

Abstract
The p53 tumor suppressor promotes cell cycle arrest or apoptosis in response to diverse stress stimuli. p53-mediated cell death depends in large part on transcriptional up-regulation of target genes. One of these targets, P53-induced protein with a death domain (PIDD), was shown to function as a mediator of p53-dependent apoptosis. Here we show that PIDD is a cytoplasmic protein, and that PIDD-induced apoptosis and growth suppression in embryonic fibroblasts depend on the adaptor protein receptor-interacting protein (RIP)-associated ICH-1/CED-3 homologous protein with a death domain (RAIDD). We provide evidence that PIDD-induced cell death is associated with the early activation of caspase-2 and later activation of caspase-3 and -7. Our results also show that caspase-2-/-, in contrast to RAIDD-/-, mouse embryonic fibroblasts, are only partially resistant to PIDD. Our findings suggest that caspase-2 contributes to PIDD-mediated cell death, but that it is not the sole effector of this pathway.
Keywords: caspase-2
The tumor suppressor p53 is a sequence-specific transcription factor that promotes cell cycle arrest or apoptosis in response to cellular stress (1). Transcriptional activation of the p21WAF1 cyclin-dependent kinase inhibitor plays a key role in the induction of cell cycle arrest by p53 (2). p53-dependent apoptosis is regulated, at least in part, by transcriptional activation of its target genes (3), and this process highly depends on cytochrome c release and the Apaf-1/caspase-9 activation pathway (4, 5). Although a number of candidate p53-effector molecules have been reported, it is yet unclear whether each contributes a part of the full response, or whether specific subsets of these genes are required for death in different cell types or in response to different signals (3).
Among the identified apoptotic effectors of p53, P53-induced protein with a death domain (PIDD)/leucine-rich DD (LRDD) is a 915-aa protein in mice (910 aa in humans) containing seven tandem LR repeats in the N terminus and a DD in the C terminus (6, 7). The dual domain structure of PIDD suggests that it may function as a key adapter protein that links additional components of the p53 apoptosis pathway. Using the method of differential display, PIDD was identified as a p53-up-regulated gene in a p53-null Friend-virus-transformed mouse erythroleukemia cell line (DP16.1/p53ts) that stably expresses a temperature-sensitive (ts) Trp-53 mutant allele. DP16.1/p53ts cells undergo apoptosis after expression of the wild-type p53 conformation at 32°C. PIDD mRNA is induced by γ-irradiation in a p53-dependent manner, and the basal level of PIDD mRNA depends on p53 gene status. Overexpression of PIDD also inhibits cell growth in a p53-like manner by inducing apoptosis. Antisense inhibition of PIDD expression was shown to attenuate apoptosis in response to p53 activation and DNA damage, suggesting that PIDD expression is required for p53-dependent death (7).
Recently, PIDD was found to be present in a large protein complex containing caspase-2 and the adapter protein receptor-interacting protein (RIP)-associated ICH-1/CED-3 homologous protein with a DD (RAIDD) (8). The DD of PIDD was shown to interact with the DD of RAIDD, which, in turn associated with caspase-2 through the caspase-recruitment domain (CARD). This study concluded that PIDD was involved in the activation of caspase-2. The role of caspase-2 in apoptosis is uncertain; caspase-2-deficient and wild-type cells (thymocytes and B and T lymphoblasts) are equally sensitive to diverse apoptotic stimuli (9), yet RNA interference experiments and caspase-2 inhibitors have implicated caspase-2 in stress-induced apoptosis in some cell types (10, 11). Caspase-2 can act upstream of the mitochondria by inducing Bid cleavage, Bax translocation to the mitochondria, and subsequent cytochrome c release (10-12). It can also directly induce the release of cytochrome c from purified mitochondria (11, 12). Thus, it appears that under certain circumstances, caspase-2 activation may be an early event that engages the mitochondrial apoptotic pathway. The role of RAIDD in apoptosis is also uncertain. RAIDD was initially identified as a death adapter protein capable of binding RIP and caspase-2 through its DD and CARD, respectively (13, 14). In the presence of RIP and TRADD, RAIDD was found to promote apoptosis by recruiting caspase-2 to TNF receptor 1. Dominant negative forms of RAIDD, however, did not abrogate TNF-mediated cell death (14).
