

Abstract
Lymphocyte chemotaxis is a complex process by which cells move within tissues and across barriers such as vascular endothelium and is usually stimulated by chemokines such as stromal cell-derived factor-1 (CXCL12) acting via G protein-coupled receptors. Because members of this receptor family are regulated (“desensitized”) by G protein-coupled receptor kinase (GRK)-mediated receptor phosphorylation and β-arrestin binding, we examined signaling and chemotactic responses in splenocytes derived from knockout mice deficient in various β-arrestins and GRKs, with the expectation that these responses might be enhanced. Knockouts of β-arrestin2, GRK5, and GRK6 were examined because all three proteins are expressed at high levels in purified mouse CD3+ T and B220+ B splenocytes. CXCL12 stimulation of membrane GTPase activity was unaffected in splenocytes derived from GRK5-deficient mice but was increased in splenocytes from the β-arrestin2- and GRK6-deficient animals. Surprisingly, however, both T and B cells from β-arrestin2-deficient animals and T cells from GRK6-deficient animals were strikingly impaired in their ability to respond to CXCL12 both in transwell migration assays and in transendothelial migration assays. Chemotactic responses of lymphocytes from GRK5-deficient mice were unaffected. Thus, these results indicate that β-arrestin2 and GRK6 actually play positive regulatory roles in mediating the chemotactic responses of T and B lymphocytes to CXCL12.
Leukocytes migrate to sites of inflammation by recognizing a gradient of chemoattractants, and moving toward the chemoattractant source. This process of ligand recognition, cell polarization and directed cell migration is complex, given that a cell needs to integrate numerous signals arising from different spatial orientations to decide in which direction to move. In lymphocytes, the signals that guide the cell in making these decisions arise from chemokine activation of heptahelical G protein-coupled receptors (GPCR) linked to Gαi proteins. Directional migration, however, requires the activity of Gβγ, but not Gα, proteins (1, 2). In addition, Rho family guanosine triphosphatases (GTPases), the phosphoinositide 3-kinases, and possibly extracellular receptor kinases each play important roles in generating the cell polarity and cytoskeletal reorganization required for directional migration (3–7).
Migration to chemokine gradients is dose dependent. Chemotaxis occurs at relatively low concentrations of chemokines, but at higher chemokine concentrations, cells become paralyzed and no longer migrate toward the chemoattractant source. The mechanisms by which cells are paralyzed at high chemokine concentrations are not clear but may involve agonist-dependent desensitization and receptor endocytosis mediated by G protein-coupled receptor kinases (GRKs) and arrestins. GRKs phosphorylate serine and threonine residues in the C terminus and intracellular loops of GPCRs, allowing for the association of arrestins that act to prevent heterotrimeric Gαβγ protein association with and activation by the GPCR (8).
Based on the current paradigm that arrestins are the primary negative regulators of GPCRs, we reasoned that they might negatively regulate chemotaxis. However, because arrestins also play positive regulatory roles for mitogen-activated protein kinase, extracellular receptor kinase, and c-jun terminal kinase activation (9–12), they might also act as positive regulators of chemotaxis. To evaluate the role of the GRK-arrestin pathway in chemotaxis, we studied the chemotactic responses of lymphocytes from β-arrestin- and GRK-deficient mice toward gradients of stromal cell-derived factor 1 (CXCL12), a well characterized chemokine whose receptor is CXCR4, a coreceptor for HIV.
Materials and Methods
Mice.
The following mouse strains were used in this study: β-arrestin2-deficient (back-crossed for six generations onto the C57/BL6 background; ref. 13); GRK5-deficient (mixed C57/BL6 × SVJ/129 background; ref. 14); and GRK6-deficient (mixed C57/BL6 x SVJ/129 background; R. R. Gainetdinov, L. M. Bohn, T. D. Sotnikova, M. Cyr, K.-M. Kim, A. D. Macrae, G. E. Torres, R. J. Lefkowitz, M. G. Caron & R. T. Premont, unpublished data). Only male mice were evaluated and controls were age- and sex-matched littermates. Requests for mice should be addressed to lefko001@mc.duke.edu.
Genotyping.
β-arrestin2, GRK5, and GRK6 animals were genotyped by using triplex PCR on DNA samples prepared from tail tips, as described (refs. 14 and 15, and R. R. Gainetdinov, L. M. Bohn, T. D. Sotnikova, M. Cyr, K.-M. Kim, A. D. Macrae, G. E. Torres, R. J. Lefkowitz, M. G. Caron & R. T. Premont, unpublished data).
Lymphocyte Isolation.
