

Abstract
Hormones, neurotransmitters, and growth factors give rise to calcium entry via receptor-activated cation channels that are activated downstream of phospholipase C activity. Members of the transient receptor potential channel (TRPC) family have been characterized as molecular substrates mediating receptor-activated cation influx. TRPC channels are assumed to be composed of multiple TRPC proteins. However, the cellular principles governing the assembly of TRPC proteins into homo- or heteromeric ion channels still remain elusive. By pursuing four independent experimental approaches—i.e., subcellular cotrafficking of TRPC subunits, differential functional suppression by dominant-negative subunits, fluorescence resonance energy transfer between labeled TRPC subunits, and coimmunoprecipitation—we investigate the combinatorial rules of TRPC assembly. Our data show that (i) TRPC2 does not interact with any known TRPC protein and (ii) TRPC1 has the ability to form channel complexes together with TRPC4 and TRPC5. (iii) All other TRPCs exclusively assemble into homo- or heterotetramers within the confines of TRPC subfamilies—e.g., TRPC4/5 or TRPC3/6/7. The principles of TRPC channel formation offer the conceptual framework to assess the physiological role of distinct TRPC proteins in living cells.
After binding to their cognate receptors on the cell membrane, many hormones, neurotransmitters, and growth factors induce increases in the intracellular free calcium concentration ([Ca2+]i) subsequent to phospholipase C (PLC) stimulation. In addition to inositol-1,4,5-trisphosphate (InsP3)-mediated calcium release from intracellular stores, plasma membrane ion channels are activated in a PLC-dependent manner in most cells either by second messenger-mediated pathways or by store depletion (1). Mammalian homologues of the Drosophila melanogaster transient receptor potential (TRP) visual transduction channels (2), the TRP channel (TRPC) proteins, have been identified (3–8) and characterized as subunits of receptor-activated cation channels (9–12). Although it has been established that TRPC proteins are subject to a gating mechanism that operates through PLC, the exact activation mechanism for distinct TRPC family members is still a controversial issue. Proposed models favoring store-dependent or InsP3 receptor-mediated channel gating (5, 6, 13, 14) were challenged (8, 15–18), and alternative hypotheses like the direct second messenger-mediated activation of TRPC3, TRPC6, and TRPC7 by diacylglycerol have been put forward (8, 16).
TRPC channels and TRP-related protein families (11, 12) belong to the superfamily of hexahelical cation channels. Therefore, several assumptions on the structure and function of TRPC channels were made based on analogous reasoning. (i) TRPC proteins are thought to span the plasma membrane 6 times with a pore loop inserted between transmembrane segments 5 and 6. (ii) Functional channel complexes are believed to be composed of tetrameric TRPC proteins. (iii) Because the canonical TRPC family comprises seven distinct members, TRPC channels are postulated to form homo- as well as heterotetramers giving rise to biophysically and functionally discernible channel entities. Whereas the first assumption has been proven experimentally for TRPC channels (19) and the second has been confirmed in the case of the vanilloid receptor (20), the crucial issue as to which TRPC proteins can form multimeric ion channels remains elusive. Considering that many TRPCs are ubiquitously expressed and often coexpressed in a given cell (9), heteromultimerization in addition to homomultimerization among members of this protein family represents an enticing possibility. A deeper understanding of the selectivity of how TRPC subunits combine to form functional ion channel complexes is an essential prerequisite to evaluate the contribution of a given TRPC channel to endogenous PLC-dependent cation currents.
Here we investigate the basic principles of TRPC channel homo- and heteromultimerization in living cells by means of four independent cell biological and biophysical approaches. First, cellular trafficking-incompetent TRPC subunits are noted to be translocated to the plasma membrane only when distinct additional TRPCs were coexpressed. Second, we find that a dominant-negative mutant of TRPC6 selectively suppresses TRPC3 and TRPC6 but not TRPC4 and TRPC5 function. Third, we show that TRPC proteins C-terminally fused to fluorescent proteins give rise to fluorescence resonance energy transfer (FRET) when coexpressed in certain combinations, thus allowing for the systematic determination of TRPC homo- and heterotetramers. Finally, a set of coimmunoprecipitations confirms the conclusions drawn from the FRET study. The systematical delineation of all permissive permutations of TRPC channel assembly sets the framework to conclusively interpret TRPC-dependent ion fluxes in living cells.
Materials and Methods
Molecular Biology.
To fuse TRPC1–6 with spectral mutants of the enhanced green fluorescent protein (GFP), the STOP codons were replaced by in-frame XhoI (TRPC1, -2, -4, -5, -6, and -7) or EcoR1 (TRPC3) restriction sites by PCR-mediated mutagenesis with the Expand High Fidelity (Roche Molecular Biochemicals) PCR polymerase system and subsequent subcloning into the appropriate pcDNA3-CFP or -YFP (CFP, cyan fluorescent protein; YFP, yellow fluorescent protein) fusion vectors as described (21). The dominant-negative TRPC6 mutant was generated by PCR-mediated mutagenesis replacing three highly conserved amino acids (L678-W680) within the putative pore region of wild-type TRPC6 channels for alanine residues and confirmed by DNA sequencing.