In this study, we generated mice carrying a null mutation in the RAIDD gene. We report that RAIDD-deficient mice have normal development and lymphocyte populations, and that RAIDD-/- thymocytes are sensitive to various cytotoxic agents. RAIDD-/- mouse embryonic fibroblasts (MEFs) are sensitive to TNF and cycloheximide; thus, RAIDD is not necessary for TNF-mediated apoptosis in embryonic fibroblasts. To determine the functional significance of the PIDD-RAIDD-caspase-2 complex in promoting cell death, we examined the apoptotic activity of PIDD in RAIDD-/- and caspase-2-/- MEFs. We show that PIDD-induced apoptosis and growth suppression depend entirely on RAIDD. We provide evidence that PIDD-induced cell death is associated with the early activation of caspase-2 and later activation of caspase-3 and -7. The early activation of caspase-2 suggests that it may act as a proximal effector of the apoptotic pathway initiated by PIDD and is consistent with recent evidence that caspase-2 functions as an initiator caspase during apoptosis. Our results also show that caspase-2-/- MEFs, in contrast to RAIDD-/- MEFs, are only partially resistant to PIDD, suggesting that, although caspase-2 contributes to PIDD-mediated cell death, it is not the sole effector of this pathway.
Experimental Procedures
Generation of RAIDD-/- Mice. For details of targeting constructs and generation of RAIDD-null mice, see Supporting Text, which is published, as supporting information on the PNAS web site.
Cell Culture. H460 cells were cultured in RPMI medium 1640 with 10% FCS. All other cells were maintained in DMEM with 10% FCS. MEFs from RAIDD-/-, caspase-2-/- (9), and caspase-8-/- (15) mice and their control littermates (+/+) were transformed by retroviral infection with pBABE-E1A/Ras, a kind gift from Maria Soengas (University of Michigan Comprehensive Cancer Center). The Fas-associated DD (FADD)-/- and corresponding +/+ MEFs were established in culture using the 3T3 protocol (16). The Apaf1-/- MEFs were transformed with Myc and Ras (4, 17).
Antibody Production. Rabbits were immunized with a His-tagged mouse PIDD fragment (residues 141-915) purified from BL-21 (DE3) bacteria transformed with the construct pET 28a-mouse PIDD (mPIDD) (Invitrogen). Briefly, after induction with isopropyl-1-thio-β-d-galactopyranoside, bacteria were collected by centrifugation and lysed, and recombinant protein was purified on a resin of Ni2+ affinity by standard affinity purification procedures. The fractions containing purified recombinant PIDD were dialyzed overnight, lyophilized to concentrate the protein, resuspended in PBS, and used to immunize rabbits. The resulting anti-PIDD rabbit serum was purified on a Protein A/G Sepharose column (Amersham Pharmacia Biosciences). Sera were mixed with borate buffer (25 mM sodium borate/100 mM boric acid/75 mM NaCl) and loaded onto the column at a slow flow rate. The column was then washed with borate buffer, and purified antibody was eluted by using 0.1 M glycine buffer. The antibody fractions were neutralized with 1 M Tris buffer and concentrated by using a centrifugal filter device (Millipore).
Plasmids. Mouse wild-type p53 cDNA was excised from pECM53 (18) and cloned into the EcoRI and XbaI sites of pcDNA3.1. Mouse PIDD cDNA was PCR-amplified from pcDNA3.1-mPIDD (7) and cloned into pcDNA3.1-Nterm-Myc/his to generate a 5′ Myc-tagged mPIDD construct. 3′ Myc-tagged mouse PIDD plasmid was generated by PCR amplification from pcDNA3.1-mPIDD using primers that included a Myc-tag before the stop codon. pMIG-IRES-GFP (IRES, internal ribosome entry site) served as the expression vector for mPIDD. PIDD cDNA was excised from pcDNA3.1-mPIDD (7) and cloned into the XhoI and HpaI sites of pMIG.
Retroviral Infection. 293TV cells (3 × 105) were plated in a 6-cm dish, incubated for 24 h, and then transfected by FuGENE 6 (Roche Applied Science, Indianapolis) with 1 μg of a retroviral plasmid and 1 μg of pCL-ECO (15 h at 37°C). After transfection, media were removed and replaced with 5 ml of DMEM-H21 supplemented with 10% FCS. After 48 h, the virus-containing medium was collected, filtered (0.45 μm, Millipore), and used for infection immediately or frozen at -80°C. Target fibroblasts were plated at 3 × 105 per 10-cm dish and incubated overnight. For infections, the culture medium was replaced with a mixture of fresh media and viral supernatant supplemented with 8 μg/ml polybrene (Sigma) and incubated at 37°C for 12 h.
DNA Transfection in Saos-2 Cells. Plasmids were transfected with Lipofectamine2000 (Invitrogen) according to the manufacturer's protocol 1 day after plating at 1.1 × 106 cells per 10-cm plate.
Immunoprecipitation, Immunoblot Analysis, and Caspase Activity Assays. For details, see Supporting Text.