Spleens from littermate wild-type and knockout male mouse siblings (8–16 wk of age) were homogenized in a Dounce homogenizer in RPMI medium 1640 containing 10 mM Hepes (GIBCO/BRL). After centrifugation at 370 × g for 10 min at 4°C, cells were resuspended in 10 ml of ice-cold RBC lysis buffer (0.14M NH4Cl/0.017M Tris), pH 7.2, and incubated for 5 min on ice. The cells were washed in RBC lysis buffer until RBCs were no longer visible. The cells were then washed three times in RPMI medium 1640 and used immediately for chemotaxis assays. Cells were stored frozen for subsequent membrane purification for GTPase activity. T and B lymphocytes were further purified by two-color cell sorting on a FACSVantage SE cell sorter (Becton Dickinson) after staining with anti-CD3 FITC and B220-TC (Caltag, South San Francisco, CA).
Immunoblotting.
Isolated splenocytes, B cells, and T cells were resuspended in 20 mM Tris (pH 7.4), 1 mM EDTA, 100 mM NaCl, and a mixture of protease inhibitors at 20 × 106 cells/ml and lysed by repeated passage through a 26-gauge needle. Lysates were mixed with an equal volume of Laemmli SDS-sample buffer, and proteins were separated on 10% polyacrylamide gels. Transferred filters were blotted with Abs raised against β-arrestin1 (16) and GRK6 (17).
GTPase Activity.
Splenocytes from genetically deficient mice and littermate controls were isolated, pooled, and frozen at −80°C until use. Membranes were prepared and assayed for GTPase activity (10 μg of membrane preparations/assay) as described (18, 19) in the presence and absence of murine CXCL12 (1 μM).
Chemotaxis Assays.
Transwell chemotaxis assays were performed in 24-well transwells (6.5-mm diameter, 5-μm pore size, Costar) as described (20). The bottom chamber was filled with 0.6 ml of RPMI 10% (vol/vol) FBS medium containing various concentrations of murine CXCL12 (R & D Systems). Murine splenocytes (1 × 106) in 0.1 ml of RPMI medium 1640 containing 10% (vol/vol) FBS were added to the top chamber. After 1.5 h of incubation at 37°C, the bottom chamber was harvested, and the transmigrated cells were stained with fluorescent Abs for cell surface markers [anti-CD3 FITC (Caltag), B220-TC (Caltag), DX5-PE (PharMingen)]. The numbers of responding splenocyte subpopulations was determined by flow cytometry with an EPICS XL flow cytometer (Beckman Coulter). Results for transwell assays are reported as chemotactic index (fold increase over baseline). For transendothelial migration, 1 × 105 Ea.hy 926 endothelial cells (21) were plated onto the 24-well transwells and incubated for 3 days. Monolayer integrity was determined by assessing diffusion of [14C]mannitol (Amersham Pharmacia) from the top chamber to the bottom chamber within the transwell (22). Confluent monolayers were defined as those transwells in which the amount of 14C cpm detected in the bottom well was <35% of the counts found in the lower chamber of a transwell containing no monolayer. The bottom chamber was then replaced with 0.6 ml of RPMI 10% (vol/vol) FBS medium containing various concentrations of murine CXCL12, and 1 × 106 murine splenocytes in 0.1 ml of RPMI medium 1640 containing 10% (vol/vol) FBS were added to the top chamber. After 4 h of incubation at 37°C, cells in the lower chamber were harvested and counted as above. Results for transendothelial migration assays were normalized to the peak migration of wild-type cells in any given experiment. All assays were performed in duplicate, and knockout animals and their littermate-matched controls were tested simultaneously.
Statistics.
Statistical analyses were performed with statistica software (StatSoft, Tulsa, OK). For individual experiments, duplicate points were averaged and treated as a single point for statistical analyses. Because all assays were performed with a littermate matched control, Student's t test for correlated samples was used.
Results
Enhanced Activation of G Proteins by CXCL12 in β-Arrestin2-Deficient Compared to β-Arrestin2-Sufficient Lymphocytes.
