD) that are encoded by exon 13, is poorly expressed on the cell surface, fails to mobilize intracellular calcium or activate Erk, and inhibits the cell surface expression of the full-length C1a isoform. Nuclear magnetic resonance- and fluorescence-activated cell sorter-based experiments showed that the residual seventh TMD of CTRΔe13 fails to partition into the lipid bilayer, resulting in an extracellular C terminus. Truncating the receptor after residue 397 to delete the cytoplasmic tail resulted in reduced cell surface expression and an inability to mobilize intracellular calcium or activate Erk, but the truncated receptor did not inhibit C1a cell surface expression. In contrast, when the receptor was truncated after residue 374 to eliminate the entire seventh TMD domain and the C-terminal domain, the resulting receptor reduced the cell surface expression of C1a in a manner similar to that of CTRΔe13. Thus, normal cell surface expression, mobilization of intracellular calcium, and Erk activation requires the cytoplasmic C-terminal tail of the CTR, whereas the absence of the seventh TMD in the transmembrane helical bundle causes the dominant-negative effect on the surface expression of C1a.
Abbreviations: aa, Amino acid; [Ca2+]i, cytosolic free Ca2+; CRH-R1, CRH receptor; CT, calcitonin; CTR, CT receptor; DPC, dodecylphosphocholine; FACS, fluorescence-activated cell sorter; GFPm green fluorescent protein; GPCR, G protein-coupled receptor; HA, hemagglutinin; HEK, human embryonic kidney; MD, molecular dynamics; NMR, nuclear magnetic resonance; NOESY, nuclear Overhauser enhancement spectroscopy; PE, phycoerythrin; sCT, salmon CT; SDS, sodium dodecyl sulfate; TMD, transmembrane domain
CALCITONIN (CT) IS a 32-amino acid polypeptide hormone originally identified as a hypocalcemic factor (1). CT acts on bone and kidney to maintain calcium homeostasis and is also present in the central nervous system, where it has anorexic and analgesic effects (2). Of the cells present in bone, osteoclasts are the main target of CT. It inhibits motility and induces marked cellular retraction of isolated osteoclasts, two effects that are thought to be responsible for the strong inhibition of bone resorption by CT (3, 4).
The CTR [calcitonin (CT) receptor] belongs to Class B of G protein-coupled receptors (GPCR) and was first cloned in 1991 (5). Recently the CTR was also identified as an amylin receptor when coexpressed with receptor activity modifying proteins (also known as RAMPs) (6). Knockout of the CTR gene in mice is embryonic lethal by a yet unidentified mechanism (7). GPCRs comprise the largest superfamily of cell receptors and are involved in the translation of various extracellular signals to the intracellular compartment. They mediate response to the majority of known hormones, neurotransmitters, and neuromodulators and members function as sensors for extracellular ions, light, and pheromones. They serve as ligand-regulated guanine nucleotide exchange factors for heterotrimeric GTP-binding proteins, which in turn regulate several downstream effectors. The GPCRs share a general topology, with an extracellular N terminus followed by seven transmembrane domains (TMDs) connected via luminal/extracellular and cytoplasmic loops and a cytoplasmic C terminus and, within classes, a significant conservation of the TMDs.
Numerous alternatively spliced variants of the CTR have been described. The most common isoform in all species corresponds to the sequence originally cloned from porcine cells (5) and the rodent C1a isoform (8). Cloning of the rabbit CTR by our group revealed a splice variant with a deletion of exon 13, designated CTRΔe13, which encodes the C-terminal part of the seventh TMD (9). The Δe13 variant showed less production of inositol phosphates, no Erk phosphorylation, and a decreased cAMP response to salmon and human CT stimulation (9–11). Recently, we demonstrated that some of these findings are caused by the fact that the Δe13 variant is poorly expressed on the cell surface (11). Moreover, we demonstrated that the CTR forms homodimers and C1a/Δe13-heterodimers and that the coexpression of the two isoforms results in decreased cell surface expression of the C1a isoform, that is, the Δe13 isoform exerts a dominant-negative effect on CTR signaling (11). Interestingly, the exon encoding the C-terminal 14 amino acids of TM 7 is highly conserved in Class B GPCR genes. Spliced variants with the deletion of the same 14 amino acids as in the Δe13 variant of the CTR have been described for three other members of the receptor family, the CRH-R1 (CRH receptor) (12), the vasoactive intestinal peptide receptor (VPAC2) (13) and the PTH/PTHrP receptor (14).
The deletion of 14 amino acids from the distal part of the seventh TMD reduces the hydrophobicity of the residual sequence that follows the third extracellular loop and might compromise the ability of that sequence to anchor in the lipid bilayer, leading to a 6-TMD receptor with a luminal/extracellular C-terminal domain. The absence of either the seventh TMD or the cytoplasmic tail could affect the transport of the receptor to the cell surface or cause the retention of coexpressed C1a isoforms within the cell. We therefore asked how the absence of the amino acids encoded by exon 13 would affect the ability of the seventh TMD to insert into the lipid bilayer, and examined the properties of CTR constructs that lack only the C terminus or both the C terminus and the seventh TMD.
We show here that the lack of the distal part of the seventh TMD in the Δe13 isoform results in a protein segment that fails to segregate into the lipid membrane phase, leading to a luminal/extracellular C terminus. A CTR construct that lacked only the cytoplasmic C-terminal domain showed identical characteristics as Δe13 in terms of intra-cellular localization, reduced cell surface expression, lack of mobilization of intracellular calcium, and failure to activate Erk, but failed to inhibit the cell surface expression of the C1a isoform. In contrast, a CTR construct that lacked the entire seventh TMD and C terminus inhibited cell surface expression of the C1a isoform, in a manner similar to the effect of Δe13. We conclude that the cytoplasmic C-terminal tail is necessary for attaining normal levels of cell surface expression of the CTR, mobilization of intracellular calcium, and MAPK activation, whereas the absence of the seventh TMD is responsible for the dominant-negative effect of the CTRΔe13 isoform.
RESULTS
The Lack of the Distal Portion of TM 7 in the Δe13 Isoform of the CTR Leads to an Extracellular C Terminus
The Δe13 isoform of the CTR is characterized by a strongly reduced expression on the cell surface and a lack of mobilization of intracellular calcium and Erk-activation after CT stimulation compared with the more abundant C1a isoform (9–11). Moreover, the Δe13 isoform was found to affect CTR signaling in a dominant-negative manner by reducing the surface expression of the coexpressed C1a isoform (11). Structurally, the Δe13 isoform lacks the distal portion of the seventh TMD, which significantly decreases the hydrophobicity of this region. The decreased hydrophobicity of the sequence raises the possibility that the helix does not anchor in the membrane. If this were true, the third extracellular loop, the remainder of the seventh TMD and the C terminus would all be within the lumen of intracellular vesicular compartments or on the outside of the cell, potentially altering the trafficking of the receptor between intracellular compartments and the cell surface or the receptor’s signaling properties. To examine the possibility that the absence of the sequence encoded by exon 13 would prevent the partitioning of the remaining sequence into the lipid phase, we examined the ability of peptides that encompass the seventh TMDs of the C1a and Δe13 isoforms to partition into the hydrophobic phase of zwitterionic dodecylphosphocholine (DPC) micelles by high-resolution nuclear magnetic resonance (NMR).
The secondary shift values of the Hα protons, averaged over triplets of amino acids, provided a first evidence of secondary structure for the two receptor domains examined here. As illustrated in Fig. 1A, both CTR (367–405) (lower panel) and CTR(362–412, Δe13) (upper panel) have helical regions, identified by a negative secondary shift (resonances shifted to high field relative to the corresponding random-coil values). For the Δe13 peptide, two helices extending from residue 365–380 and 399–412 subtend the region of the exon 13 deletion (residues 384–397). The results for CTR (367–405) clearly indicate that the residues corresponding to exon 13 adopt a helical structure, in accordance with the putative assignment of the seventh TMD.
Fig. 1. The Absence of the Distal Part of the Seventh TMD in the Δe13 Isoform of the CTR Abolishes the Ability of the Seventh TMD to Insert into the Lipid Phase.