Cell Culture and Transfection.
HEK293 cells were cultured in Eagle's minimal essential medium supplemented with 10% FCS. For transfections, we used 4 μl of FuGene6 transfection reagent (Roche Molecular Biochemicals) and 3 μg of DNA per 30-mm dish. When no other fluorescent construct was expressed, we added 100 ng of peGFP-C1-encoding enhanced GFP (CLONTECH) to allow detection of transfected cells. Cells were used 24–36 h after transfection.
An inducible cell line stably expressing hTRPC6 was generated with an HEK293 cell line that stably expresses the Drosophila ecdysone receptor under Zeocin selection (EcR293, Invitrogen). hTRPC6 was subcloned into the pIND(SP1)/Hygro plasmid (Invitrogen), transfected into these cells, and selected by hygromycin B (400 μg/ml). Clones were induced by 10 μM ponasterone A for 1 day and tested for receptor-activated Mn2+ entry and AlF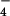 -induced whole-cell currents. Clone no. 6-3 was chosen for further studies.
-induced whole-cell currents. Clone no. 6-3 was chosen for further studies.
Chinese hamster ovary (CHO)-K1 cells were microinjected with mixtures of 5 ng/μl of enhanced GFP-C1, 5 ng/μl of guinea pig H1 histamine receptor in pCMV, 100 ng/μl of the respective wild-type channel cDNA in pcDNA3, and 400 ng/μl of a variable mixture of empty pcDNA3 vector with the dominant-negative TRPC6 mutant in pcDNA3.
Confocal Fluorescence Microscopy.
Living cells grown and transfected on glass coverslips were examined with an LSM 510 laser-scanning microscope (Zeiss). For detection of GFP or YFP fluorescence, the sample was excited with the 488-nm line of an argon laser applied through a 488-nm beamsplitter. A 505-nm long pass filter filtered emitted light. Pinhole settings were adjusted to allow sections thinner than 0.8 μM.
Assessment of Mn2+ Entry.
Measurements of Mn2+ quenching were performed as described (16). Briefly, cells were loaded in Hepes-buffered saline (HBS; 140 mM NaCl/6 mM KCl/1.25 mM MgCl2/1.25 mM CaCl2/10 mM Hepes/5 mM glucose/BSA 0.1%, pH 7.4) at 37°C for 30 min. After a 15-min incubation at room temperature, cells were rinsed with HBS and used for experiments within 2 h. Experiments were carried out with a Polychrome IV monochromator and an IMAGO II peltier-cooled charge-coupled device (CCD) camera (TILL Photonics, Planegg, Germany) coupled to an inverted Olympus (New Hyde Park, NY) IX70 microscope. Fluorescence signals were recorded at the isosbestic point of fura-2 (358 nm in our system) at 330-ms intervals and normalized to 100% of the initial value. For quantification, the point-by-point difference was run-averaged by 11 data points, and the maximum fluorescence decay rate expressed as percentage per time was used for further analysis. Statistical analysis was performed with the Wilcoxon–Mann–Whitney U test.
FRET.
C-terminal fusion cDNAs of different TRPC channels with either CFP or YFP were prepared as described above and coexpressed with a cDNA ratio of 1:3 (CFP:YFP) to minimize the probability of CFP-only multimers. The cells were examined with the setup used for calcium imaging except for a dual band-pass dichroic mirror and an emission-filter wheel (Lambda 10–2, Sutter Instruments, Novato, CA) included in the emission path. For the acceptor-bleach protocol applied here, cells were excited in 5-s intervals for 50 ms at 440 nm for CFP detection and for 50 ms at 500 nm for YFP detection. After 10 excitation cycles used as baseline, an additional 4,500-ms bleach pulse at 512 nm was added to each cycle. This procedure resulted in more than 90% of YFP photobleached within less than 2 min. We quantified the percentage increase of CFP (donor) fluorescence after bleach of a potential acceptor, YFP. Statistical analysis was performed with a Student's t test.
Electrophysiology.
Whole-cell patch clamp experiments were performed with an EPC-7 patch clamp amplifier (HEKA Electronics, Lambrecht/Pfalz, Germany) essentially as described (22). The pipette solution contained 120 mM CsCl, 1 mM MgCl2, 10 mM Hepes, and 10 mM 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA; pH = 7.4) and was supplemented with 10 mM NaF and 30 μM AlCl3. The bath solution contained 120 mM NaCl, 5 mM CsCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM Hepes, and 5 mM glucose and was adjusted to pH 7.4 and 290–310 milliosmoles/liter.
Coimmunoprecipitation.
