

Abstract
We develop a dynamic programming algorithm for haplotype block partitioning to minimize the number of representative single nucleotide polymorphisms (SNPs) required to account for most of the common haplotypes in each block. Any measure of haplotype quality can be used in the algorithm and of course the measure should depend on the specific application. The dynamic programming algorithm is applied to analyze the chromosome 21 haplotype data of Patil et al. [Patil, N., Berno, A. J., Hinds, D. A., Barrett, W. A., Doshi, J. M., Hacker, C. R., Kautzer, C. R., Lee, D. H., Marjoribanks, C., McDonough, D. P., et al. (2001) Science 294, 1719–1723], who searched for blocks of limited haplotype diversity. Using the same criteria as in Patil et al., we identify a total of 3,582 representative SNPs and 2,575 blocks that are 21.5% and 37.7% smaller, respectively, than those identified using a greedy algorithm of Patil et al. We also apply the dynamic programming algorithm to the same data set based on haplotype diversity. A total of 3,982 representative SNPs and 1,884 blocks are identified to account for 95% of the haplotype diversity in each block.
Single nucleotide polymorphisms (SNPs) are promising markers for population genetic studies and for localizing genetic variations responsible for complex diseases. They are preferred to other genetic markers, such as microsatellites, because of their high abundance (SNPs with minor allele frequency greater than 0.1 occur once about every 600 base pairs) (1), relatively low mutation rate, and easy adaptability to automatic genotyping. It is known that studies using haplotype information generally outperform those using single-marker analysis. Thus it is important to know the haplotype structure of the whole genome in the populations under study. Several groups have carried out studies of the haplotype structure in specific genes and populations (2, 3), and the results vary significantly for different regions of the chromosomes. In an effort to identify genetic variations for Crohn's disease in a candidate region on human chromosome 5q31, Daly et al. (4) and Rioux et al. (5) genotyped 103 common (≥5% minor allele frequency) SNPs in an 500-kb region on 129 parents–offspring trios. Based on the data, they found that the region can be divided into blocks (referred as haplotype blocks) spanning from tens up to about 92 kb. There is limited diversity within each block.
In a recent paper, Patil et al. (6) studied the global haplotype structure on chromosome 21. There are two general classes of methods to infer haplotype frequencies. One is based on genotypes on large pedigrees and the other is based on statistical methods such as the EM algorithm (7–11). Those methods deal with genotype data on diploid copies of the chromosomes. Uncertainties of haplotype phase are generally unavoidable for such data. Unlike previous studies, Patil et al. (6) studied haplotypes of haploid copies of chromosome 21 isolated in rodent–human somatic cell hybrids, allowing the determination of full haplotypes of those chromosomes. They also found that the haplotypes can be divided into blocks of limited haplotype diversity. Only a small fraction of the SNPs in a block are sufficient to uniquely identify the common haplotypes in each block. Those SNPs are referred as representative SNPs. The techniques to perform the haplotype block partitioning to minimize the total number of representative SNPs for the entire chromosome is the topic of this paper.
One of the major objectives of Patil et al. (6) is to characterize the common haplotypes. To define the common haplotypes in a block, we need to first introduce the concept of ambiguous and unambiguous haplotypes as in Patil et al. (6) when missing data are present. Two haplotypes are said to be compatible if the alleles are identical at all loci for which there are no missing data; otherwise the two haplotypes are said to be incompatible. As in Patil et al. (6), we define the ambiguous haplotypes as those haplotypes compatible with at least two haplotypes that are themselves incompatible. We will give a rigorous definition of ambiguous and unambiguous haplotypes in Methods. In the rest of the paper, we only study the unambiguous haplotypes. It should be noted that when there are no missing data, the concept of ambiguous and unambiguous haplotypes is not needed because in that case all of the haplotypes are unambiguous.
We define the common haplotypes as those haplotypes that are represented more than once in a block. We are mainly interested in the common haplotypes. Therefore we require that, in the final block partition result, a significant fraction of the haplotypes in each block are common haplotypes. In Patil et al. (6), they require that at least α = 70%, 80%, and 90%, respectively, of the unambiguous haplotypes are represented more than once.