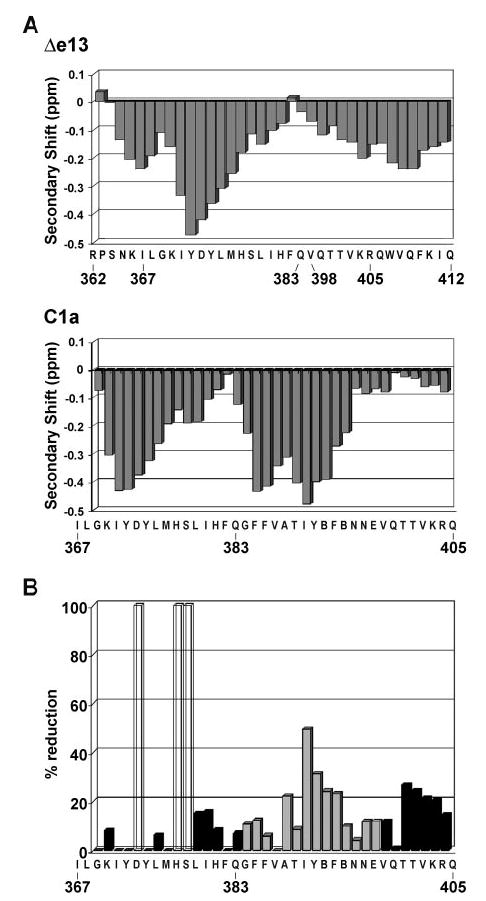
A, Secondary shifts for the H7agr; protons of the region of the seventh TMD of the C1a isoform (residues 367–405, lower panel) and the corresponding region of the Δe13 isoform (residues 362–383,398–412, upper panel). (In the C1a peptide sequence, B represents aminobutyric acid, an isostereic replacement for cysteine.) Values are averaged over triplets of amino acids. The negative values are consistent with α-helices. The removal of the 14 amino acids of exon 13 does not produce a drastic change in the secondary structural features. B, Effect of the addition of 16-doxylstearate to the DPC micelle-associated peptide containing the CTR seventh TMD (residues 367–405 of the C1a isoform). 16-Doxylstearate places the radical in the center of the micelle. The region passing through the micelle displays appreciable radical-induced reduction in the 1H NMR signals. The residues encoded by exon13 (384–397) are shown in gray. Accurate values could not be measured for three residues, denoted with empty bars. No reduction of the 1H signal of the Δe13 isoform peptide was observed upon titration with 16-doxylstearate.
The nuclear Overhauser enhancement spectroscopy (NOESY) spectra present the characteristic patterns of a highly helical structure in the corresponding regions of the sequence. Typical NOEs Hα(i)-HN(i+3), Hα(i)-HN(i+4), Hα (i)-Hβ (i+3), HN(i)-HN(i+2) and long stretches of continuous HN(i)-HN(i+1) NOEs are spread throughout the sequences in accord with the secondary shifts. The HN(i)-HN(i+2) and Hα (i)-HN(i+4) are located prevalently in the N-terminal helices of both peptides and in the region corresponding to exon 13. A number of medium-range side-chain to side-chain NOEs contributes to the overall fold. The distance geometry calculations produced 100 structures, all with low penalty functions. The φ and ψ dihedral angle order parameters were large for the helical regions, with decreasing values toward the helical termini, consistent with experimentally well-defined helices with structural variations at the termini.
To examine the ability of the peptides to partition into a lipid bilayer, we measured the change in 1H signal intensity that was induced by titration with 16-doxylstearic acid. 16-Doxylstearic acid places the nitroxide radical near the center of the zwitterionic micelle, and is therefore expected to significantly affect only those residues in the hydrophobic core. For CTR (367–405) there was a significant decrease in 1H signal intensity for the central portion of the peptide, corresponding to the residues coded by exon 13 (Fig. 1B). This clearly indicates that the putative seventh TMD of the CTR, including the residues coded by exon 13, passes through the micelle. In stark contrast, only a small background broadening was observed throughout the sequence of the Δe13 peptide (data not shown), in agreement with the entire molecule lying at the surface. Consistent with this observation, addition of 5-doxylstearic acid, which places the nitroxide radical at the level of the phosphate head-groups, produced a noticeable decrease in NMR signal intensity of Δe13 (data not shown). The 5-doxylstearic acid-induced relaxation pattern corresponds to the periodicity of the two α-helices lying on the micelle surface, as previously observed for domains of another receptor (15). To examine the energetics of the conformations and topological arrangements, extensive molecular dynamics (MD) simulations were carried out for the receptor domains. The starting structure was placed at the water/decane interface with a topological orientation corresponding to results from the 5-and 16-doxyl stearate titration. The starting topological orientation is preserved during the MD simulation, showing the contemporary agreement of the experimentally derived structure and topological orientation, with the partition between the hydrophobic/hydrophilic phases and a low energy conformation.
The NMR analysis of the peptide conformation suggested that the absence of the sequence encoded by exon 13 leads to the loss of a functional seventh TMD and therefore an extracellular C-terminal domain of the Δe13 isoform. We therefore examined the locations of the C-terminals of the C1a and Δe13 isoforms. C-terminally green fluorescent protein (GFP)-tagged constructs of the two isoforms were expressed in human embryonic kidney (HEK) 293 cells, and the cells were analyzed for the presence of extracellular GFP by fluorescence-activated cell sorter (FACS) analysis using anti-GFP antibody, as described in Materials and Methods. As expected, the cell-associated fluorescence of the cells transfected with the C1a isoform (Fig. 2, lower panel) was the same in preparations stained with anti-GFP and those stained with secondary antibody only, indicating that the C-terminal of the C1a isoform is intracellular. In contrast, when the cells expressed the Δe13 isoform (Fig. 2, upper panel), the anti-GFP-dependent fluorescence (dark trace) of all the cells was slightly increased relative to the fluorescence of cells stained with secondary antibody only (light trace), and the fluorescence of a small population of the cells was increased by about 5- to 10-fold. These results suggest that the C-terminal domains of the small number of Δe13 receptors that reach the cell surface are located extracellularly.
Fig. 2. The Absence of the Distal Part of the Seventh TMD in the Δe13 Isoform of the CTR Leads to an Extracellular C-Terminal Tail.
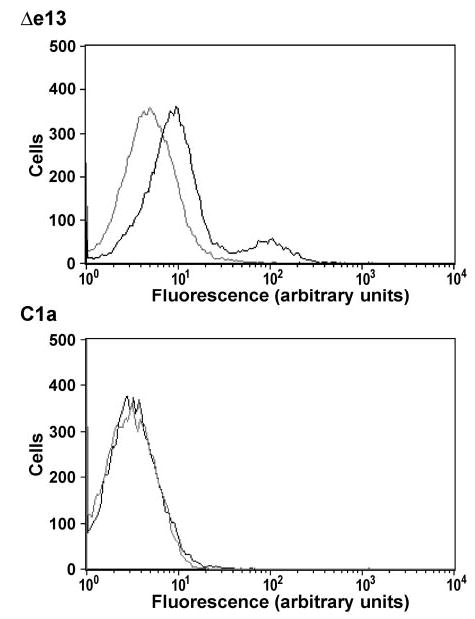
FACS analysis of cells transfected with C-terminally GFP-tagged C1a or Δe13 CTR isoforms and stained with anti-GFP antibody (dark traces) showed that the GFP tag was exposed on the surface of the cells transfected with the Δe13 isoform (upper panel) but not on the surface of the cells transfected with the C1a isoform (lower panel). The light traces are cells treated with PE-conjugated secondary antibody only. Similar results were obtained in three independent experiments.
The Absence of a Cytoplasmic C Terminus of the CTR Is Associated with a Strongly Reduced Cell Surface Expression and an Altered Intracellular Localization of the CTR
Having provided evidence that the absence of the distal portion of the seventh TMD in Δe13 leads to a failure of the residual seventh TMD residues to anchor in the lipid bilayer, and therefore to a luminal/extracellular C-terminal cytoplasmic tail and residual seventh TMD, we asked whether CTR truncation mutants that lack only the cytoplasmic tail (C1aΔ397) or the cytoplasmic tail plus the seventh TMD (C1aΔ374) show the same characteristics as the Δe13 isoform. First, we compared the cellular localization of the naturally occurring isoforms (C1a and Δe13) and the truncation mutants (C1aΔ397 and C1aΔ374). C-terminally GFP-tagged constructs were transfected into HEK 293 cells and living cells were examined by confocal microscopy 36 h after transfection (Fig. 3, left panels). As described before, the C1a-GFP chimera was clearly localized at the cell membrane and in a perinuclear compartment (Fig. 3, upper left panel) that we recently classified as a recycling compartment for the constitutively internalized CTR (16). In contrast, also as reported before (11), the Δe13-GFP chimera showed no clear expression at the cell surface, but accumulated intracellularly (Fig. 3, second micrograph from top). The C1aΔ397-GFP and the C1aΔ374-GFP chimeras were also poorly expressed on the cell surface and showed a similar intracellular distribution as the Δe13-GFP chimera (Fig. 3, third micrograph from top and bottom micrograph, respectively). To verify the differences in the expression of the different CTR constructs on the cell surface, we quantified the surface expression of an hemagglutinin (HA)-epitope in the extracellular N terminus of all the CTR constructs using FACS (Fig. 3, middle and right panels). All isoforms were expressed at similar levels (compare GFP fluorescence, Fig. 3, middle panels). As expected and consistent with our earlier results (11, 16) and the micrographs, the specific surface labeling [PE (phycoerythrin) fluorescence] of the C1a-expressing cells was up to 10-fold higher than the specific surface labeling of the Δe13-expressing cells. The surface labeling of the Δe13-expressing cells was distinctly higher than the nonspecific labeling with the PE-conjugated secondary only, however, consistent with the presence of some Δe13 isoform at the cell surface, as indicated by the staining for the GFP-tagged C-terminal tail (Fig. 2). The specific fluorescence of the cells transfected with the C1aΔ374 and C1aΔ397 constructs was similar to the fluorescence of the Δe13-expressing cells. The markedly reduced surface expression of both the C1aΔ397 and C1aΔ374 mutants relative to that of the C1a isoform indicates that achieving the normal level of expression of the CTR on the cell surface requires the presence of a cytoplasmic C-terminal tail. This suggests that the inefficient expression of the Δe13 isoform on the cell surface is a consequence of the failure of the seventh TMD to anchor in the membrane and the resulting failure of the C-terminal tail to remain within the cytoplasm, as demonstrated by the detection of extracellular GFP by FACS analysis (Fig. 2).