HEK293 cells coexpressing TRPC channels containing either a hemagglutinin (HA) or myc tag were solubilized in 1 ml of PBS supplemented with 1% Triton X-100 and a protease inhibitor mixture (Roche Molecular Biochemicals) and centrifuged at 4°C (10 min at 9,000 × g, 1 h at 150,000 × g). Of the supernatants, 400 μl were incubated overnight with 2 μg of anti-myc Ab (Upstate Biotechnology, Lake Placid, NY), precipitated for 2 h with 10 μl of protein A-Sepharose, and washed 3 times in 700 μl of solubilization buffer. The cell lysates (20 μl) and 1/3 of the immunoprecipitates were subjected to SDS/PAGE, blotted, and probed with an anti-HA Ab (Sigma).
Results
Cellular Trafficking of TRPC Channels.
The subcellular localization of a C-terminal fusion protein of hTRPC1A (13) with enhanced YFP was assessed by confocal laser-scanning microscopy after transient expression in HEK293 cells. In analogy to previous observations on mTRPC2 expression in heterologous cell systems (22), we observed hTRPC1A to be localized within intracellular membrane compartments including the nuclear membrane, whereas the cell membrane was devoid of TRPC1-YFP fluorescence (Fig. 1a). On the contrary, a discrete plasma membrane staining became visible after TRPC3, -4, -5, and -6 expression (22–24; see Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). In light of previous reports suggesting a potential heteromultimerization of TRPC1 with TRPC3 (25, 26), we tested the hypothesis that correct TRPC1 trafficking might depend on the presence of TRPC3. Thus, we coexpressed TRPC1-YFP with a 3-fold excess of hTRPC3 cDNA. Yet, this maneuver did not change the cellular TRPC1-YFP fluorescence pattern (Fig. 1b). However, when TRPC1-YFP was co-expressed with mTRPC4β (24), a distinct fluorescence lining of the plasma membrane was discernible, indicative of correctly inserted TRPC1-YFP (Fig. 1c). Identical results were obtained with a longer splice variant of hTRPC1 [hTRPC1B (27), data not shown]. Thus, TRPC4 but not TRPC3 is capable of ushering TRPC1 into the plasma membrane, most probably by forming mixed multimers.
Figure 1.

Co-trafficking of TRPC1 and TRPC6Δ131 by members of the TRPC family. C-terminally YFP-tagged hTRPC1A was expressed in HEK293 cells alone (a) or together with untagged hTRPC3 (b) or mTRPC4β (c), and the cellular localization was assessed by confocal laser-scanning microscopy. d shows a cell expressing TRPC6-GFP. An N-terminally truncated TRPC6 mutant C-terminally tagged with GFP (TRPC6Δ131-GFP) was expressed alone (e) or together with untagged hTRPC3 (f), hTRPC6 (g), mTRPC4β (h), or mTRPC5 (i). Typical examples from three independent transfections are shown. ne, nuclear envelope; pm, plasma membrane.
To exploit this experimental approach as a means to delineate heteromultimerization of TRPC subunits in living cells further, we generated a truncated TRPC6 mutant (TRPC6Δ131) by removing the N-terminal 131 amino acid residues corresponding to the stretch of amino acids upstream of the ankyrin repeat domains. As opposed to wild-type TRPC6-GFP (Fig. 1d), TRPC6Δ131-GFP expression did not result in fluorescence of the plasma membrane (Fig. 1e). In analogy to the aforementioned experiments with TRPC1, TRPC6Δ131-GFP was coexpressed with untagged TRPC3, -4, -5, or -6. We noted that TRPC6 as well as TRPC3 coexpression was able to restore plasma membrane trafficking of TRPC6Δ131-GFP (Fig. 1 f and g), whereas both TRPC4 and TRPC5 left the TRPC6Δ131-GFP fluorescence pattern unchanged (Fig. 1 h and i).
Selective Suppression of TRPC3 and TRPC6 Activity by a Dominant-Negative TRPC6 Mutant.
To investigate whether the different subunits of interacting TRPC proteins indeed contribute to the formation of a common channel pore and to determine the degree of cooperativity within a TRPC channel pore complex, we generated a dominant-negative construct of TRPC6 (TRPC6DN) by exchanging three highly conserved residues, L678, F679, and W680, in the putative pore region for alanine residues. The resulting channel construct was correctly inserted into the plasma membrane and was functionally silent (data not shown). An inducible HEK293 cell line was incubated with 10 μM ponasterone A to express TRPC6 under steady-state conditions such that perfusion of the cell with the direct G protein-activator aluminum fluoride (AlF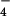 ) consistently resulted in peak whole-cell currents of 100–200 pA at a holding potential of −60. The current-voltage relations of the AlF
) consistently resulted in peak whole-cell currents of 100–200 pA at a holding potential of −60. The current-voltage relations of the AlF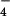 -induced currents displayed a dual inward and outward rectification and a reversal potential close to 0 mV (Fig. 2A, thin trace) indistinguishable from the properties of transiently expressed TRPC6 (16, 22). Transient expression of TRPC6DN in these cells nearly abrogated TRPC6-dependent currents (Fig. 2 A, bold trace, and B at different time points after transfection).