For each block, we want to minimize the number of SNPs that distinguish at least α percent of the unambiguous haplotypes in the block. Those SNPs can be thought of as a signature of the haplotype block partition. Let f(Bi) be the minimum number of SNPs required to uniquely distinguish at least α percent of the unambiguous haplotypes in the ith block, Bi. α is referred to as the coverage. The objective is to find a partition to minimize the total number of representative SNPs required to distinguish at least α percent of unambiguous haplotypes in each block for the entire chromosome, Σ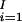 f(Bi), where I is the number of blocks in the partitioning. The number of blocks, I, is unknown before the partitioning.
f(Bi), where I is the number of blocks in the partitioning. The number of blocks, I, is unknown before the partitioning.
Patil et al. (6) developed a greedy algorithm to achieve this objective. The greedy algorithm can be briefly described as follows. They considered all potential blocks of consecutive SNPs and selected the block that maximizes the ratio of the number of SNPs in the block, B, with f(B), the minimum number of SNPs required to distinguish the unambiguous haplotypes represented more than once. The process was repeated until the entire chromosome was covered.
Although the greedy algorithm gives an approximate solution to the problem, it does not guarantee an optimal solution. In this paper, we present a dynamic programming algorithm for haplotype partitioning that gives an optimal solution to the problem. In addition, we also minimize the number of blocks among all of the block partitions with the minimum number of representative SNPs.
For each block, we measure the block quality by a function of the haplotypes defined by the SNPs in the block. The program can be easily adapted to any measures of haplotype block quality. For example, Clayton (12) defined haplotype diversity, D, in a block. He also defined the proportion of haplotype diversity explained by a subset of SNPs in a block. Let f*(Bi) be the minimum number of SNPs required to explain β percent of the haplotype diversity in the ith block. It is reasonable to partition the chromosome into haplotype blocks such that Σ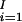 f*(Bi) is minimized.
f*(Bi) is minimized.
Methods
In this section, we mathematically formulate the problem and provide a dynamic programming algorithm to minimize the number of representative SNPs. We also minimize the number of blocks among those block partitions with the minimum number of representative SNPs.
Assume that we are given K haplotypes and a sequence of n consecutive SNPs. Let ri, i = 1, 2, . . . , n be a K-dimensional vector with the kth component ri(k) = 0, 1, or 2 being the allele of the kth haplotype at the ith SNP locus, where 0 indicates missing data, and 1 and 2 are the two alleles.
Consider a block defined by ri, . . . , rj. Let us first define ambiguous and unambiguous haplotypes. Two haplotypes, the kth and k′th haplotypes, are compatible if the alleles are the same for the two haplotypes at the loci with no missing data, that is, rl(k) = rl(k′) for any l, i ≤ l ≤ j such that rl(k)rl(k′) ≠ 0. A haplotype in the block is ambiguous if it is compatible with two other haplotypes that are themselves incompatible. For example, consider three haplotypes h1 = (1, 1, 0, 2), h2 = (1, 1, 2, 0), and h3 = (1, 1, 1, 2). Haplotype h1 is compatible with haplotypes h2 and h3, but h2 is not compatible with h3 because they differ at the third locus. Thus, h1 is an ambiguous haplotype, whereas h2 and h3 are not ambiguous.
Using compatibility as a criterion, we can classify the unambiguous haplotypes into disjoint groups. Two unambiguous haplotypes are in the same group if they are compatible with each other and will be treated as identical in the reminder of the paper.
With the above definition of unambiguous haplotypes, we then define a boolean function block(ri, . . . ,rj) = 1 if at least α percent of the unambiguous haplotypes in the block are represented more than once. As discussed above, the blocks in the final block partition should satisfy this condition.
Let f(⋅) be the minimum number of SNPs required to uniquely distinguish at least α percent of the unambiguous haplotypes within a block. Given a block partition—i.e., B1, . . . . , BI—the total number of representative SNPs for these blocks is f(B1) + . . . + f(BI). The optimal block partition is defined to be the one that minimizes the total number of representative SNPs. Our goal is to find the optimal block partition for all n SNPs.