Based on current understanding of the role of arrestins in regulating GPCR activity, arrestins should down-regulate signaling through chemokine receptors. Which arrestins might be the critical regulators of this activity in lymphocytes, however, is not clear. Visual arrestins expression is limited primarily to the visual system (23). Both β-arrestin1 and β-arrestin2 are expressed in lymphocytes (Fig. 1A), as determined by immunoblotting with a β-arrestin1 antiserum that crossreacts with β-arrestin2. B cells contain substantially less β-arrestin2 than T cells. Given the lower affinity of this antiserum for β-arrestin2 compared to β-arrestin1, it is likely that β-arrestin2 is the predominant arrestin protein in T cells. β-arrestin2 has been reported to associate well with CXCR4 and mediates its internalization (24). To determine whether β-arrestin2 is important for regulating signals through a typical chemokine receptor in lymphocytes, we tested CXCL12-stimulated GTPase activity in splenocyte cell membranes from β-arrestin2-deficient and sex-matched littermate control mice. As would be predicted if β-arrestin2 is a critical regulator of CXCR4 activity in lymphocytes, CXCL12-stimulated GTPase activity was higher in β-arrestin2-deficient compared to control splenocytes (Fig. 1B).
Figure 1.

β-arrestin expression in lymphocyte subsets and role of β-arrestin2 in regulating G-protein-dependent signaling in response to CXCL12. (A) β-arrestin expression in lymphocyte subsets. Equivalent amounts of protein extracts from unsorted wild-type and β-arrestin2-deficient splenocytes and sorted CD3+ T cells and B220+ B cells from wild-type mice were immunoblotted with an antiserum to β-arrestin1 that crossreacts with β-arrestin2. Shown is a representative immunoblot of three performed. (B) GTPase activity in wild-type and β-arrestin2−/− membranes. Membranes from wild-type and β-arrestin2-deficient splenocytes were stimulated with CXCL12 and assayed for GTPase activity. Shown is a representative experiment of three performed. Error bars represent the SEM of triplicate samples.
CXCL12-Stimulated Chemotaxis of β-Arrestin2-Deficient T and B Lymphocytes Is Impaired.
To examine the role of β-arrestin2 in regulating chemokine signaling in the context of the physiologic functioning of the intact cell, we next measured the chemotactic responses of wild-type and β-arrestin2-deficient splenocytes to CXCL12. The numbers and percentages of T and B lymphocytes were comparable in β-arrestin2-deficient and control mice (Table 1). Surprisingly, the chemotactic responses of both T and B lymphocytes to CXCL12 in the transwell migration assay were significantly decreased in β-arrestin2-deficient cells compared to wild-type cells (0.49- and 0.61-fold of control, P = 0.00016 and 0.00004, for T and B cells, respectively, when all points are evaluated together by using the chemotactic index; Fig. 2A). The result was not caused by CXCL12-induced cell aggregation as determined by microscopy and flow cytometry. This chemotactic defect was unexpected, and we reasoned that it may in part be because of the inconsistently higher baseline chemokinesis of β-arrestin2-deficient lymphocytes (1.4- and 1.5-fold of control, P = not significant, for T and B cells, respectively). Thus, to confirm this unexpected result, we also tested chemotactic responses in the more rigorous transendothelial migration assay. As in the transwell assay, T lymphocyte migration, and to a lesser extent B lymphocyte migration, to CXCL12 was decreased (0.45- and 0.77-fold, P = 0.00005 and 0.021, for T and B cells, respectively) with β-arrestin2-deficiency (Fig. 2B). The above results suggest a positive regulatory role for β-arrestin2 in CXCL12-induced chemotaxis.
Table 1.
Comparison of lymphocyte subsets in β-arrestin2-deficient, GRK6-deficient, GRK5-deficient, and control mouse spleens
| Cell number, millions | CD3+ T cells, % | B220+ B cells, % | |
|---|---|---|---|
| Barr2-WT | 85 ± 28 | 29 ± 4 | 56 ± 4 |
| Barr2-KO | 72 ± 17 | 24 ± 7 | 59 ± 6 |
| GRK5-WT | 110 ± 16 | 34 ± 5 | 47 ± 0.3 |
| GRK5-KO | 88 ± 27 | 36 ± 8 | 51 ± 6 |
| GRK6-WT | 88 ± 18 | 29 ± 3 | 49 ± 2 |
| GRK6-KO | 101 ± 16 | 27 ± 10 | 49 ± 5 |
Shown are the means ± SD of splenocyte numbers and percentages of CD3+ T and B220+ B cells from at least three knockout mice and littermate-matched controls. There were no statistically significant differences between genetically deficient mice and controls (P > 0.05). WT, wild type; KO, knockout.
Figure 2.

Directional migration of β-arrestin2−/− and control T and B lymphocytes toward gradients of CXCL12 was tested by standard transwell chemotaxis assays (A) and the more rigorous transendothelial migration assays (B) as described in Materials and Methods. Shown are the means and SEs of at least three independent experiments. Statistically significant differences are indicated.
GRK6 Is a Critical Regulator of CXCR4 in Lymphocytes.