Fig. 3. The Absence of the Intracellular Cytoplasmic Tail of the CTR Leads to Strongly Reduced Expression of the Receptor on the Cell Surface.
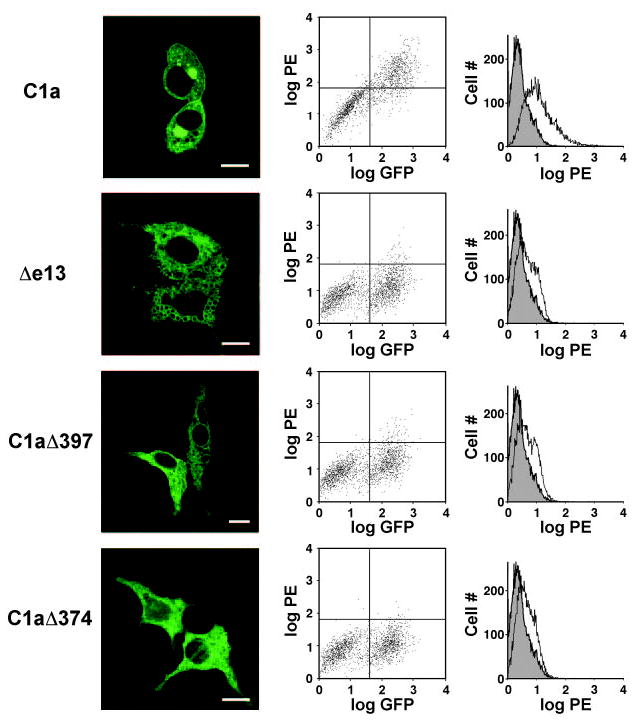
HEK 293 cells, seeded on glass coverslips, were transfected with a cDNA for N-terminally HA-tagged C1a-GFP, Δe13-GFP, C1aΔ397-GFP, or C1aΔ374-GFP. Thirty-six hours after transfection, cells were examined by confocal microscopy (left panels). The white bar represents 5 μm. To analyze the cell surface expression of the receptor constructs by FACS, the N-terminally HA-tagged receptors on the surface were labeled using anti-HA and a PE-conjugated secondary antibody, as described in Materials and Methods. FACS results are presented both as plots of GFP fluorescence vs. PE fluorescence (dot-plots, middle panels) and plots of PE fluorescence vs. cell number (right panels). Cells that are both GFP- and PE-positive are located in the upper right quadrant of the dot plots and correspond to the GFP-tagged receptor at the cell surface. The gray peaks in the right panels represents nonspecific fluorescence of cells stained with only the PE-conjugated secondary antibody. Similar results were obtained in four independent experiments.
The defective trafficking of some mutated membrane proteins (e.g. the CFTR chloride channel, the P2X7 nucleotide receptor) to the cell surface can be rescued by culturing the cells at a reduced temperature (17, 18). To test whether the aberrant trafficking of the CTRΔe13 isoform might be similarly rescued, cells transiently expressing the C1a and Δe13 isoforms were cultured at 27 C and 37 C, and the amounts of the receptors on the cell surface were measured by FACS (Fig. 4). The amounts of both the C1a isoform and the Δe13 isoform at the cell surface were essentially unchanged at the lower temperature, suggesting that the retention of the Δe13 isoform is not related to some temperature-dependent property, such as misfolding, and providing additional evidence to support the conclusion that the luminal/extracellular location of the normally cytoplasmic C-terminal domain is responsible for the inefficient translocation of the Δe13 isoform to the cell surface.
Fig. 4. The Surface Expression of the Δe13 Isoform Is Not Affected by Temperature.
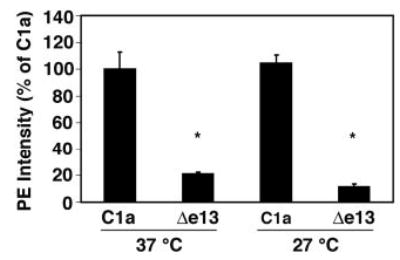
HEK 293 cells were transfected with N-terminally HA-tagged C1a-GFP or Δe13-GFP cDNAs and cultured at 27 C or 37 C for 24 h. The cell surface expression of the receptor isoforms was determined by FACS, as described in Materials and Methods and Fig. 3. The mean fluorescence intensities were normalized to the fluorescence of the C1a-expressing cells cultured at 37 C. The mean values and sem of cell-associated PE from three independent samples are shown (*, P < 0.05 relative to temperature-matched C1a-expressing cells, determined by Student’s t test).
Mobilization of Intracellular Calcium and Phosphorylation of Erk Are Inhibited by Removing the Cytoplasmic Tail of the CTR
We previously reported that the Δe13 isoform, in contrast to the C1a isoform, failed to mediate the CT-induced activation of phospholipase C and production of inositol phosphates, whereas the cAMP response to salmon and human CT stimulation was preserved, albeit reduced (9, 11). To determine whether these signaling properties of the Δe13 isoform are a consequence of the absence of a cytoplasmic C-terminal tail or of the absence of a seventh TMD, we compared CT-induced signaling in HEK 293 cells transfected with the C-terminally truncated C1aΔ397 and C1aΔ374 constructs with signaling in cells transfected with the C1a and Δe13 isoforms. As expected, we found a strong dose-dependent cAMP response to salmon CT (sCT) stimulation of C1a-expressing HEK 293 cells (Fig. 5A), whereas untransfected HEK 293 cells did not respond to sCT stimulation (data not shown). HEK 293 cells transfected with Δe13, C1aΔ397, or C1aΔ374 also responded to stimulation with sCT with dose-dependent increases in cAMP that were indistinguishable from one another but significantly lower than the response by the C1a-expressing cells (Fig. 5A), consistent with the lower cell surface expression of these constructs found in the FACS analysis (Fig. 4). This result suggests that the generation of a cAMP response via coupling of the CTR to Gαs requires neither a cytoplasmic C-terminal tail nor the seventh TMD.
Fig. 5. CTR Constructs that Lack a Cytoplasmic C-Terminal Tail Couple to Adenylyl Cyclase but Fail to Increase [Ca2+]i or Erk Phosphorylation.