Subcellular Localization of PIDD by Immunofluorescence. Saos-2 cells were seeded onto Lab-Tek Chamber Slides (Invitrogen) at 2.4 × 105 cells per chamber. The next day, cells were transfected with pcDNA3.1-N-Myc/his or pcDNA3.1-N-Myc/his-mPIDD by using Lipofectamine2000. At 24 h after transfection, cells were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature (RT). Fixed cells were washed with PBS and then permeabilized with 1% Triton X-100 in PBS for 10 min. The chambers were treated with anti-Myc monoclonal antibody and/or anti-PIDD polyclonal antibody, diluted in 3% BSA/PBS for 1 h at RT. They were then washed twice with 0.02% Tween 20 and 1% BSA in PBS, followed by incubation with a Cy3-conjugated anti-mouse and/or FITC-conjugated anti-rabbit (Jackson ImmunoResearch) for 30 min at RT. Cells were examined under a Zeiss LSM 410 laser-scanning fluorescence microscope.
Clonogenic Assays. For long-term colony assays, established MEFs derived from RAIDD-/-, FADD-/-, caspase-2-/-, and caspase-8-/- mice were plated at 3 × 105 cells per 10-cm plate. The next day, MEFs were coinfected with pMIG-IRES-GFP or pMIG-PIDD-IRES-GFP together with pBabe-Hygro using viral supernatants at a volume ratio of 10:1. At 12 h after infection, media was replaced with DMEM with 10% FCS. After a recovery period of 12 h, selection in hygromycin (Invitrogen) was started. Resistant colonies were counted 7 or 8 days after the start of drug selection. For clonogenic assays involving Apaf1-/- MEFs, cells were plated at 9 × 105 per 10-cm plate. The next day, cells were transfected overnight (FuGENE 6) with pcDNA3.1 or pcDNA3.1-PIDD together with a plasmid expressing zeomycin resistance. The next day, cells were split and incubated overnight at 37°C, and then selection with zeocin (Invitrogen) was started. Resistant colonies were counted 7 days after the start of selection.
Apoptosis Assay. Two assays were used for detection of apoptotic cells, annexin V staining and assessment of morphology. DP16.1 cells were cultured at 3 × 106 per ml and transfected by using FuGENE 6 for 3 h with pcDNA3.1, pcDNA3.1-mp53, or pcDNA3.1-mPIDD together with a plasmid expressing CD20. After 48 h, cells were stained with anti-CD20-FITC (BD Biosciences) according to the manufacturer's protocol, and apoptosis was assessed by annexin V-phycoerythrin staining of CD20-positive cells.
For assessment of apoptosis by morphology, E1A/ras-transformed RAIDD-/- and RAIDD+/+ MEFs were plated at 2 × 104 cells per chamber. The next day, MEFs were infected for 12 h with pMIG or pMIG-mPIDD. At 48 h after infection, MEFs were fixed and permeabilized as described above and visualized by Hoechst 33258 staining. MEFs were also treated with a combination of 2 μg/ml cycloheximide (Sigma) and 10 ng/ml recombinant murine TNF-α (R & D Systems) before assessment of apoptotic morphology. Cells were examined for apoptotic morphology under a Leica DM LB microscope.
Results
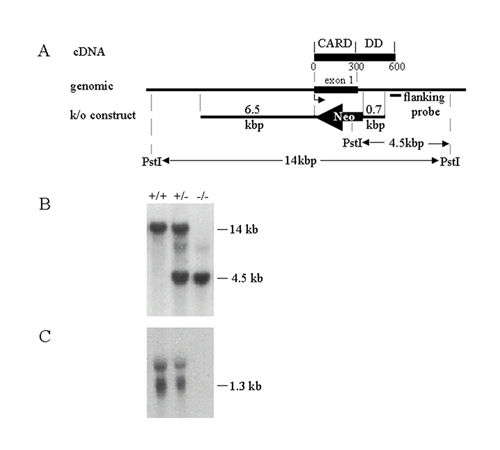
Generation of RAIDD-/- Mutant Mice. The targeting strategy to generate RAIDD-deficient mice is described in Supporting Text. Southern and Northern blots were performed to confirm the presence of the rearranged allele and lack of RAIDD RNA expression in mouse tissues (see Fig. 6, which is published as supporting information on the PNAS web site). RAIDD-deficient mice have normal development and lymphocyte populations (data not shown). Absence of RAIDD is also not detrimental to normal lymphocyte proliferation in response to various mitogens, including Con A, anti-CD3, anti-CD3 + anti-CD28, and LPS (data not shown). In addition, we found that RAIDD-/- thymocytes were sensitive to various cytotoxic agents, including dexamethasone, γ-radiation, etoposide, and staurosporine (data not shown).