Define Sj to be the number of representative SNPs for the optimal block partition of the first j SNPs, r1, r2, . . . , rj and set S0 = 0. Then, applying dynamic programming theory,
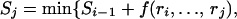 |
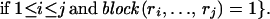 |
Using this recursion, we can design a dynamic programming algorithm to compute the minimum number of representative SNPs for the optimal block partition of all of the n SNPs.
In practice, there may exist several block partitions that give the minimum number of representative SNPs. We want to find the partition with the minimum number of blocks. Let Cj be the minimum number of blocks of all of the block partitions requiring Sj representative SNPs in the first j SNPs and set C0 = 0. Then, applying dynamic programming theory again,
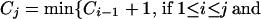 |
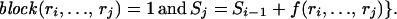 |
By this recursion, the minimum number of blocks in the partition, Cn, can be computed.
The above dynamic programming algorithm can easily be adapted to other measures of block quality. Corresponding to the haplotype diversity measure of Clayton (12), we let f(⋅) be the minimum number of SNPs required to explain β percent of the total haplotype diversity in the block. The dynamic programming algorithm described above is still valid.
The problem of finding the minimum number of representative SNPs within a block to uniquely distinguish all of the haplotypes is known as the MINIMUM TEST SET problem, which has been proven to be NP-Complete (13). This is to say that there is no polynomial time algorithm that guarantees one's finding the optimal solution for any input. However, in this application, the minimum number of representative SNPs in a block is generally small, so we can enumerate all of the possible SNP combinations to find the minimum set.
Results
Haplotype Block Partition Based On Coverage.
We apply our dynamic programming algorithm to the haplotype data for chromosome 21 of Patil et al. (6). The data contain 20 haplotypes and each haplotype contains 24,047 SNPs spanning 32.4 Mb of chromosome 21. The minor allele frequency at each marker locus is at least 10%. Using our algorithm with the same criteria as in Patil et al. (6) with α = 80%, a total of 3,582 representative SNPs and a total of 2,575 blocks are required to define the haplotype patterns. In contrast, Patil et al. (6) identified a total of 4,563 representative SNPs and a total of 4,135 blocks to define the haplotype structure. Our dynamic programming algorithm reduces these numbers by 21.5% and 37.7%, respectively. The properties of blocks are given in Table 1. For comparison, we also give the results of Patil et al. (6) in the same table. The dynamic programming algorithm defines a total of 742 blocks containing more than 10 SNPs per block. The blocks with more than 10 SNPs account for 28.8% of all of the blocks. The average number of SNPs for these blocks is 24.10. The largest block contains 128 common SNPs spanning 140 kb along the chromosome, which is larger than the largest block (containing 114 SNPs spanning 115 kb) identified by Patil et al. (6). The number of blocks that contain more than 10 SNPs is increased by 26.0% (from 589 to 742). The number of blocks that contain less than 3 SNPs is reduced by 56.8% (from 2,138 to 924). On average, there are 3.05 common haplotypes per block, which is slightly larger than the 2.72 common haplotypes defined by Patil et al. (6). Fig. 1 gives the number of blocks as a function of (a) the number of SNPs in a block and (b) the genomic length of the block. Note that if at least α percent of the unambiguous haplotypes in a block are the same, no SNPs are required to distinguish them.
Table 1.
The comparison of properties of haplotype blocks defined by the dynamic programming algorithm and the greedy algorithm of Patil et al. (6) with 80% coverage
| Method | Common SNPs/block | No. of blocks | No. of blocks requiring ≥1 SNPs | Average size/block, kb | Average no. common haplotypes/block | All blocks, % | Common SNPs, % |
|---|---|---|---|---|---|---|---|
| Dynamic programming | >10 | 742 | 738 | 24.27 | 4.23 | 28.8 | 75.5 |
| 3–10 | 909 | 842 | 7.30 | 3.03 | 35.3 | 19.5 | |
| <3 | 924 | 274 | 0.73 | 2.12 | 35.9 | 5.0 | |
| Total | 2,575 | 1,854 | 12.58 | 3.05 | 100.0 | 100.0 | |
| Greedy | >10 | 589 | 589 | 23.90 | 3.75 | 14.2 | 56.8 |
| 3–10 | 1,408 | 1,396 | 8.52 | 2.92 | 34.1 | 30.7 | |
| <3 | 2,138 | 1,776 | 2.96 | 2.30 | 51.7 | 12.4 | |
| Total | 4,135 | 3,761 | 7.83 | 2.72 | 100.0 | 100.0 |
Common SNPs are the 24,047 SNPs with minor allele frequency ≥0.1. Common haplotypes are the haplotypes in each block present more than once in the sample of 20 chromosomes.