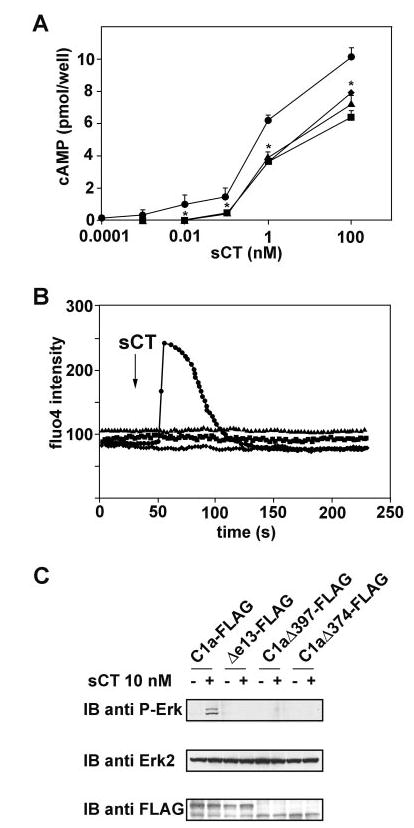
A, Twenty-four hours after transfection of HEK 293 cells with FLAG-tagged C1a (●), Δe13 (♦), C1aΔ397 (▴), or C1aΔ374 (▪) cDNAs, cells were seeded in 96-well plates, cultured for 24 h and incubated with different concentrations of sCT for 10 min in the presence of a phosphodiesterase inhibitor. cAMP was measured as described in Materials and Methods. The data represent the means of three independent experiments. The error bars represent the sem of the different experiments (*, P < 0.05 relative to C1a at the same concentration of sCT). No increase in cAMP concentration was seen after treatment of untransfected cells with sCT (data not shown). B, HEK 293 cells were transfected with the FLAG-tagged C1a (●), Δe13 (♦), C1aΔ397 (▴), or C1aΔ374 (▪) cDNAs. Forty-eight hours later, cells were loaded with the calcium-sensitive fluorescent dye FLUO-4 and changes in fluorescence after stimulation with 1 nm sCT were measured as described in Materials and Methods. The same results were obtained in eight independent experiments for each construct. No sCT-induced increase in fluorescence was seen in untransfected cells (data not shown). C, To examine sCT-induced Erk phosphorylation, HEK 293 cells were transfected with different C-terminal FLAG-tagged CTR constructs. After 24 h, cells were serum-starved for 12 h. Cells were stimulated with 1 nm sCT for 5 min, lysed and immunoblotted (IB) for phosphorylated-Erk1/2, using a specific monoclonal antibody (upper panel). Cell lysates were also blotted for total Erk2 to ensure equal loading (middle panel) and for FLAG to show relative expression levels of the CTR constructs (lower panel). Similar results were obtained in three independent experiments.
In contrast to its ability to mediate CT-induced cAMP production, the Δe13 isoform lacks the ability of the C1a isoform to induce the production of inositol phosphates and the subsequent increase in cytosolic free Ca2+ concentration ([Ca2+]i) or the phosphorylation and activation of Erk1/2 (9, 11). We therefore examined the abilities of the C1aΔ397 and the C1aΔ374 truncation mutants to mediate these responses to CT. Changes in [Ca2+]i HEK 293 cells that were transiently transfected with the various CTR constructs were measured using a calcium-sensitive fluorescent dye (Fig. 5B), as described in Materials and Methods. No mobilization of intracellular calcium was seen in untransfected HEK 293 cells (data not shown). As previously reported (10), a clear mobilization of intracellular calcium was observed in C1a-transfected cells treated with 1 nm (Fig. 5B) or 10 nm sCT (data not shown). In contrast, neither concentration of sCT induced a mobilization of intracellular calcium in cells that expressed the Δe13 isoform or either of the C-terminally truncated constructs. Similarly, sCT failed to induce the phosphorylation of Erk1/2 in cells that expressed the Δe13 isoform or the C1aΔ397 and the C1aΔ374 truncation mutants, whereas C1a-expressing cells showed a clear phosphorylation of Erk1/2 after sCT stimulation (Fig. 5C). Thus, the presence of a cytoplasmic C-terminal tail appears to be essential for CT-induced mobilization of intracellular calcium and Erk1/2-phosphorylation, but not for coupling to adenylyl cyclase.
The Absence of the Seventh TMD Is Responsible for the Dominant-Negative Effect of the Δe13 Isoform on the Surface Expression of the C1a Isoform
We previously reported that the Δe13 isoform exerts a dominant-negative effect on CTR signaling by inhibiting the cell surface expression of the C1a isoform, and hypothesized that the heterodimerization of the C1a isoform with the Δe13 isoform leads to the retention of the C1a isoform in an intracellular compartment (11). We therefore sought to determine whether coexpression of either or both of the C-terminally truncated C1aΔ397 and C1aΔ374 mutants with the C1a isoform would reduce the surface expression of the C1a isoform. We first examined the abilities of the C-terminally truncated CTR constructs to heterodimerize with the C1a isoform because dimerization is presumably a requirement for the dominant-negative effect of the Δe13 isoform on the cell surface expression of the C1a isoform. HEK 293 cells were cotransfected with a FLAG-tagged C1a construct and with GFP-tagged C1a, Δe13, C1aΔ397, or C1aΔ374 CTR constructs. Both the C-terminally truncated GFP-tagged CTR constructs were coimmunoprecipitated with the FLAG-tagged C1a isoform (Fig. 6), indicating that like the Δe13 isoform, they bind to the C1a isoform.
Fig. 6. The Presence of the Seventh TMD and the C-Terminal Tail of the CTR Is Not Necessary for Dimerization of the CTR.
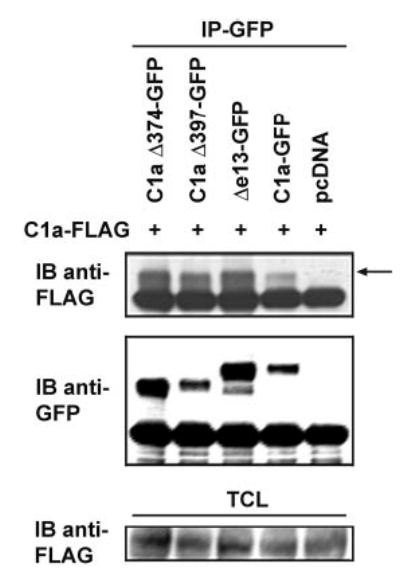
HEK 293 cells were transfected with the C1a-FLAG cDNA alone or in combination with GFP-tagged C1a, Δe13, C1aΔ397, or C1aΔ374, as indicated. Thirty-six to 48 h after transfection, the cells were lysed and the CTR-GFP construct was immunoprecipitated (IP) using the monoclonal anti-GFP antibody. The immune complexes were immunoblotted (IB) with the anti-FLAG antibody (upper panel). The membrane was stripped and reblotted with the anti-GFP antibody (middle panel). To show equal amounts of the FLAG-tagged CTR construct in the different lysates, total cell lysates (TCL) were immunoblotted with the anti-FLAG antibody (lower panel). Identical results were obtained in three independent experiments.
Because C1aΔ397 and C1aΔ374 were indistinguishable from the Δe13 isoform in terms of cell surface expression, cell signaling, and dimerization with the C1a isoform, we predicted that both truncation mutants would have a dominant-negative effect on C1a surface expression, like the Δe13 isoform. To examine this hypothesis, we quantified the amount of C1a receptor expressed on the cell surface in the absence or presence of Δe13, C1aΔ397, or C1aΔ374 using FACS analysis (Fig. 7A). As already demonstrated, the expression of the C1a isoform on the cell surface was much greater that that for the Δe13, C1aΔ397, or C1aΔ374 receptor constructs when individually expressed. The C1a surface expression was reduced by about half when the Δe13 isoform was coexpressed, as previously reported (11), and C1aΔ374, which lacks both the cytoplasmic C-terminal domain and the seventh TMD, reduced the surface expression of C1a to the same degree as seen for the Δe13 isoform. Contrary to our prediction, however, coexpression of C1aΔ397, which lacks the cytoplasmic C-terminal domain but not the seventh TMD, did not reduce the surface expression of the full-length C1a isoform.
Fig. 7. The Absence of the Seventh TMD, But Not the Absence of the Cytoplasmic C-Terminal Tail of the CTR, Leads to a Dominant-Negative Effect on the Surface Expression of the C1a Isoform.
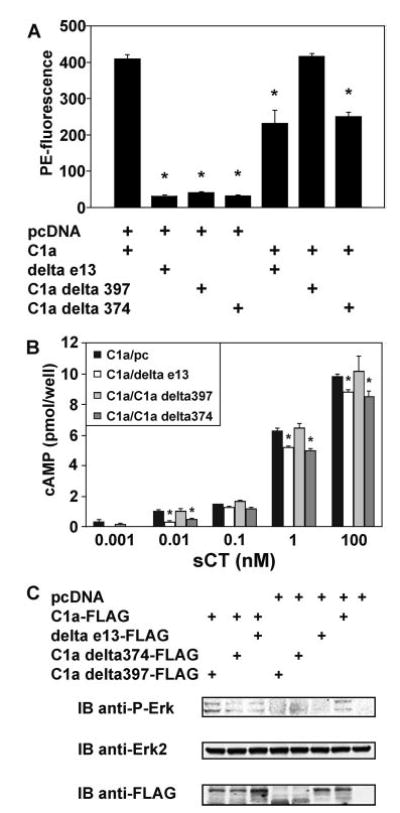
A, HEK 293 cells were transfected singly with C1a-GFP, Δe13-GFP, C1aΔ397-GFP, and C1aΔ374-GFP or with C1a-GFP in combination with the FLAG-tagged truncated CTRs as shown. The cell surface expression of the C1a-GFP chimera was measured by FACS, as described in Materials and Methods, using the anti-HA primary antibody. The mean values and sem of cell-associated PE from three independent samples are shown (*, P < 0.05, as determined by ANOVA followed by a Bonferroni test). B, HEK 293 cells were transfected with C1a-FLAG alone or in combination with Δe13-FLAG, C1aΔ397-FLAG, or C1aΔ374-FLAG. Twenty-four hours after transfection, cells were seeded in 96-well plates and cultured for 24 h before being treated with different concentrations of sCT for 10 min in the presence of a phosphodiesterase inhibitor. cAMP was measured as described in Materials and Methods. The data represent the means of three independent experiments, the error bars represent the sem of the different experiments. Significant differences between the response in cells transfected with C1a alone and cells cotransfected with C1a and one other CTR construct are indicated (*, P < 0.05). C, HEK 293 cells were transfected with C1a-FLAG, Δe13-FLAG, C1aΔ397-FLAG, and C1aΔ374-FLAG in the combinations shown. After 24 h, cells were serum-starved for 12 h, then treated with 0.1 nm sCT for 5 min and lysed. The lysates were immunoblotted (IB) for phosphorylated-Erk1/2 (upper panel), as well as for total Erk2 to ensure equal loading (middle panel) and for FLAG to demonstrate comparable expression of the CTR constructs (lower panel). Identical results were obtained in three independent experiments. The relative intensities of the phospho-Erk bands determined by densitometric analysis of the three experiments were: C1a alone, 100% ± 4.8%; C1a cotransfected with Δe13, 56.4% ± 6.6% (P < 0.05); C1a cotransfected with C1aΔ374, 60% ± 5.5% (P < 0.05); C1a cotransfected with C1aΔ397, 109.1% ± 16.7% (not significant).