PIDD Protein Expression. To investigate PIDD protein expression, we generated a polyclonal antibody against a portion of the mouse PIDD protein (residues 141-915). Antibody specificity was validated by the results of Western blotting and immunoprecipitation presented in Fig. 1. Ectopically expressed Myc-tagged PIDD was detected both with a Myc monoclonal antibody and our PIDD polyclonal antibody (Fig. 1 A). In addition, endogenous PIDD, which migrated as a protein species of ≈110 kDa, was detected with the polyclonal antibody. We examined PIDD protein induction in DP16.1/p53ts cells in response to p53 activation at 32°C by immunoprecipitation/Western blot analysis. Endogenous PIDD protein levels increased within 8 h of p53 activation and remained elevated for 16 h (Fig. 1B). This pattern of expression is similar to that of PIDD mRNA in DP16.1/p53ts cells (7). Immunofluorescent staining of ectopically expressed Myc-PIDD protein using our PIDD antibody or Myc antibody revealed cytoplasmic localization in Saos-2 cells (Fig. 1C).
Fig. 1.

PIDD protein expression. (A) Saos-2 cells were transfected with pcDNA3.1-N-myc/his (vector), pcDNA3.1-N-myc/his-mPIDD (5′Myc-PIDD), or pcDNA3.1-mPIDD-Myc (3′Myc-PIDD). After 48 h, cell extracts were resolved by SDS/PAGE and analyzed by immunoblotting by using Myc monoclonal or PIDD polyclonal antibodies. The blot was reprobed with an antibody against β-actin as a loading control. (B) DP16.1/p53ts cells were incubated at 32°C to activate p53 for the times indicated. Cell extracts were immunoprecipitated with PIDD antibodies and analyzed by immunoblotting with the same antibodies. (C) Saos-2 cells were transiently transfected with 5′Myc-PIDD or empty vector. Immunostaining was carried out 24 h later with PIDD or Myc antibodies and analyzed by confocal microscopy. (D) H460 cells were treated with 200 ng/ml adriamycin for the indicated time periods. The level of endogenous PIDD protein was determined by immunoblotting with PIDD antibodies. The blot was reprobed with an antibody against β-actin as a loading control. (E) MEFs were exposed to γ radiation (6 Gy) and at the indicated times after irradiation, cell extracts were prepared and subjected to immunoblotting with PIDD antibodies.
We next examined endogenous PIDD expression in response to DNA damage in p53-expressing human H460 lung cancer cells and in MEFs. Treatment of H460 cells with adriamycin resulted in PIDD protein accumulation by 16 h (Fig. 1D). γ-Irradiated MEFs showed PIDD accumulation within 4 h of treatment (Fig. 1E). Interestingly, a smaller fragment of ≈55 kDa was observed in both H460 cells and in MEFs after DNA damage and likely represents a specific cleavage fragment similar to one reported previously (8).
PIDD-Induced Apoptosis Depends on RAIDD. We showed previously that ectopically expressed PIDD promotes apoptosis in K562 cells (7). PIDD also promotes apoptosis in p53-null DP16.1 murine erythroleukemia cells (Fig. 2A) on the basis of Annexin V staining as well as in Saos-2 cells by nuclear morphology (data not shown). PIDD was shown recently to interact with RAIDD and to promote the activation of caspase-2 (8). To determine whether the apoptotic function of PIDD depends on its interaction with RAIDD, we compared PIDD-induced apoptosis in RAIDD+/+ and RAIDD-/- MEFs. MEFs from RAIDD-deficient and littermate controls were first transformed with the E1A and Ras oncogenes to increase their sensitivity to undergo apoptosis. Transformed MEFs were then retrovirally infected with empty vector (pMIG) or pMIG-PIDD and assessed for apoptosis on the basis of morphological changes, including chromatin condensation and nuclear fragmentation as revealed by staining with Hoechst 33258 (Fig. 2B). The data are summarized in Fig. 2C and show that RAIDD-/- MEFs are completely resistant to PIDD-induced apoptosis. We confirmed that RAIDD protein was expressed in RAIDD+/+ MEFs, that it was absent in RAIDD-/- MEFs (Fig. 3A), and that the level of ectopic PIDD expression was similar in both cell types (Fig. 3B). A similar pattern of PIDD cleavage fragments was also observed in pMIG-PIDD-infected RAIDD+/+ and RAIDD-/- MEFS (Fig. 3B). Both RAIDD+/+ and RAIDD-/- MEFs were equally sensitive to TNF-α-induced apoptosis (Fig. 2C).