Figure 1.

The number of blocks versus the size of blocks, using 80% coverage. (a) The number of blocks as a function of the number of SNPs in a block using a window of two SNPs. The data points are drawn at the the center of the window, except that the last point is the number of blocks containing at least 25 SNPs. (b) The number of blocks as the function of the genomic length of the block, using a window of 2,000 kb. The data points are drawn at the center of the window except that the last point is the number of blocks spanning at least 25,000 kb along the chromosome.
We statistically test whether the break points of the blocks follow a Poisson process. If they follow a Poisson process, the break points should be uniform samples along the region given the number of break points. The SNPs form four contigs (Fig. 2): (a) NT_002836, (b) NT_001035, (c) NT_003545, and (d) NT_002835. Fig. 2 gives the Q-Q plot for the uniform distribution on each contig and the positions of the break points. If the break points follow a uniform distribution, the Q-Q plot should be close to the line y = x. There does seem to be a general agreement of the curve with the line y = x. However, we test this hypothesis by using the two-sided Kolomogorov–Smirnov test (14) on the four contigs. The corresponding P values for the four contigs are 0.0002, 0.7294, 10−6, and 0.0083, respectively. The nonsignificant result for the second contig may be due to the relatively small number of block break points (24) in this contig. In the future, we will use r-scan statistics to identify statistically significant regions of dense and sparse block break points.
Figure 2.

The Q-Q plots for the uniform distribution and the positions of the block break points for four contigs, using 80% coverage for contigs (a) NT_002836, (b) NT_0 01035, (c) NT_00354.5, and (d) NT_002835.
To test whether the results for the block partition are statistically significant, we generate 1,000 samples of 20 chromosomes by permuting the unambiguous alleles at each SNP locu. We apply the dynamic programming algorithm to each sample of 20 permuted chromosomes and record the number of representative SNPs required, the number of blocks, the size of the largest block, and the number of blocks containing more than ten SNPs. The histograms for these quantitates for the 1,000 simulated samples are given in Fig. 3. The four quantities for the real haplotype data are far out of the range of the corresponding quantities for the 1,000 simulated samples.
Figure 3.

The histograms for (a) the number of blocks, (b) the number of representative SNPs, (c) the size of the largest block, and (d) the number of blocks with more than ten SNPs for the 1,000 permuted samples. α = 80%.
We investigate the influence of coverage α in the dynamic programming algorithm on the block patterns. We change the coverage from 80% to 90% and 70%. When the coverage is increased to 90%, the total number of representative SNPs required to define these blocks increases to 7,536. The total number of blocks increases to 3,573. The size of the largest block decreases to 97 SNPs. When the coverage is decreased to 70%, the total number of SNPs required to define these blocks decreases to 1,977 and the total number of blocks decreases to 2,381 with the largest block containing 177 SNPs spanning 177 kb. The properties of the blocks for α = 90% and α = 70% are given in Tables 2 and 3, respectively.
Table 2.
The properties of haplotype blocks defined by the dynamic programming algorithm with 90% coverage
| Common SNPs/block | No. of block | No. of blocks requiring ≥1 SNPs | Average size/block, kb | Average no. common haplotypes/block | All blocks, % | Common SNPs, % |
|---|---|---|---|---|---|---|
| >10 | 669 | 668 | 17.41 | 4.45 | 18.7 | 52.9 |
| 3–10 | 1,724 | 1,722 | 6.28 | 3.51 | 48.3 | 40.0 |
| <3 | 1,180 | 1,127 | 0.84 | 2.23 | 33.0 | 7.1 |
| Total | 3,573 | 3,517 | 9.07 | 3.27 | 100.0 | 100.0 |
Common SNPs are the 24,047 SNPs with minor allele frequency ≥ 0.1. Common haplotypes are the haplotypes in each block present more than once in the sample of 20 chromosomes.