A reduction of the cell surface expression of C1a by Δe13 and C1aΔ374 should be reflected by a reduction in downstream signaling events. Therefore, we compared the cAMP generation after stimulation with different concentrations of sCT when HEK 293 cells expressed C1a alone or in combination with Δe13, C1aΔ397, or C1aΔ374, respectively. At most sCT concentrations, we found small but significant reductions in CT-induced cAMP production when the C1a isoform was coex-pressed with Δe13 or C1aΔ374, whereas no reduction was seen when the C1a isoform was coexpressed with C1aΔ397 (Fig. 7B), consistent with the effects of the constructs on the C1a surface expression. Although significant differences were seen at both low and high sCT concentrations, the fractional reductions were more dramatic at low sCT concentrations, for example with only about 50% of cAMP generated at 0.01 nm sCT by co-expressing Δe13 or C1aΔ374 with the C1a isoform (Fig. 7B). To further confirm the dominant-negative effect of Δe13 and C1aΔ374, but not C1aΔ397 on signaling by the CTR, we examined the effect on Erk1/2 phosphorylation of coexpressing of the Δe13 isoform or the truncation mutants with the C1a isoform. We found substantially less phosphorylated Erk1/2 when C1a was coexpressed with Δe13 and C1aΔ374, whereas the co-expression of C1a with C1aΔ397 had little effect (Fig. 7C).
DISCUSSION
The data presented here strongly suggest that the Δe13 splice variant of the CTR gene forms a receptor that traverses the membrane six times rather than seven, resulting in a CTR with an extracellular C-terminal tail and seventh TMD. Consistent with the demonstration by NMR that the Δe13 seventh TMD fails to insert into the lipid bilayer, FACS analysis detected low levels of a C-terminal GFP tag on the surface of cells transfected with the GFP-tagged Δe13 isoform, but not in the surface of cells transfected with a similarly tagged C1a isoform. Furthermore, the functional properties of C-terminally truncated CTR mutants that lack either the cytoplasmic tail alone or the cytoplasmic tail plus the seventh TMD sequence are entirely consistent with our conclusion that the Δe13 isoform has only six transmembrane helices and consequently that the normally cytoplasmic C-terminal domain of this isoform is located luminally/extracellularly.
Comparing the Δe13 isoform with C-terminally truncated CTR constructs demonstrates that the absence of a cytoplasmic C-terminal domain and a seventh TMD have different functional consequences: absence of the cytoplasmic C-terminal tail causes a strong reduction of the expression of the receptor on the cell surface and the inability to mobilize intracellular calcium and activate Erk, whereas the absence of the seventh TMD confers the dominant-negative effect of the Δe13 isoform on the cell surface expression and signaling of the C1a isoform. The intracellular retention of the Δe13 isoform is not reduced by culturing the cells at a lower temperature, as is true for some mutated membrane proteins (17, 18), consistent with the conclusion that the luminal/extracellular location of the normally cytoplasmic C-terminal domain is responsible for the inefficient translocation of the Δe13 isoform to the cell surface.
To our knowledge, no GPCR with only six TMDs and a luminal/extracellular C terminus has been previously described, although naturally occurring splice isoforms similar to the CTRΔe13 isoform have been reported for three other class B GPCRs: CRH-R1, VPAC2, and the PTH/PTHrP receptor (12–14), and it is likely that the conformations of these splice isoforms relative to the membrane are similar to that of the CTRΔe13 isoform. There are, however, several reports of the functional consequences of the absence of cytoplasmic tails of GPCRs. The effects vary, depending on the GPCR examined. The GnRH receptor is the only naturally occurring GPCR that completely lacks a cytoplasmic C-terminal tail. The GnRH receptor does not show rapid desensitization and the rates of internalization are slow relative to other GPCRs, properties that correlated with the absence of a cytoplasmic tail (19). C-terminal truncation mutants have been generated for various GPCRs, with various receptor-specific results: a reduction of ligand-induced internalization (20, 21), an increase in the activation of signaling (22), or no apparent effect on trafficking and signaling of the GPCR (23, 24). Our data demonstrate that the absence of a cytoplasmic C-terminal tail on the rabbit CTR significantly reduces the transport of the receptor to the cell surface, presumably due to the presence in the C-terminal tail of an amino acid motif that links the CTR to protein components of the transport mechanism, possibly filamin (16). The Δe13 isoform retains all the amino acids present in the cytoplasmic tail, but an extracellular location of the C terminus would render that putative binding site inaccessible to intracellular proteins. Amino acid motifs that may be involved in the transport of other GPCRs to the cell surface have been described (25, 26), but none of them is present in the CTR.
The decrease in cAMP production in the cells transfected with the Δe13, C1aΔ397, or C1aΔ374 receptors (to 60–80% of the cAMP production in the C1a-expressing cells at 1 nm and 10 nm sCT) is not proportional to the marked reduction in the numbers of receptors on the cell surface, raising a question of whether the receptors with the truncations or deletion couple more efficiently to adenylyl cyclase than the C1a receptor. Other studies have shown that the coupling of human and porcine CTRs to adenylyl cyclase is quite sensitive to changes in the cytoplasmic domains. The maximum cAMP production mediated by an isoform of the human CTR that contains a 16-residue insert in the first cytoplasmic loop (27) is about 30% less than that of the human CTR isoform that is equivalent of the C1a isoform, whereas the EC50 is about 100-fold higher (28). Like the rabbit Δe13 isoform, the human isoform with the loop 1 insert fails to induce a change in [Ca2+]i. Progressive truncation of the porcine receptor cytoplasmic C-terminal domain changes the coupling to adenylyl cyclase in a complex manner; truncating after residues 399 (near the cytoplasmic face of the membrane) or 438 eliminates 95–99% of the cAMP production, whereas an intermediate truncation after residue 418 is essentially indistinguishable from the intact receptor (29). Finally, replacing the cytoplasmic domains of the insert-containing human CTR isoform with various combinations of the homologous domains of the porcine receptor, which in some cases changes as few as four or five residues, results in changes in coupling to adenylyl cyclase that differ by as much as 4-fold and are in some cases unique to the specific combination of domains exchanged (30). In all of these studies, the changes in coupling to adenylyl cyclase were not correlated with changes in the coupling to phospholipase C.
A possible alternative explanation for the apparently increased coupling efficiency of the Δe13 isoform to adenylyl cyclase is suggested by the relative ligand binding and dose-response curves of the rabbit C1a isoform (9). Nearly 50% of the maximal cAMP response occurs at a CT concentration (0.01 nm) at which little binding is detected, demonstrating that a substantial cAMP response can be achieved by activating a relatively small number of receptors, even in the case of the unspliced C1a isoform. Elucidating the reasons for the differences in the apparent molar coupling efficiency of the CTR isoforms is the goal of future investigation.