Fig. 2.

PIDD expression induces apoptosis. (A) DP16.1 cells were transfected with pcDNA3.1, pcDNA3.1-PIDD, or pcDNA3.1-p53, together with a plasmid expressing the CD20 receptor. Forty-eight hours later, cells were stained with an anti-CD20-FITC antibody and Annexin V-phycoerythrin. Apoptosis was then assessed by flow cytometry. CD20-positive cells were gated, and the data shown (mean ± SEM in three independent experiments) represent the percentage of CD20-positive cells that are also Annexin V-positive. (B) E1A/Ras-transformed RAIDD+/+ and RAIDD-/- MEFs were infected with pMIG or pMIGPIDD or treated with TNF (10 ng/ml) and cycloheximide (2 μg/ml); infected cells were fixed 48 h later, stained with Hoechst 33258, and visualized by microscopy. The infection efficiency was determined by the proportion of cells that became GFP-positive 48 h after infection and ranged between 83% and 94%. TNF-treated cells were examined 3 h after treatment. Cells with condensed chromatin or nuclear fragmentation were scored as apoptotic, and the data are summarized in C.
Fig. 3.

Activation of caspase-2, -3, and -7 in PIDD-expressing MEFs depends on RAIDD. Western blots were performed with the indicated antibodies on lysates prepared from E1A/Ras-transformed RAIDD+/+ and RAIDD-/- MEFs after infection with pMIG or pMIG-PIDD (A and B). The infection efficiency was determined by the proportion of cells that became GFP-positive 24 h after infection. The infection efficiency of RAIDD+/+ MEFs was 98% with pMIG and 91% with pMIG-PIDD; the infection efficiency of RAIDD-/- MEFs was 97% with pMIG and 92% with pMIG-PIDD. (C) Caspase activity assays were performed as described in Experimental Procedures. (Upper) The histograms show the fold increase in activity measured in RAIDD+/+ cells 48 h after infection with pMIG-PIDD compared with pMIG infection. (Lower) The change in activity at different time points after infection is shown.
Activation of Caspase-2, -3, and -7 in PIDD-Expressing MEFs. The resistance of RAIDD-/- MEFS to PIDD-induced apoptosis represents the only phenotype we have been able to measure thus far in cells that lack RAIDD and highlights an important role for RAIDD in PIDD-induced apoptosis. We proceeded, therefore, to characterize further the apoptotic response of PIDD-expressing MEFS. Western blots were performed to monitor the processing of procaspase-2, -3, -7, -8, and -9 in RAIDD+/+ and RAIDD-/- MEFs at different times after infection with pMIG-PIDD or pMIG (Fig. 3B). Elevated PIDD expression was observed in the pMIG-PIDD-infected cells within 24 h after infection and remained high after 72 h. Strikingly, procaspase-2 processing occurred early after PIDD expression only in RAIDD+/+ MEFs and not in RAIDD-/- MEFs and preceded the processing of procapsase-3 and -7 (Fig. 3B). Processing of procaspase-8 and -9 was not detected in PIDD-expressing MEFs of either genotype (Fig. 3B). Hence, PIDD expression leads to the processing of procaspase-2, -3, and -7, and this processing depends on RAIDD. Complementary experiments performed with fluorogenic peptide substrates demonstrated an increase (2.6-fold) in caspase-3/-7 activation (DEVD cleavage) in lysates prepared from PIDD-expressing +/+ MEFs and a lack of caspase-8 (IETD cleavage) and caspase-9 (LEHD cleavage) activity (Fig. 3C). We detected only a slight increase (1.2-fold) in caspase-2 activity (VDVAD cleavage) in the PIDD-expressing +/+ MEFs (Fig. 3C), even though procaspase-2 was completely processed, as reflected by the disappearance of the full length protein on Western blots. As a control, we treated +/+ MEFs with TNF-α and detected activation of caspase-2 (3.3-fold), caspase-3/-7 (8.9-fold), caspase-8 (1.8-fold), and caspase-9 (2.0-fold).
PIDD-Dependent Growth Suppression Depends on RAIDD and Partially on Caspase-2 and -8. To investigate further the involvement of RAIDD in PIDD-induced cell death, we compared the ability of PIDD to inhibit the growth of E1A/Ras-transformed RAIDD+/+ and RAIDD-/- MEFs. We used a colony-forming assay, because long-term clonogenic assays provide a more stringent measurement of cell survival than short-term assays. MEFs (RAIDD+/+ and RAIDD-/-) were coinfected with retroviruses expressing resistance to hygromycin (pBabe-hygro) together with pMIG or pMIG-PIDD. Cells were placed under hygromycin selection 1 day after infection, and we enumerated the drug-resistant colonies ≈1 week later. In multiple independent experiments, PIDD-dependent growth suppression was completely abolished in the absence of RAIDD (Fig. 4A). The requirement of RAIDD for PIDD-dependent apoptosis (Figs. 2 and 3) and growth suppression (Fig. 4) indicates that the RAIDD adapter molecule plays an essential role in mediating PIDD-dependent death in MEFs.