-induced currents displayed a dual inward and outward rectification and a reversal potential close to 0 mV (Fig. 2A, thin trace) indistinguishable from the properties of transiently expressed TRPC6 (16, 22). Transient expression of TRPC6DN in these cells nearly abrogated TRPC6-dependent currents (Fig. 2 A, bold trace, and B at different time points after transfection).
Figure 2.

Analysis of TRPC multimerization properties with a dominant-negative TRPC6 mutant (TRPC6DN). (A) Current-voltage relationships of AlF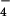 -induced TRPC6 currents in HEK293 cells stably expressing wild-type hTRPC6 (thin trace) and additionally expressing TRPC6DN (bold trace) were recorded. (B) Means ± SEM (n = 6) of peak TRPC6 whole-cell currents at −60 mV holding potential from 6 experiments each with (filled bars) or without (open bars) TRPC6DN coexpressed are shown at different time points after transfection of TRPC6DN-cDNA. Statistical significance is indicated (*, P < 0.05; **, P < 0.01). (C) CHO-K1 cells were comicroinjected with a fixed amount of wild-type TRPC6 and variable amounts of TRPC6DN cDNA as indicated on the abscissa. The rate of receptor-activated manganese influx relative to cells expressing only wild-type TRPC6 is plotted as a function of the relative content of TRPC6DN in the cDNA mixture. Each point represents the mean of 1 independent Mn2+ quenching experiment. The extent of suppression expected for the indicated levels of cooperativity are represented. (D) The relative suppression of Mn2+ influx rates (mean ± SEM of n = 5 independent experiments) in CHO-K1 cells microinjected with cDNAs encoding the wild-type TRPC proteins and equal amounts of TRPC6DN cDNA (filled bars) or vector control (open bars) is depicted. Statistical significance is indicated (*, P < 0.05; **, P < 0.01).
-induced TRPC6 currents in HEK293 cells stably expressing wild-type hTRPC6 (thin trace) and additionally expressing TRPC6DN (bold trace) were recorded. (B) Means ± SEM (n = 6) of peak TRPC6 whole-cell currents at −60 mV holding potential from 6 experiments each with (filled bars) or without (open bars) TRPC6DN coexpressed are shown at different time points after transfection of TRPC6DN-cDNA. Statistical significance is indicated (*, P < 0.05; **, P < 0.01). (C) CHO-K1 cells were comicroinjected with a fixed amount of wild-type TRPC6 and variable amounts of TRPC6DN cDNA as indicated on the abscissa. The rate of receptor-activated manganese influx relative to cells expressing only wild-type TRPC6 is plotted as a function of the relative content of TRPC6DN in the cDNA mixture. Each point represents the mean of 1 independent Mn2+ quenching experiment. The extent of suppression expected for the indicated levels of cooperativity are represented. (D) The relative suppression of Mn2+ influx rates (mean ± SEM of n = 5 independent experiments) in CHO-K1 cells microinjected with cDNAs encoding the wild-type TRPC proteins and equal amounts of TRPC6DN cDNA (filled bars) or vector control (open bars) is depicted. Statistical significance is indicated (*, P < 0.05; **, P < 0.01).
Assuming that a single dominant-negative subunit is sufficient to completely inactivate the pore complex to which it contributes, the relative extent of dominant-negative suppression is a function of the probability of a given pore complex to contain at least one dominant-negative subunit. To determine the cooperativity of the dominant-negative modulation of TRPC6, we microinjected different cDNA ratios of wild-type and TRPC6DN together with a cDNA construct encoding the H1 histamine receptor into the nuclei of CHO-K1 cells and registered the histamine-activated Mn2+ influx rate normalized to cells expressing wild-type TRPC6 and H1 histamine receptor only (Fig. 2C). The calculated extent of suppression assuming a two-, four-, or six-fold cooperativity are displayed. Nonlinear approximation of the experimental data obtained yielded a cooperativity factor of n = 3.89 ± 0.3, indicating that TRPC channels form tetrameric pore complexes.
Next, we set out to quantify the suppressive effect of TRPC6DN on other members of the TRPC family. Equal amounts of expression plasmids coding for TRPC6DN were microinjected into nuclei of CHO-K1 cells together with cDNA constructs encoding wild-type TRPC3, TRPC4, TRPC5, or TRPC6 and the H1 histamine receptor. Normalized maximal receptor-activated manganese influx rates compared with the wild-type control in the absence of TRPC6DN are shown in Fig. 2D. These data illustrate that TRPC6DN efficiently suppresses TRPC3 and TRPC6 currents but does not compromise TRPC4 or TRPC5 activity.
FRET Between CFP/YFP-Tagged TRPCs.