In contrast to the ability of the Δe13, C1aΔ397, and C1aΔ374 receptors to couple to adenylyl cyclase, none of these constructs mediated the mobilization of intracellular calcium or Erk phosphorylation by CT when expressed alone. Neither 1 nm nor 10 nm sCT elicited a calcium response, indicating that the effect is not simply a change in the potency of the response. [We previously found that even 10 μm sCT failed to induce the activation of phospholipase C-catalyzed production of inositol phosphates by the Δe13 isoform (9).] These results indicate that activation of Gαs is at least partly independent of the C-terminal tail of the CTR, but that mobilization of intracellular calcium via activation of Gαq and phospholipase C requires the presence of a cytoplasmic C-terminal domain. Similar findings have been reported for another GPCR, the δ-opioid receptor (31), suggesting that at least some GPCRs share this structural requirement for activation of Gαq and phospholipase C. The partial dependence of the CTR-mediated activation of Erk on Gαi (32) suggests another possible reason for the absence of Erk phosphorylation when the cytoplasmic tail of the CTR is absent. Coupling of the β2-adrenergic receptor to Gαi was recently found to be induced by protein kinase A-dependent phosphorylation of the receptor, which switches the coupling specificity of the activated receptor from Gαs to Gαi (33). If a similar mechanism regulates the coupling of the CTR to Gαi, the absence of a potential protein kinase A target site (residue 401) in the proximal C-terminal tail could prevent the induction of coupling of the CTR to Gαi and activation of Erk. On the other hand, the presence of some pertussis toxin-insensitive activation of Erk by the CTR (32) suggests that the cytoplasmic C terminus of the CTR also activates another mechanism that stimulates Erk.
Our data clearly demonstrate that the absence of the seventh TMD in Δe13 and C1aΔ374 is responsible for their dominant-negative effect on the cell surface expression of the C1a isoform, but the mechanism remains to be elucidated. Removing the seventh TMD from its usual position in the cluster of transmembrane helices may expose a cryptic endoplasmic reticulum retention signal or otherwise make it appear that the receptor is misfolded, thereby activating a mechanism that prevents the transport of the receptor to the cell surface. Further investigation will be required to elucidate the mechanism.
Several pieces of evidence suggest that the generation of the Δe13 isoform by alternative splicing is a physiologically significant mechanism for modulating the response of CTR-expressing cells to CT. The Δe13 isoform is found in all tissues that express the CTR. These include classic CT target tissues (brain, kidney, and osteoclasts) where CTR expression is relatively high, as well as lung and skeletal muscle, which are not typically thought of as responding to CT and contain significantly lower levels of CTR mRNA (9). The proportion of Δe13 mRNA varies from 10–15% in brain, kidney, and osteoclasts to 60% or more in muscle and lung (9). Analysis of the relative expression levels in individual osteoclasts (11) revealed that the ratio of the two isoforms is highly variable even in a single cell type, with some cells expressing mostly the C1a mRNA, others expressing mostly the Δe13 mRNA and still others expressing both mRNAs in various proportions. Together, the change in the coupling properties of the alternatively spliced Δe13 isoform and its ability to reduce the number of coexpressed C1a receptors at the cell surface indicate that a cell can regulate both the nature and the intensity of CT-induced signaling by altering the relative numbers of the two isoforms that are expressed. Elucidation of the way in which this splicing event is regulated could yield important insights into the physiology of CT. Moreover, because similar splice isoforms have been described for other members of the CTR family [the CRH-R1 (12), the VPAC2 vasoactive intestinal peptide receptor (13) and the PTH/PTHrP receptor (14)], alternative splicing of exon 13 in class B G protein-coupled receptors is likely to have a significance beyond the CTR.
In summary, we demonstrate for the first time the existence of a naturally occurring six TMD receptor derived from a splice variant of the CTR gene. The consequent luminal/extracellular localization of the normally cytoplasmic C-terminal domain results in markedly reduced levels of the receptor at the cell surface and prevents the ligand-induced mobilization of intracellular calcium and activation of Erk. However, the absence of a properly localized seventh TMD is responsible for the dominant-negative effect of the Δe13 isoform on cell surface expression and signaling by the common C1a isoform, demonstrating a prominent role of a seventh TMD in the regulation of trafficking of a GPCR.
MATERIALS AND METHODS
Reagents and Antibodies
sCT was purchased from Peninsula Laboratories, Inc. (Belmont, CA). The monoclonal anti-HA antibody (F-7), the monoclonal anti-Myc antibody (9E10), and the antibody against Erk2 were from Santa Cruz Biotechnology (Santa Cruz, CA), the monoclonal anti-GFP antibody was from CLONTECH (Palo Alto, CA), the phosphorylated Erk1/2 antibody was from New England Biolabs, Inc. (Beverly, MA), and the monoclonal anti-FLAG antibody (M2) was from Sigma (St. Louis, MO). Enhanced chemiluminescence solutions and nitrocellulose membranes were from Amersham (Piscataway, NJ) and Schleicher & Schuell (Keene, NH), respectively.
Peptide Synthesis
Peptides CTR (367–405), consisting of residues 367–405 of rabbit CTR, and CTR(363–412, Δe13), consisting of residues 363–383,398–412 of rabbit CTR, were synthesized and purified in the Peptide Facility of Tufts Medical School. Peptide identity and purity were verified by mass spectrometry and NMR. For the CTR (367–405) peptide, the two cysteine residues, C392 and C394, were replaced with aminobutyric acid, a common isostere for Cys and represented by a B in the figures.
NMR Methods
CTR (367–405) and CTR(363–412, Δe13) were examined (1.4 mm, 9:1 H2O/2H2O, pH 4.3) in the presence of DPC micelles [200 mm DPC-d38 from Cambridge Isotope Laboratories, Inc. (Andover, MA)] and sodium dodecyl sulfate (SDS) (160 mm SDS-d25). All experiments were recorded on a Bruker (Billerica, MA) AVANCE spectrometer (600 MHz) and at temperatures varying between 12 and 45 C. Data processing utilized XWIN-NMR software (Bruker) or NMRPipe (34). Chemical shifts were referenced to the signal of tetramethyl silane (TMS, 0.0 ppm). The proton resonances were identified following standard procedures using double quantum-filtered correlation spectroscopy, total correlation spectroscopy (mixing times = 30, 70 msec), and NOESY (mixing times = 100, 150, 200 msec). Suppression of the solvent signal was achieved by WATERGATE (35).
Radical-Induced Relaxation
The 5- and 16-doxylstearic acid were solubilized in methanol-d4 to a final concentration of 53 mm. Aliquots of this solution were added to the solution of peptide and DPC to obtain 0.25–0.94 mm concentrations of the spin-label. The titrations with 5- and 16-doxylstearic acid were carried out separately on two equivalent peptide solutions. TOCSY experiments (mixing time 35 msec) were recorded under identical conditions before and after the addition of the doxylstearic acid. The intensities of cross-peaks involving both backbone (HN-Hα) and side chain protons (Hα-Hβ, Hγ-Hδ) were compared.
Distance Geometry
The NOESY spectra acquired with a mixing time of 200 msec (temperature = 25, 35 C) were used to measure cross-peak volumes that were converted to distances using the two-spin approximation and a β1/β2 cross-peak set to a distance of 1.78 Å. Addition and subtraction of 10% to the calculated distances yielded upper and lower bounds used in the distance geometry calculations. A home-written program, based on the random metrization algorithm of Havel (36), was used to calculate an ensemble of 100 structures following previously published procedures (15).
Molecular Dynamics
MD simulations were performed with GROMACS (37) interactive modeling using Insight II (Biosym Technologies Inc., San Diego, CA). A two-phase box of water and decane was used to mimic the aqueous/hydrophobic phases of the lipid-micelle environment used for the spectroscopic studies, following previously published procedures (15). One of the low-violation distance geometry structures was used as the starting structure for the MD simulation. The molecule was placed in the (periodic) two-phase box of H2O/decane, containing 992 water and 123 decane molecules in a volume of 101.6 nm3. The system was energy-minimized for 100 steps (steepest descent). In the next 10 psec of MD at 27 C, the peptide was restrained to its original position with a force constant of 1000 kJ mol−1 nm−1. Experimental distance restraints were then introduced with a force constant of 10,000 kJ mol−1 nm−1 for the 400 psec of the simulation.
Cell Culture and Transient Transfections
DMEM and fetal bovine serum were purchased from Invitrogen (Carlsbad, CA). Media were supplemented with 100 μg/ml streptomycin and 100 U/ml penicillin. HEK 293 cells were cultured as described before (38). For transient transfections, cells were grown to 60–70% confluence and then transfected with Fugene 6 (Roche, Mannheim, Germany) according to the protocol of the manufacturer. When not otherwise described, experiments that involved transfection of CTR isoforms alone or in combination were performed with constant amounts of each cDNA and adding empty vector DNA when only one CTR isoform was expressed to keep the total amount of DNA constant.