Fig. 4.

PIDD suppresses cell growth. Transformed RAIDD-/- MEFs (A), caspase-2-/- MEFs (B), FADD-/- MEFs (D), caspase-8-/- MEFs (E), and corresponding MEFs from +/+ littermate controls were coinfected with pMIG-IRES-GFP (pMIG) or pMIG-PIDD-IRES-GFP (PIDD) together with retroviruses expressing hygromycin resistance (pBabe-Hygro); 24 h later, MEFs were treated with hygromycin, and hygromycin-resistant colonies were enumerated 7-8 days after the start of drug selection. The results represent the mean of at least three independent experiments (infections) and are presented as a percentage of the colonies obtained after coinfection with pMIG and pBabe-Hygro. Error bars indicate SEM In A, the number of colonies varied between 341 and 531 colonies per plate in the pMIG-infected +/+ MEFs and between 363 and 664 colonies per plate in the pMIG-infected RAIDD-/- MEFs. Infection efficiency was determined by the proportion of cells that became GFP-positive 48 h after infection with pMIG alone (+/+, 59%; RAIDD-/-, 54%). In B, the number of colonies varied between 450 and 860 per plate in the pMIG-infected +/+ MEFs and between 360 and 800 per plate in the pMIG-infected caspase-2-/- MEFs. In D, the number of colonies varied between 140 and 200 per plate in the pMIG-infected +/+ MEFs and between 180 and 260 per plate in the pMIG-infected FADD-/- MEFs. The infection efficiency was 27% for the +/+ MEFs and 34% for the FADD-/- MEFs. In E, the number of colonies varied between 770 and 1,080 per plate in the pMIG-infected +/+ MEFs and between 360 and 640 per plate in the pMIG-infected caspase-8-/- MEFs. The infection efficiency was 24% for the +/+ MEFs and 52% for the caspase-8-/- MEFs. (C) Apaf-1-/- and Apaf-1+/+ MEFs were cotransfected with pcDNA3.1 or pcDNA3.1-mPIDD along with a zeomycin selection plasmid. Zeomycin-resistant colonies were enumerated 7 days after the start of drug selection. The number of colonies varied between 170 and 200 per plate in the empty vector-transfected Apaf-1+/+ MEFs and between 120 and 180 per plate in the empty vector-transfected Apaf-1-/- MEFs. Error bars indicate SEM (n = 3).
Having shown that PIDD expression leads to RAIDD-dependent procaspase-2 processing and activation, we proceeded to evaluate the role of caspase-2 in PIDD-mediated growth suppression using caspase-2+/+ and caspase-2-/- MEFs. The ability of PIDD to suppress colony formation was attenuated but not abolished in caspase-2-/- MEFs (Fig. 4B). This suggests that caspase-2 contributes to PIDD-mediated cell death, but that it is not the sole effector of this pathway. Because RAIDD and caspase-2 appear to act upstream of the mitochondria, we sought to determine whether PIDD acted predominantly through the mitochondrial apoptotic pathway. Apaf-1 is considered a central element in the mitochondrial pathways of apoptosis, in response to a wide range of apoptotic stimuli (17). Because Apaf-1-/- MEFs were shown to be partially resistant to a variety of apoptotic stimuli, we examined the susceptibility of these cells to PIDD-induced growth inhibition in a colony assay. Apaf-1-/- MEFs were partially resistant to PIDD-dependent colony suppression (Fig. 4C).