Table 3.
The properties of haplotype blocks defined by the dynamic programming algorithm with 70% coverage
| Common SNPs/block | No. of block | No. of blocks requiring ≥1 SNPs | Average size/block, kb | Average no. common haplotypes/block | All blocks, % | Common SNPs, % |
|---|---|---|---|---|---|---|
| >10 | 650 | 617 | 30.78 | 3.93 | 27.3 | 79.3 |
| 3–10 | 703 | 440 | 7.47 | 2.81 | 29.5 | 14.6 |
| <3 | 1,028 | 117 | 0.60 | 2.17 | 43.2 | 6.1 |
| Total | 2,381 | 1,174 | 13.61 | 2.84 | 100.0 | 100.0 |
Common SNPs are the 24,047 SNPs with minor allele frequency ≥0.1. Common haplotypes are the haplotypes in each block present more than once in the sample of 20 chromosomes.
We compare the block break points for α = 80% with the block break points for α = 90%. Of the block break points for α = 80%, 48.1% (total of 1,238) are conserved for α = 90%, and 76.9% (total of 1,979) of the block break points are within a distance of one SNP. This result means that the block structures derived from these two criteria are comparable to each other.
Haplotype Block Partition Based On Haplotype Diversity.
To show that the dynamic programming algorithm can be easily adapted to other measures of haplotype quality in a block, we apply our dynamic programming algorithm to define a set of blocks with the minimum total number of SNPs required to explain 95% of haplotype diversity in each block as developed by Clayton (12). It is worth noting that the criteria used in Patil et al. (6) and Clayton (12) are related. For the same set of haplotypes, if a subset of SNPs can distinguish all of the different types of haplotypes in a block, the proportion of diversity explained by the subset of SNPs is 100%. The properties of blocks defined based on haplotype diversity are given in Table 4. As above, we require that at least 80% of the unambiguous haplotypes are represented more than once in each block. A total of 3,982 SNPs and a total of 1,884 blocks are required to explain 95% of the haplotype diversity in each block. The largest block contains 124 SNPs spanning 125 kb along chromosome 21. More than 10 SNPs are found in 42.7% of the blocks, and together they cover 78.6% of all of the SNPs in the study. Only 15.6% of the blocks contain less than three SNPs and together they cover only 1.6% of the SNPs. Comparing the block break points by using this criterion with those based on coverage for α = 80%, 64.6% (total of 1,218) of the block break points based on haplotype diversity are conserved and 83.4% (total of 1,571) are within a distance of one SNP.
Table 4.
The properties of haplotype blocks defined by the dynamic programming algorithm with 80% coverage and 95% proportion of diversity
| Common SNPs/ block | No. of blocks | Average size/ block, kb | Average no. common haplotypes/block | All blocks, % | Common SNPs, % |
|---|---|---|---|---|---|
| >10 | 804 | 24.60 | 4.48 | 42.7 | 78.6 |
| 3–10 | 786 | 8.88 | 3.49 | 41.7 | 19.8 |
| <3 | 294 | 0.85 | 2.15 | 15.6 | 1.6 |
| Total | 1,884 | 17.20 | 3.70 | 100.0 | 100.0 |
Common SNPs are the 24,047 SNPs with minor allele frequency ≥ 0.1. Common haplotypes are the haplotypes in each block present more than once in the sample of 20 chromosomes.
We generate 1,000 samples of 20 chromosomes by permutation as above and apply the criterion for haplotype diversity to the randomized samples. The results are similar to the results based on coverage (data not shown) and are statistically significant.