DNA Constructs
cDNAs encoding the C1a and Δe13 splice isoforms of the rabbit CTR with a 3-fold FLAG-tag or GFP at the C terminus were generated as described before (16). A fragment spanning the complete receptor including the seventh TMD but lacking the cytoplasmic C-terminal tail of the receptor [amino acids (aa) 1–397] was cloned in the KpnI/HindIII site of the pEGFP-N1 vector and designated C1aDelta;397-GFP. Similarly, a fragment spanning the complete receptor up to the N-terminal of the seventh TMD (aa 1–374) was generated by PCR and cloned in the KpnI/HindIII site of the pEGFP-N1 vector and the p3XFLAG-CMV-13 vector and designated C1aDelta;374-GFP and C1aDelta;374-FLAG, respectively. All constructs contained an HA-tag in the extracellular N terminus of the receptor (after aa 29 of the original sequence). All PCR-derived constructs were sequenced by the Yale Keck Sequencing Facility.
Coimmunoprecipitation and Western Blotting
Cells were lysed in modified radioimmunoprecipitation assay buffer [50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.1% Igepal CA-630 (Sigma) 1% sodium deoxycholate, 10 mm NaF, 1 μg/ml pepstatin, and 1 mm phenylmethanesulfonyl fluoride] and incubated at 4 C for 30 min. Lysates were then centrifuged for 30 min at 4 C, 16,000 × g, the protein concentrations were measured with the bicinchoninic acid protein assay kit (Pierce, Rockford, IL) and equal amounts of protein were used for immunoprecipitation. Thirty microliters of protein G-agarose slurry and typically 5 μg of antibody were suspended in 500 μl PBS and incubated for 1 h at 4 C. The beads were washed three times in modified radioimmunoprecipitation assay buffer, then 500 μg of protein lysate and BSA (0.2% wt/vol) were added, and the mix was incubated for 2 h at 4 C. The immune complexes on the beads were washed four times with washing buffer containing 300 mm NaCl and 0.1% Triton X-100, and once with PBS. Beads were boiled in 2× SDS-PAGE sample buffer and samples were electrophoresed on precast 10% SDS-PAGE gels (Invitro-gen). Proteins were transferred to nitrocellulose membranes and the transfer was verified by staining with 0.2% Ponceau S in 3% trichloroacetic acid. Nonspecific binding was blocked by incubating the membranes in 5% nonfat milk in TBST buffer [50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.1% Tween 20) for 1 h. Membranes were incubated in the primary antibody in TBST for 2 h, washed three times for 15 min in TBST, and incubated for 1 h in 1:10,000 diluted horseradish peroxidase-conjugated antimouse IgG or antirabbit IgG antibody (Promega) in TBST. Blots were developed using the enhanced chemiluminescence system from Amersham.
Measurement of Receptor Cell Surface Expression by FACS Analysis
Cells in six-well plates were incubated with trypsin-EDTA solution at room temperature. All further steps were performed at 4 C. The trypsin activity was neutralized by addition of growth medium containing 10% dialyzed fetal bovine serum. Cells were collected by centrifugation at 800 × g for 3 min. The cell pellet was resuspended in 100 μl ice-cold PBS. Usually about 3 × 105 cells were used for each experiment. Normal goat IgG was added to a final concentration of 200 μg/ml. After 10 min, the antibody against the N-terminal HA-tag or C-terminal GFP, as indicated in the text, was added to a final concentration of 10 μg/ml. A 30-min incubation step was followed by resuspending in 100 μl PBS containing 50 μg/ml PE-conjugated goat antimouse IgG antibody (Molecular Probes, Eugene, OR). After a final washing step, cells were resuspended in PBS containing 2% formaldehyde to fix the sample. Bound antibody was analyzed by fluorescence flow cytometry (FACSCalibur, Becton Dickinson, Franklin Lakes, NJ). Win MDI software version 2.8 was used for data analysis.
cAMP Measurement
cAMP was measured as described before (9). Cells transfected with the CTR constructs were plated in a 96-well plate (8000 per well). Cells were preincubated with 1 mm of 3-isobutyl-1-methyl-xanthine (Sigma) for 10 min before stimulation with sCT for 10 min at 37 C. The reaction was stopped with 95% ethanol containing 3 mm HCl. cAMP was measured by scintillation proximity assay (Amersham) following the manufacturer’s instructions.
Measurement of [Ca2+]i
Transfected cells were seeded on glass-bottom dishes. Twenty-four hours later, cells were loaded with 6 μm Fluo-4 by incubation for 30 min at 37 C in conditioned medium. Cells were transferred to a heated perfusion chamber in a confocal microscope and perfused with Na+-HEPES-buffer [135 mm NaCl, 5 mm KCl, 1 mm MgCl2, 1 mM CaCl2, 10 mM glucose, and 20 mm HEPES (pH 7.3), 290 mOsmol/liter]. The chamber was maintained at 37 C and perfused with 1 nm or 10 nm sCT to stimulate the CTR. Intracellular calcium mobilization was monitored using excitation at 468 nm and detection of the emission at 505–530 nm. The fluorescence of at least three cells was measured for each construct in each of eight identical experiments.
Statistics
For the FACS analyses, the statistical significance of the differences in PE fluorescence values relative to the fluorescence of cells expressing only C1a was determined by Student’s t test or by ANOVA followed by a Bonferroni test, as indicated in the figure legends. For the measurements of cAMP production, the statistical significance was determined by ANOVA followed by a Tukey’s test.
Acknowledgments
This work was supported by Grant SE 999/3-1 from the Deutsche Forschungsgemeinschaft (to T.S.) and Grants DE-04724 (to R.B.), GM-54083 (to D.F.M.) and AR-49879 (to W.C.H.) from the National Institutes of Health.
References
- 1.Copp DH. Calcitonin and parathyroid hormone. Annu Rev Pharmacol. 1969;9:327–344. doi: 10.1146/annurev.pa.09.040169.001551. [DOI] [PubMed] [Google Scholar]
- 2.Azria M 1989 The calcitonins: physiology and pharmacology. Basel, Switzerland: Karger; 43–58
- 3.Zaidi M, Datta HK, Moonga BS, MacIntyre I. Evidence that the action of calcitonin on rat osteoclasts is mediated by two G proteins acting via separate post-receptor pathways. J Endocrinol. 1990;126:473–481. doi: 10.1677/joe.0.1260473. [DOI] [PubMed] [Google Scholar]
- 4.Su Y, Chakraborty M, Nathanson MH, Baron R. Differential effects of the 3′,5′-cyclic adenosine mono-phosphate and protein kinase C pathways on the response of isolated rat osteoclasts to calcitonin. Endocrinology. 1992;131:1497–1502. doi: 10.1210/endo.131.3.1324163. [DOI] [PubMed] [Google Scholar]
- 5.Lin HY, Harris TL, Flannery MS, Aruffo A, Kaji EH, Gorn A, Kolakowski Jr LF, Lodish HF, Goldring SR. Expression cloning of an adenylate cyclase-coupled calcitonin receptor. Science. 1991;254:1022–1024. doi: 10.1126/science.1658940. [DOI] [PubMed] [Google Scholar]
- 6.Christopoulos G, Perry KJ, Morfis M, Tilakaratne N, Gao Y, Fraser NJ, Main MJ, Foord SM, Sexton PM. Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol Pharmacol. 1999;56:235–242. doi: 10.1124/mol.56.1.235. [DOI] [PubMed] [Google Scholar]
- 7.Dacquin R, Davey RA, Laplace C, Levasseur R, Morris HA, Goldring SR, Gebre-Medhin S, Galson DL, Zajac JD, Karsenty G. Amylin inhibits bone resorption while the calcitonin receptor controls bone formation in vivo. J Cell Biol. 2004;164:509–514. doi: 10.1083/jcb.200312135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albrandt K, Mull E, Brady EMG, Herich J, Moore CX, Beaumont K. Molecular cloning of two receptors from rat brain with high affinity for salmon calcitonin. FEBS Lett. 1993;325:225–232. doi: 10.1016/0014-5793(93)81078-e. [DOI] [PubMed] [Google Scholar]
- 9.Shyu J-F, Inoue D, Baron R, Horne WC. The deletion of 14 amino acids in the seventh transmembrane domain of a naturally occurring calcitonin receptor isoform alters ligand binding and selectively abolishes coupling to phospholipase C. J Biol Chem. 1996;271:31127–31134. doi: 10.1074/jbc.271.49.31127. [DOI] [PubMed] [Google Scholar]