The observation that caspase-2-/- and Apaf-1-/- MEFs are only partially resistant to PIDD-induced colony suppression, whereas RAIDD-/- MEFs are fully resistant, raised the possibility that other initiator caspases may act downstream of PIDD and RAIDD to promote cell death. We therefore tested the ability of PIDD to inhibit the growth of FADD-/- and caspase-8-/- MEFs in a colony assay. Both FADD-/- and caspase-8-/- MEFs were partially resistant to PIDD-dependent colony suppression (Fig. 4 D and E). Partial resistance to PIDD-induced growth suppression was also observed in wild-type MEFs and human SkBr3 cells expressing FADD dominant negative (FADD-DN) (data not shown). These data suggest that FADD and caspase-8, two components of the extrinsic pathway of apoptosis, may contribute to the ability of PIDD to suppress the growth of MEFs. The potential involvement of FADD is notable, because PIDD can also interact with FADD (6) (Fig. 5). We expressed Myc-tagged PIDD in Saos-2 cells and detected binding to endogenous FADD by immunoprecipitation/Western blotting (Fig. 5A). We also detected an interaction between endogenous PIDD and endogenous FADD in H460 cells either left untreated or treated with adriamycin (Fig. 5B). These data demonstrate that PIDD and FADD interact under physiological conditions, and that this interaction is not affected by DNA damage. We estimate that a small proportion of PIDD protein in the H460 cell extract is bound to FADD based on quantitative immunoprecipitation of FADD (data not shown) and the total amount of PIDD protein present in these cells (Fig. 1D).
Fig. 5.

Interaction between PIDD and FADD. (A) Saos-2 cells were transfected with 5′Myc-PIDD or empty vector. Cell extracts were prepared 48 h later and immunoprecipitated with a FADD polyclonal antibody. Immunoprecipitated proteins were separated by SDS/PAGE, and PIDD was detected by Western blotting by using a Myc monoclonal antibody. (B) H460 cells were left untreated or incubated with 200 ng/ml adriamycin for 16 h, and extracts were prepared and immunoprecipitated with purified mouse IgG1 antibody as a control or FADD monoclonal antibody before immunoblotting with PIDD polyclonal antibodies.
To evaluate the possibility that PIDD-induced growth suppression of caspase-2-/- and Apaf-1-/- MEFs is mediated through a pathway that involves FADD and caspase-8, we generated dominant negative FADD (FADD-DN)-expressing caspase-2-/- MEFs and tested these cells in a colony assay. Caspase-2-/- MEFs and caspase-2-/- MEFs expressing FADD-DN were equally sensitive to PIDD-induced cell death (data not shown). Hence, the combined loss of caspase-2 and FADD (by expression of FADD-DN in caspase-2-/- cells) provided no additional survival advantage over that seen with the loss of caspase-2 alone.
Discussion
In this study, we have generated RAIDD-/- mice and used embryonic fibroblasts from these mice to investigate the functional significance of the recently reported interaction between RAIDD and PIDD (8). Several important observations arise from these studies. First, RAIDD-/- mice have normal development and normal lymphocyte populations; TNF-induced killing of embryonic fibroblasts is normal in the absence of RAIDD; and thymocytes from RAIDD-/- mice respond normally to various apoptosis-inducing agents. Hence, these studies failed to identify a critical role for RAIDD either in mouse development or cellular apoptosis. In a second series of experiments designed to test the functional significance of a protein complex containing RAIDD, caspase-2, and PIDD, we found that the apoptotic activity of PIDD in embryonic fibroblasts depended completely on RAIDD and only partly on caspase-2. The resistance of RAIDD-/- MEFs to PIDD-induced apoptosis represents the only phenotype that we have been able to measure thus far in cells that lack RAIDD and highlights an important role for RAIDD in PIDD-induced apoptosis.
PIDD was identified as a p53-up-regulated gene involved in mediating p53-dependent apoptosis (7). The ability of PIDD to interact with two cytoplasmic adaptor molecules RAIDD and FADD suggests that PIDD may engage the apoptotic machinery at different levels. Despite the overwhelming evidence indicating that p53 promotes apoptosis through the mitochondrial pathway (4, 5), compelling evidence supports a relationship between p53 and the death receptor pathway of apoptosis (19). Interestingly, p53-mediated cell death can be attenuated by overexpression of c-Flip-s, a specific inhibitor of caspase-8, and by pharmacological inhibition of caspase-8, at least in some cell types (20, 21). Our findings indicate that enforced PIDD expression promotes cell death through the activation of both the RAIDD/caspase-2 and FADD/caspase-8 pathways. Therefore, PIDD may represent yet another link between the extrinsic and the intrinsic apoptotic pathways.
The absence of phenotype associated with RAIDD and caspase-2 deficiency in mice raises a question regarding the role of PIDD in vivo. The physiological role of PIDD remains unknown; it may be important for apoptosis in some cell types and in response to specific apoptotic signals. Once we have a better understanding of PIDD function in vivo, the importance of RAIDD and caspase-2 as physiological effectors of PIDD-dependent apoptosis can be investigated by using the mouse lines and reagents developed in this study.
Supplementary Material
Acknowledgments
We thank Junying Yuan (Harvard Medical School, Boston) for the caspase-2-/- MEFs. This work was supported by grants from the Terry Fox Foundation and the National Cancer Institute of Canada.