To demonstrate the direct protein–protein interaction of pore-forming channel subunits in living cells, we generated C-terminal fusion constructs of TRPC1, -2, -3, -4, -5, -6, and -7 with the CFPs or YFPs and assessed the proximity of coexpressed TRPC subunits differentially tagged on their C-termini by FRET. The quantitative FRET signal was recorded under static conditions by measuring the increase in donor (CFP) emission during selective photobleach of the acceptor fluorophore YFP at 512 nm. The recovery of donor fluorescence emission, which is a measure of steady-state FRET efficiency, was monitored at 480 nm and was expressed as percentage of CFP emission after acceptor bleach.
Fig. 3 illustrates our approach to monitor static FRET between TRPC3-CFP and TRPC6-YFP at the subcellular level. Direct recording of CFP and YFP fluorescence signals at successive time points after the onset of YFP photobleaching reveals a recovery of CFP fluorescence concomitant with progressive YFP bleaching in intracellular membranous compartments as well as in the plasma membrane (Fig. 3A). The time courses of emitted CFP and YFP fluorescence monitored over regions of interest (Fig. 3A) are resolved in Fig. 3B. When CFP fluorescence (FCFP at various time points after/FCFP before onset of photobleaching) was plotted against normalized YFP fluorescence (Fig. 3C), we noticed that during the initial phase of YFP photobleaching the extent of CFP recovery linearly correlated with decaying YFP fluorescence. The extrapolated intersection of the linear regression line with the y axis at FYFP/FYFP,0 = 0 is indicative of the actual FRET efficiency in the experimental system. In summary, our results show that fluorescent TRPC3 and TRPC6 fusion proteins display substantial FRET, indicating direct protein–protein interaction in cellular membranes.
Figure 3.

Determination of FRET between TRPC channel subunits. (A) Fluorescence of a cell coexpressing TRPC3-CFP and TRPC6-YFP. The images represent CFP (Upper) or YFP (Lower) fluorescences at different time points during photobleaching of YFP. (B) Time courses of the relative CFP and YFP fluorescence of regions of interest defined in A are shown. Circles represent intracellular fluorescence, and triangles denote fluorescence signals over the plasma membrane. (C) Kinetic correlation of the relative amount of YFP photobleaching and the concomitant increase in CFP fluorescence in the same cell. An extrapolation of the linear regression to FYFP = 0 indicates the actual extent of CFP fluorescence recovery. (D) Different combinations of TRPC channels tagged with either CFP or YFP were coexpressed in HEK293 cells as indicated on the abscissa, and the recovery of CFP fluorescence (expressed as percentage of final CFP fluorescence) during YFP photobleaching was quantified. Each bar represents means ± SEM of at least 3 independent cotransfection experiments. Expression of soluble CFP and YFP served as controls (c), and combinations showing a significant FRET signal (P < 0.05) are indicated by filled bars. The phylogenetic relationships within the TRPC channel family are given (Inset).
We extended our protocol to other members of the TRPC family and found that each TRPC-CFP channel when tested against its TRPC-YFP counterpart consistently displayed FRET, albeit with different efficiencies (Fig. 3D, bars 3–9). These data indicate that each of the TRPCs investigated is capable of forming homomultimers. In the case of TRPC3, we observed FRET between N-terminally tagged constructs, between a combination of N- and C-terminally tagged constructs, and between the termini of a TRPC3 subunit CFP-tagged on its N terminus and YFP-tagged on its C terminus. In the latter case, the cDNA was 10-fold diluted with wild-type TRPC3 to isolate the intramolecular FRET signal from FRET between different subunits within the tetramers. When CFP and YFP fusion constructs of distinct TRPC channels were systematically coexpressed, a FRET signal significantly elevated above the controls [i.e., soluble, nonfused CFP and YFP (Fig. 3D, bar 1) or hTRPC6-CFP and rat AT2-receptor (Fig. 3D, bar 2) coexpressed in the same cell] was observed in the following combinations: TRPC1/TRPC4 TRPC1/TRPC5, TRPC4/TRPC5, TRPC3/TRPC6, TRPC6/TRPC7, and TRPC3/TRPC7. TRPC1 coexpressed with TRPC3 or TRPC6, TRPC2 coexpressed with TRPC3, TRPC4, or TRPC6 as well as any combination beyond the TRPC4/5 or TRPC3/6/7 subfamily confines did not exhibit any significant FRET signals.
Coimmunoprecipitation of TRPC Channel Subunits.