Discussion
We develop a dynamic programming algorithm for haplotype block partitioning with the minimum number of representative SNPs required to account for most of the haplotype block quality in each block. The algorithm can be used for any measures of block quality. The measure should depend on the purpose of a specific application. Using the same criteria as in Patil et al. (6), the dynamic programming algorithm reduces the numbers of representative SNPs and the number of blocks by 21.5% and 37.7%, respectively, for the haplotype data on chromosome 21. The results for the block partition are highly statistically significant. As indicated in Patil et al. (6), knowing the haplotype structure is extremely important for whole-genome association studies. In association studies, we compare the haplotype frequencies in unrelated cases to those in the controls. Instead of genotyping all of the SNP markers, one may wish to use only the genotype information on the representative SNPs. Only about 14.9% (3,582) of all of the SNPs (24,047) can account for 80% of the common haplotypes in each block. Thus, studying the representative SNPs can dramatically reduce the time and effort for genotyping, without losing much haplotype information. Haplotype diversity is widely used in population genetics studies. We also partition chromosome 21 into haplotype blocks based on haplotype diversity. Of all of the SNPs (24,047), 16.6% (3,982) can account for 95% of the haplotype diversity in each block. As in Patil et al. (6), we partition the chromosome into blocks based on the genetic information without referring to the biological basis for generating such blocks. Thus, blocks may not have definite boundaries and caution should be used when inferring the biological meaning of the break points.
Acknowledgments
We thank Perlegen Sciences for making their chromosome 21 data available to the public. We are greatly indebted to Dr. David A. Hinds of Perlegen Sciences for help in organizing the data, explaining the concept of ambiguous and unambiguous haplotypes, and pointing out some inconsistencies between the criteria used in our original program and the criteria used in their paper (6). This research was partially supported by National Institutes of Health Grant DK53392, National Science Foundation Information Technology Research Grant EIA-0112934, and the University of Southern California.
Abbreviation
- SNP
single nucleotide polymorphism
References
- 1.Kruglyak L, Nickerson D A. Nat Genet. 2001;27:234–236. doi: 10.1038/85776. [DOI] [PubMed] [Google Scholar]
- 2.Abecasis G R, Noguchi E, Heinzmann A, Traherne J A, Bhattacharyya S, Leaves N I, Anderson G G, Zhang Y M, Lench N J, Carey A, et al. Am J Hum Genet. 2001;68:191–197. doi: 10.1086/316944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reich D E, Cargill M, Bolk S, Ireland J, Sabeti P C, Richter D J, Lavery T, Kouyoumjian R, Farhadian S F, Ward R, Lander E S. Nature (London) 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- 4.Daly M J, Rioux J D, Schaffner S E, Hudson T J, Lander E S. Nat Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- 5.Rioux J D, Daly M J, Silverberg M S, Lindblad K, Steinhart H, Cohen Z, Delmonte T, Kocher K, Miller K, Guschwan S, et al. Nat Genet. 2001;29:223–228. doi: 10.1038/ng1001-223. [DOI] [PubMed] [Google Scholar]
- 6.Patil N, Berno A J, Hinds D A, Barrett W A, Doshi J M, Hacker C R, Kautzer C R, Lee D H, Marjoribanks C, McDonough D P, et al. Science. 2001;294:1719–1723. doi: 10.1126/science.1065573. [DOI] [PubMed] [Google Scholar]
- 7.Clark A G, Weiss K M, Nickerson D A, Taylor S L, Buchanan A, Stengard J, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, et al. Am J Hum Genet. 1998;63:595–612. doi: 10.1086/301977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Excoffier L, Slatkin M. Mol Biol Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- 9.Hawley M E, Kidd K K. J Hered. 1995;86:409–411. doi: 10.1093/oxfordjournals.jhered.a111613. [DOI] [PubMed] [Google Scholar]
- 10.Stephens M, Smith N J, Donnelly P. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niu T H, Qin Z S, Xu X P, Liu J S. Am J Hum Genet. 2001;70:157–169. doi: 10.1086/338446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clayton, D. (2001) http://www.nature.com/ng/journal/v29/n2/extref/ng1001-233-S10.pdf.
- 13.Garey M R, Johnson D S. Computers and Intractability. New York: Freeman; 1979. p. 222. [Google Scholar]
- 14.Bickel P J, Doksum K A. Mathematical Statistics, Basic Ideas and Selected Ideas. Englewood Cliffs, NJ: Prentice–Hall; 1977. [Google Scholar]