Author contributions: C.B., T.W.M., and S.B. designed research; C.B., L.-M.B., W.M., and A.W. performed research; C.B., L.S., R.H., and W.-C.Y. contributed new reagents/analytic tools; C.B. and W.-C.Y. analyzed data; C.B. wrote the paper; and T.W.M. and S.B. supervised and discussed experimental work.
Abbreviations: DD, death domain; PIDD, p53-induced protein with a DD; mPIDD, mouse PIDD; RAIDD, receptor-interacting protein-associated ICH-1/CED-3 homologous protein with a DD; LRDD, leucine-rich DD; ts, temperature sensitive; MEF, mouse embryonic fibroblast; FADD, Fas-associated DD; IRES, internal ribosome entry site.
References
- 1.Levine, A. J. (1997) Cell 88, 323-331. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein, B., Lane, D. & Levine, A. J. (2000) Nature 408, 307-310. [DOI] [PubMed] [Google Scholar]
- 3.Vousden, K. H. (2000) Cell 103, 691-694. [DOI] [PubMed] [Google Scholar]
- 4.Soengas, M. S., Alarcon, R. M., Yoshida, H., Giaccia, A. J., Hakem, R., Mak, T. W. & Lowe, S. W. (1999) Science 284, 156-159. [DOI] [PubMed] [Google Scholar]
- 5.Schuler, M., Bossy-Wetzel, E., Goldstein, J. C., Fitzgerald, P. & Green, D. R. (2000) J. Biol. Chem. 275, 7337-7342. [DOI] [PubMed] [Google Scholar]
- 6.Telliez, J. B., Bean, K. M. & Lin, L. L. (2000) Biochim. Biophys. Acta 1478, 280-288. [DOI] [PubMed] [Google Scholar]
- 7.Lin, Y., Ma, W. & Benchimol, S. (2000) Nat. Genet. 26, 122-127. [DOI] [PubMed] [Google Scholar]
- 8.Tinel, A. & Tschopp, J. (2004) Science 304, 843-846. [DOI] [PubMed] [Google Scholar]
- 9.Bergeron, L., Perez, G. I., Macdonald, G., Shi, L., Sun, Y., Jurisicova, A., Varmuza, S., Latham, K. E., Flaws, J. A., Salter, J. C., et al. (1998) Genes Dev. 12, 1304-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lassus, P., Opitz-Araya, X. & Lazebnik, Y. (2002) Science 297, 1352-1354. [DOI] [PubMed] [Google Scholar]
- 11.Robertson, J. D., Enoksson, M., Suomela, M., Zhivotovsky, B. & Orrenius, S. (2002) J. Biol. Chem. 277, 29803-29809. [DOI] [PubMed] [Google Scholar]
- 12.Guo, Y., Srinivasula, S. M., Druilhe, A., Fernandes-Alnemri, T. & Alnemri, E. S. (2002) J. Biol. Chem. 277, 13430-13437. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad, M., Srinivasula, S. M., Wang, L., Talanian, R. V., Litwack, G., Fernandes-Alnemri, T. & Alnemri, E. S. (1997) Cancer Res. 57, 615-619. [PubMed] [Google Scholar]
- 14.Duan, H. & Dixit, V. M. (1997) Nature 385, 86-89. [DOI] [PubMed] [Google Scholar]
- 15.Salmena, L., Lemmers, B., Hakem, A., Matysiak-Zablocki, E., Murakami, K., Au, P. Y., Berry, D. M., Tamblyn, L., Shehabeldin, A., Migon, E., et al. (2003) Genes Dev. 17, 883-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeh, W. C., Pompa, J. L., McCurrach, M. E., Shu, H. B., Elia, A. J., Shahinian, A., Ng, M., Wakeham, A., Khoo, W., Mitchell, K., et al. (1998) Science 279, 1954-1958. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida, H., Kong, Y. Y., Yoshida, R., Elia, A. J., Hakem, A., Hakem, R., Penninger, J. M. & Mak, T. W. (1998) Cell 94, 739-750. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, P., Gray, D., Mowat, M. & Benchimol, S. (1991) Mol. Cell. Biol. 11, 1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fridman, J. S. & Lowe, S. W. (2003) Oncogene 22, 9030-9040. [DOI] [PubMed] [Google Scholar]
- 20.Kovar, H., Jug, G., Printz, D., Bartl, S., Schmid, G. & Wesierska-Gadek, J. (2000) Oncogene 19, 4096-4107. [DOI] [PubMed] [Google Scholar]
- 21.Burns, T. F., Bernhard, E. J. & El-Deiry, W. S. (2001) Oncogene 20, 4601-4612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}