Various TRPC constructs fused with either the HA tag or the c-myc tag at their C termini were co-expressed at different combinations in HEK293 cells, solubilized, and immunoprecipitated with an α-myc Ab. Both cell lysates and immunoprecipitates were subjected to SDS-gel electrophoresis and subsequent immunoblotting with an α-HA Ab (Fig. 4). With this approach, we detected HA immunoreactivity in the α-myc immunoprecipitates within the following combinations: (i) all homomultimeric combinations tested (TRPC2/TRPC2, TRPC3/TRPC3, TRPC4/TRPC4, and TRPC6/TRPC6), (ii) between TRPC3 and TRPC6, (iii) TRPC4 and TRPC5 and (iv) TRPC1 and TRPC4. We found that combinations beyond subfamilies, TRPC1 with TRPC3/TRPC6 as well as any combination with TRPC2, did not observe protein interactions. Moreover, we observed that when a mixture of TRPC4-HA and TRPC6-HA was used in a coimmunoprecipitation experiment with TRPC1-myc, only TRPC4, distinguishable from TRPC6 by its inherent lack of N-glycosylation, was found in the α-myc immunoprecipitate, thus excluding an incorporation of TRPC6 into TRPC1/TRPC4 multimeric assemblies. Taken together, the findings observed in FRET studies were confirmed by the analysis of TRPC channel immunocomplexes and can be summarized as follows: Although TRPC2 does not interact with any known TRPC protein except itself, and TRPC1 has the unique ability to form channel complexes with TRPC4 and TRPC5, other TRPC proteins generally assemble to heteromultimers only within a given structural subfamily—i.e., TRPC4/5 or TRPC3/6/7.
Figure 4.

Co-immunoprecipitation of TRPC channel subunits. TRPC channels were C-terminally tagged with an HA tag or an myc-tag and coexpressed in HEK293 cells in the combinations indicated below the panels. Anti-HA immunoreactivity was detected by immunoblotting (IB) before (Upper, lysate) or after immunoprecipitation with an anti-myc Ab (Lower, IP-myc).
Discussion
Several observations strongly indicate that Drosophila TRP family members as well as mammalian TRPC proteins function as subunits of multimeric channels (12). Thus, we set out to decipher the principal combinatory rules governing the assembly of different TRPC family members to functional, regulated cation channel complexes in living cells. Coimmunoprecipitation and Western blot analyses have previously proven instrumental to the detection of multimers of cellular signaling proteins. However, when examining hydrophobic, membrane-spanning proteins like TRPC channels, which may in addition interact with cytoskeletal elements or be enriched in detergent-insoluble membrane provinces (28), the latter techniques require protein solubilization, leaving researchers with the conundrum of how to rule out with certainty that the observed multimers do not result from technical limitations—i.e., incomplete solubilization. Therefore, we primarily focused on cell biological and biophysical approaches to study the multimerization properties of TRPC proteins in living cells. These results were then confirmed by additional coimmunoprecipitation experiments.
First, we noted that it is possible to restore the plasma membrane localization of intracellularly retained TRPC1 by coexpression of TRPC4, but not TRPC3. Similarly, an N-terminally truncated targeting-deficient TRPC6 was correctly cotrafficked to the plasma membrane by wild-type TRPC6 and TRPC3, but not by TRPC4 or TRPC5. Second, a dominant-negative mutant of TRPC6 was able to suppress cation entry mediated by TRPC3 and TRPC6, but not by TRPC4 or TRPC5. By systematically varying the ratio of wild-type vs. dominant-negative TRPC6 expression, we provide evidence that TRPC6 channels assemble into tetramers composed of four pore-forming subunits. Third, we adapted a static FRET approach to monitor TRPC complex formation. Steady-state FRET signals between TRPC channel subunits tagged either with CFP or YFP were recorded. All homomultimeric TRPC channel combinations displayed a FRET signal. When extending the FRET acceptor bleaching method to the entire TRPC family, we realized that TRPC channels assemble into FRET-competent complexes only in the narrow confines of the TRPC3/6/7 and TRPC4/5 subfamilies, the only exception being TRPC1, which coassembles with TRPC4 and TRPC5. Performing additional coimmunoprecipitation studies, we noticed selective protein–protein interactions among all homomultimeric combinations tested, and between TRPC3 with TRPC6, TRPC4 with TRPC5, and TRPC1 with TRPC4. In aggregate, all four independent experimental setups unequivocally show that TRPC channels can form homo- and strictly defined heterotetramers.
Initial studies on the biophysical and functional properties of channels composed of single TRPCs were carried out with overexpression models. Although there is overt controversy in the literature as to how a given TRPC channel is activated under these conditions, most researchers agree that there are two major structural and functional TRPC channel subfamilies representing receptor-activated cation channels: TRPC4/5 on the one hand and TRPC3/6/7 on the other (9, 11, 12, 29). Ion currents carried by members of both subfamilies are augmented by receptor-dependent PLC activity. In addition to an inositol-1,4,5-trisphosphate (InsP3)-receptor-dependent or InsP3-receptor-independent yet store-operated activation mechanism for TRPC3 (14, 30), a membrane-delimited channel gating by diacylglycerols has been proposed for TRPC3, -6, and -7 (8, 16, 18). Most TRPC proteins are characterized by a fairly broad and chiefly overlapping expression pattern (9). Therefore, only in very few instances is a single TRPC protein expressed singularly or in conspicuous excess over other TRPC family members in a given tissue or cell. Rare examples for this scenario are the expression of TRPC2 in the rodent vomeronasal organ (7, 22) and the prevalence of TRPC6 in certain types of smooth muscle (31). In most instances, however, comparable amounts of several TRPC proteins are coexpressed in the same cell. Therefore, little is known about the physiological role of distinct TRPC proteins.