- 10.Santhanagopal A, Chidiac P, Horne WC, Baron R, Dixon SJ. Calcitonin (CT) rapidly increases Na+/H+ exchange and metabolic acid production: effects mediated selectively by the C1a CT receptor isoform. Endocrinology. 2001;142:4401–4413. doi: 10.1210/endo.142.10.8411. [DOI] [PubMed] [Google Scholar]
- 11.Seck T, Baron R, Horne WC. The alternatively spliced Δe13 transcript of the rabbit calcitonin receptor dimerizes with the C1a isoform and inhibits its surface expression. J Biol Chem. 2003;278:23085–23093. doi: 10.1074/jbc.M211280200. [DOI] [PubMed] [Google Scholar]
- 12.Grammatopoulos DK, Dai Y, Randeva HS, Levine MA, Karteris E, Easton AJ, Hillhouse EW. A novel spliced variant of the type 1 corticotropin-releasing hormone receptor with a deletion in the seventh transmembrane domain present in the human pregnant term myometrium and fetal membranes. Mol Endocrinol. 1999;13:2189–2202. doi: 10.1210/mend.13.12.0391. [DOI] [PubMed] [Google Scholar]
- 13.Grinninger C, Wang W, Oskoui KB, Voice JK, Goetzl EJ. A natural variant type II G protein-coupled receptor for vasoactive intestinal peptide with altered function. J Biol Chem. 2004;279:40259–40262. doi: 10.1074/jbc.C400332200. [DOI] [PubMed] [Google Scholar]
- 14.Ding C, Racusen L, Wilson P, Burrow C, Levine MA. J Bone Miner Res. 1995;10(Suppl 1):S484. [Google Scholar]
- 15.Pellegrini M, Bisello A, Rosenblatt M, Chorev M, Mierke DF. Binding domain of human parathyroid hormone receptor: from conformation to function. Biochemistry. 1998;37:12737–12743. doi: 10.1021/bi981265h. [DOI] [PubMed] [Google Scholar]
- 16.Seck T, Baron R, Horne WC. Binding of filamin to the C-terminal tail of the calcitonin receptor controls recycling. J Biol Chem. 2003;278:10408–10416. doi: 10.1074/jbc.M209655200. [DOI] [PubMed] [Google Scholar]
- 17.Loffing-Cueni D, Loffing J, Shaw C, Taplin AM, Govindan M, Stanton CR, Stanton BA. Trafficking of GFP-tagged ΔF508-CFTR to the plasma membrane in a polarized epithelial cell line. Am J Physiol Cell Physiol. 2001;281:C1889–C1897. doi: 10.1152/ajpcell.2001.281.6.C1889. [DOI] [PubMed] [Google Scholar]
- 18.Denlinger LC, Sommer JA, Parker K, Gudipaty L, Fisette PL, Watters JW, Proctor RA, Dubyak GR, Bertics PJ. Mutation of a dibasic amino acid motif within the C terminus of the P2X7 nucleotide receptor results in trafficking defects and impaired function. J Immunol. 2003;171:1304–1311. doi: 10.4049/jimmunol.171.3.1304. [DOI] [PubMed] [Google Scholar]
- 19.Heding A, Vrecl M, Bogerd J, McGregor A, Sellar R, Taylor PL, Eidne KA. Gonadotropin-releasing hormone receptors with intracellular carboxyl-terminal tails undergo acute desensitization of total inositol phosphate production and exhibit accelerated internalization kinetics. J Biol Chem. 1998;273:11472–11477. doi: 10.1074/jbc.273.19.11472. [DOI] [PubMed] [Google Scholar]
- 20.Huang Z, Chen Y, Nissenson RA. The cytoplasmic tail of the G-protein-coupled receptor for parathyroid hormone and parathyroid hormone-related protein contains positive and negative signals for endocytosis. J Biol Chem. 1995;270:151–156. doi: 10.1074/jbc.270.1.151. [DOI] [PubMed] [Google Scholar]
- 21.Le Gouill C, Parent J-L, Rola-Pleszczynski M, Stankova J. Structural and functional requirements for agonist-induced internalization of the human platelet-activating factor receptor. J Biol Chem. 1997;272:21289–21295. doi: 10.1074/jbc.272.34.21289. [DOI] [PubMed] [Google Scholar]
- 22.Alblas J, van Etten I, Moolenaar WH. Truncated, desensitization-defective neurokinin receptors mediate sustained MAP kinase activation, cell growth and transformation by a Ras-independent mechanism. EMBO J. 1996;15:3351–3360. [PMC free article] [PubMed] [Google Scholar]
- 23.Hashido K, Adachi M, Gamou T, Watanabe T, Furuichi Y, Miyamoto C. Identification of specific intracellular domains of the human ETA receptor required for ligand binding and signal transduction. Cell Mol Biol Res. 1993;39:3–12. [PubMed] [Google Scholar]
- 24.Hipkin RW, Liu X, Ascoli M. Truncation of the C-terminal tail of the follitropin receptor does not impair the agonist- or phorbol ester-induced receptor phosphorylation and uncoupling. J Biol Chem. 1995;270:26683–26689. doi: 10.1074/jbc.270.44.26683. [DOI] [PubMed] [Google Scholar]
- 25.Schulein R, Hermosilla R, Oksche A, Dehe M, Wiesner B, Krause G, Rosenthal W. A dileucine sequence and an upstream glutamate residue in the intracellular carboxyl terminus of the vasopressin V2 receptor are essential for cell surface transport in COS. M6 cells Mol Pharmacol. 1998;54:525–535. doi: 10.1124/mol.54.3.525. [DOI] [PubMed] [Google Scholar]
- 26.Bermak JC, Li M, Bullock C, Zhou Q-Y. Regulation of transport of the dopamine D1 receptor by a new membrane-associated ER protein. Nat Cell Biol. 2001;3:492–498. doi: 10.1038/35074561. [DOI] [PubMed] [Google Scholar]
- 27.Gorn AH, Lin HY, Yamin M, Auron PE, Flannery MR, Tapp DR, Manning CA, Lodish HF, Krane SM, Goldring SR. Cloning, characterization, and expression of a human calcitonin receptor from an ovarian carcinoma cell line. J Clin Invest. 1992;90:1726–1735. doi: 10.1172/JCI116046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore EE, Kuestner RE, Stroop SD, Grant FJ, Matthewes SL, Brady CL, Sexton PM, Findlay DM. Functionally different isoforms of the human calcitonin receptor result from alternative splicing of the gene transcript. Mol Endocrinol. 1995;9:959–968. doi: 10.1210/mend.9.8.7476993. [DOI] [PubMed] [Google Scholar]
- 29.Findlay DM, Houssami S, Lin HY, Myers DE, Brady CL, Darcy PK, Ikeda K, Martin TJ, Sexton PM. Truncation of the porcine calcitonin receptor cytoplasmic tail inhibits internalization and signal transduction but increases receptor affinity. Mol Endocrinol. 1994;8:1691–1700. doi: 10.1210/mend.8.12.7708057. [DOI] [PubMed] [Google Scholar]
- 30.Nussenzveig DR, Thaw CN, Gershengorn MC. Inhibition of inositol phosphate second messenger formation by intracellular loop one of a human calcitonin receptor. Expression and mutational analysis of synthetic receptor genes. J Biol Chem. 1994;269:28123–28129. [PubMed] [Google Scholar]
- 31.Hirst RA, Smart D, Devi LA, Lambert DG. Effects of C-terminal truncation of the recombinant δ-opioid receptor on phospholipase C and adenylyl cyclase coupling. J Neurochem. 1998;70:2273–2278. doi: 10.1046/j.1471-4159.1998.70062273.x. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, Shyu J-F, Santhanagopal A, Inoue D, David J-P, Dixon SJ, Horne WC, Baron R. The calcitonin receptor stimulates Shc tyrosine phosphorylation and Erk1/2 activation. Involvement of Gi, protein kinase C, and calcium. J Biol Chem. 1998;273:19809–19816. doi: 10.1074/jbc.273.31.19809. [DOI] [PubMed] [Google Scholar]
- 33.Lefkowitz RJ, Pierce KL, Luttrell LM. Dancing with different partners: protein kinase A phosphorylation of seven membrane-spanning receptors regulates their G protein-coupling specificity. Mol Pharmacol. 2002;62:971–974. doi: 10.1124/mol.62.5.971. [DOI] [PubMed] [Google Scholar]
- 34.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 35.Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 36.Havel TF. An evaluation of computational strategies for use in determination of protein structure from distance geometry constraints obtained by nuclear magnetic resonance. Prog Biophys Mol Biol. 1991;56:43–78. doi: 10.1016/0079-6107(91)90007-f. [DOI] [PubMed] [Google Scholar]
- 37.Lindahl E, Hess B, Van der Spoel D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J Mol Mod. 2001;7:306–317. [Google Scholar]
- 38.Zhang Z, Hernandez-Lagunas L, Horne WC, Baron R. Cytoskeleton-dependent tyrosine phosphorylation of the p130Cas family member HEF1 downstream of the G protein-coupled calcitonin receptor. Calcitonin induces the association of HEF1, paxillin, and focal adhesion kinase. J Biol Chem. 1999;274:25093–250938. doi: 10.1074/jbc.274.35.25093. [DOI] [PubMed] [Google Scholar]