For hexahelical cation channels other than TRPCs, heteromultimerization of pore-forming subunits has been demonstrated. Voltage-operated potassium channels, for instance, have been shown to form heterotetrameric channel complexes mainly restricted to structural subfamilies (32, 33). Recent evidence highlights that accessory pore-forming subunits, not functionally expressed alone, can coassemble with them into mixed tetramers (34). In either case, parameters like biophysical properties and surface expression are modified (34, 35).
The known spectrum of biophysically and functionally distinct receptor-activated or store-operated channel entities (9) exceeds by far what can be derived from data obtained with single, heterologously overexpressed TRPC channels. Moreover, some members of the TRPC family seem to be poorly expressed in heterologous expression systems. For these reasons, heteromultimerization of pore-forming subunits among TRPCs represents an enticing possibility, which could indeed be demonstrated for single combinations. Coassembly of TRPL and TRPγ, members of the Drosophila TRPC family that are constitutively active alone, gives rise to a tightly regulated, PLC-operated channel (36). Lintschinger et al. (26) extended previous coimmunoprecipitation data (25) and observed that coexpression of TRPC1 and TRPC3 resulted in a constitutively active cation conductance significantly higher than what could be observed in the case of singular TRPC1 or TRPC3 expression. These findings were interpreted to mean that TRPC1 and TRPC3 form heteromultimers with functional properties distinct from either channel alone. Our results presented here do not lend credence to the latter conclusion but support a recent report (37) that shows that TRPC1 interacts with TRPC5 in vivo to give rise to a novel, store-independent nonselective cation channel clearly set apart from known TRPC5 function (6, 24).
Because the composition of functional TRPC complexes is still largely unknown, it has proven difficult to unequivocally ascribe receptor-activated cation currents to molecularly defined TRPC proteins. We performed a comprehensive analysis on the entire TRPC family to describe the multimerization potential of TRPC proteins in living cells. The present study provides a conceptual framework necessary to reliably interpret future detailed studies to resolve discrete biophysical differences emanating from the heterotetramerization of TRPC proteins. Defining the combinatorial rules that govern the assembly of TRPC proteins in living cells is a crucial step to unravel the bewildering diversity of PLC-dependent cation conductances encoded by this gene family.
Supplementary Material
Acknowledgments
We are grateful to Dr. Tullio Pozzan and his laboratory for helpful advice with the FRET technique. The mTRPC7 cDNA was kindly provided by Dr. Yasuo Mori (National Institute for Physiological Sciences, Okazaki, Japan). This work was supported by grants from the Deutsche Forschungsgemeinschaft, Fonds der Chemischen Industrie, and by Stiftung P. E. Kempkes (Marburg).
Abbreviations
- TRPC
transient receptor potential channel
- PLC
phospholipase C
- FRET
fluorescence resonance energy transfer
- CFP
cyan fluorescent protein
- YFP
yellow fluorescent protein
- GFP
green fluorescent protein
- CHO
Chinese hamster ovary
- HA
hemagglutinin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Berridge M J. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 2.Montell C, Rubin M R. Neuron. 1989;2:1313–1323. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- 3.Wes P, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C. Proc Natl Acad Sci USA. 1995;92:9652–9656. doi: 10.1073/pnas.92.21.9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu X, Jiang M, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L. Cell. 1996;85:661–671. doi: 10.1016/s0092-8674(00)81233-7. [DOI] [PubMed] [Google Scholar]
- 5.Philipp S, Cavalie A, Freichel M, Wissenbach U, Zimmer S, Trost C, Marquart A, Murakami M, Flockerzi V. EMBO J. 1996;15:6166–6171. [PMC free article] [PubMed] [Google Scholar]
- 6.Philipp S, Hambrecht J, Braslavski L, Schroth G, Freichel M, Murakami M, Cavalie A, Flockerzi V. EMBO J. 1998;17:4274–4282. doi: 10.1093/emboj/17.15.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liman E R, Corey D P, Dulac C. Proc Natl Acad Sci USA. 1999;96:5791–5796. doi: 10.1073/pnas.96.10.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y. J Biol Chem. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- 9.Hofmann T, Schaefer M, Schultz G, Gudermann T. J Mol Med. 2000;78:14–25. doi: 10.1007/s001099900070. [DOI] [PubMed] [Google Scholar]
- 10.Harteneck C, Plant T D, Schultz G. Trends Neurosci. 2000;23:159–166. doi: 10.1016/s0166-2236(99)01532-5. [DOI] [PubMed] [Google Scholar]
- 11.Clapham D E, Runnels L W, Strübing C. Nat Rev Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- 12.Montell C. Science's STKE. 2001. www.stke.org/cgi/content/full/OC_sigtrans , www.stke.org/cgi/content/full/OC_sigtrans;2001/90/re1. ;2001/90/re1. [DOI] [PubMed] [Google Scholar]
- 13.Zitt C, Zobel A, Obukhov A G, Harteneck C, Kalkbrenner F, Lückhoff A, Schultz G. Neuron. 1996;16:1189–1196. doi: 10.1016/s0896-6273(00)80145-2. [DOI] [PubMed] [Google Scholar]
- 14.Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Nature (London) 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- 15.Zitt C, Obukhov A G, Strübing C, Zobel A, Kalkbrenner F, Lückhoff A, Schultz G. J Cell Biol. 1997;138:1333–1341. doi: 10.1083/jcb.138.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hofmann T, Obukhov A G, Schaefer M, Harteneck C, Gudermann T, Schultz G. Nature (London) 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 17.Okada T, Shimizu S, Wakamori M, Maeda A, Kurosaki T, Takada N, Imoto K, Mori Y. J Biol Chem. 1998;273:10279–10287. doi: 10.1074/jbc.273.17.10279. [DOI] [PubMed] [Google Scholar]
- 18.Venkatachalam K, Ma H T, Ford D L, Gill D L. J Biol Chem. 2001;276:33980–33985. doi: 10.1074/jbc.C100321200. [DOI] [PubMed] [Google Scholar]
- 19.Vannier B, Zhu X, Brown D, Birnbaumer L. J Biol Chem. 1998;273:8675–8679. doi: 10.1074/jbc.273.15.8675. [DOI] [PubMed] [Google Scholar]
- 20.Kedei N, Szabo T, Lile J D, Treanor J J, Olah Z, Iadarola M J, Blumberg P M. J Biol Chem. 2001;276:28613–28619. doi: 10.1074/jbc.M103272200. [DOI] [PubMed] [Google Scholar]
- 21.Schaefer M, Albrecht N, Hofmann T, Gudermann T, Schultz G. FASEB J. 2001;15:1634–1636. doi: 10.1096/fj.00-0824fje. [DOI] [PubMed] [Google Scholar]
- 22.Hofmann T, Schaefer M, Schultz G, Gudermann T. Biochem J. 2000;351:115–122. doi: 10.1042/0264-6021:3510115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKay R R, Szymeczek-Seay C L, Lievremont J P, Bird G S, Zitt C, Jüngling E, Lückhoff A, Putney J W., Jr Biochem J. 2000;351:735–746. [PMC free article] [PubMed] [Google Scholar]
- 24.Schaefer M, Plant T D, Obukhov A G, Hofmann T, Gudermann T, Schultz G. J Biol Chem. 2000;275:17517–17526. doi: 10.1074/jbc.275.23.17517. [DOI] [PubMed] [Google Scholar]
- 25.Xu X Z, Li H S, Guggino W B, Montell C. Cell. 1997;89:1155–1164. doi: 10.1016/s0092-8674(00)80302-5. [DOI] [PubMed] [Google Scholar]
- 26.Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier W F, Romanin C, Zhu M X, Groschner K. J Biol Chem. 2000;275:27799–27805. doi: 10.1074/jbc.M002705200. [DOI] [PubMed] [Google Scholar]
- 27.Zhu X, Chu P B, Peyton M, Birnbaumer L. FEBS Lett. 1995;373:193–198. doi: 10.1016/0014-5793(95)01038-g. [DOI] [PubMed] [Google Scholar]
- 28.Lockwich T P, Liu X, Singh B B, Jadlowiec J, Weiland S, Ambudkar I S. J Biol Chem. 2000;275:11934–11942. doi: 10.1074/jbc.275.16.11934. [DOI] [PubMed] [Google Scholar]
- 29.Birnbaumer L, Boulay G, Brown D, Jiang M, Dietrich A, Mikoshiba K, Zhu X, Qin N. Recent Prog Horm Res. 2000;55:127–161. [PubMed] [Google Scholar]
- 30.Vazquez G, Lievremont J P, St J, Bird G, Putney J W., Jr Proc Natl Acad Sci USA. 2001;98:11777–11782. doi: 10.1073/pnas.201238198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, Ito Y, Mori Y. Circ Res. 2001;88:256–259. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- 32.Isacoff E Y, Jan Y N, Jan L Y. Nature (London) 1990;345:530–534. doi: 10.1038/345530a0. [DOI] [PubMed] [Google Scholar]
- 33.Covarrubias M, Wei A A, Salkoff L. Neuron. 1991;7:763–773. doi: 10.1016/0896-6273(91)90279-9. [DOI] [PubMed] [Google Scholar]
- 34.Patel A J, Lazdunski M, Honore E. EMBO J. 1997;12:6615–6625. doi: 10.1093/emboj/16.22.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manganas L N, Trimmer J S. J Biol Chem. 2000;275:29685–29683. doi: 10.1074/jbc.M005010200. [DOI] [PubMed] [Google Scholar]
- 36.Xu X Z, Chien F, Butler A, Salkoff L, Montell C. Neuron. 2000;26:647–657. doi: 10.1016/s0896-6273(00)81201-5. [DOI] [PubMed] [Google Scholar]
- 37.Strübing C, Krapivinsky G, Krapivinsky L, Clapham D E. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}