For β-arrestin to bind CXCR4, it is expected that the CXCR4 cytoplasmic tail needs to be phosphorylated by GRKs. GRK expression and function is cell type- and receptor-specific, providing a high level of discrimination for signaling through GPCRs. Again, it is unclear which GRKs may be important for chemokine receptors in lymphocytes. To begin addressing this question, expression of GRKs in splenocytes and in isolated T and B lymphocytes was examined by immunoblotting. Immunoblotting with a GRK6 Ab that crossreacts with the other GRK4-subfamily members shows that all of the cells tested contain GRK6, as well as a substantial amount of GRK5. All these cell types have essentially equivalent levels of GRKs 5 and 6 (Fig. 3A). Based on these findings, GRK5 and GRK6 could potentially phosphorylate CXCR4 to induce β-arrestin binding.
Figure 3.
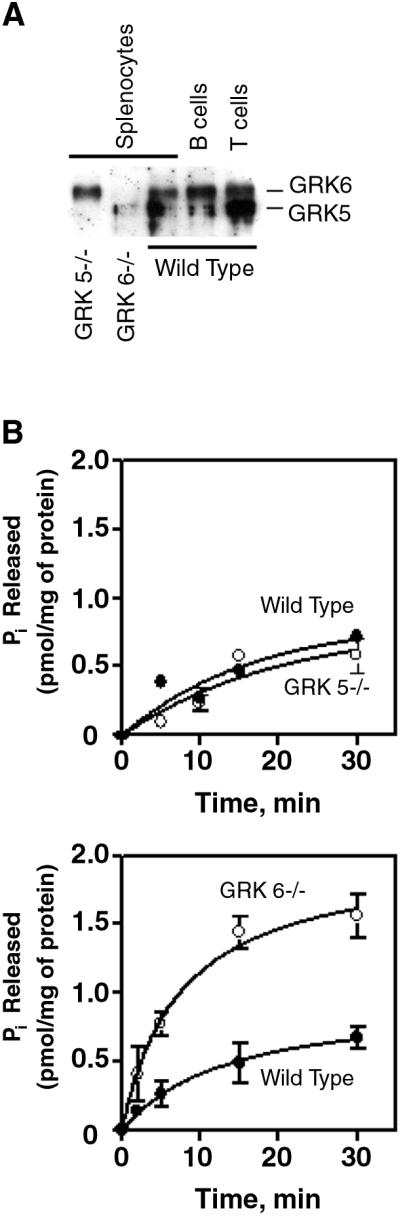
GRK5 and GRK6 expression in lymphocyte subsets and their roles in regulating G-protein-dependent signaling in response to CXCL12. (A) GRK5/6 expression in lymphocyte subsets. Equivalent amounts of protein extracts from unsorted wild-type, GRK5-deficient, and GRK6-deficient splenocytes, and sorted CD3+ T cells and B220+ B cells from wild-type mice were immunoblotted with an antiserum to GRK6 that crossreacts with GRK5. Shown is a representative immunoblot of three performed. (B) GTPase activity in GRK5−/−, GRK6−/−, and control membranes in response to CXCL12. Membranes from wild-type and β-arrestin2-deficient splenocytes were stimulated with CXCL12 and assayed for GTPase activity. Shown are representative experiments of three performed. Error bars represent the SEM of triplicate samples.
We studied splenocytes from GRK5- and GRK6-deficient mice and littermate controls. As for the β-arrestin2-deficient mice, the numbers and percentages of T and B lymphocytes were comparable in GRK5- and GRK6-deficient and control mice (Table 1). Whereas GTPase activity in response to CXCL12 was unaffected in GRK5-deficient splenocytes, it was dramatically increased in GRK6-deficient splenocytes (Fig. 3B), indicating a role for GRK6 in regulating CXCR4 activity.
Deficiency of GRK5 had no effect on lymphocyte chemotactic responses to CXCL12, but chemotaxis of T lymphocytes was markedly impaired in GRK6-deficient mice (0.45- and 0.57-fold, P = 0.0003 and 0.00004, for chemotaxis and transendothelial migration, respectively) (Fig. 4). Whereas chemotaxis of GRK6-deficient B lymphocytes was not impaired in the transwell assay, it was impaired in the transendothelial migration assay (0.56-fold, P = 0.016). Thus, GRK6 is an important regulator of CXCR4 activity in T and perhaps B lymphocytes.
Figure 4.

Directional migration of GRK5−/−, GRK6−/−, and control lymphocytes. Chemotaxis of GRK5-deficient (A), GRK6-deficient (B), and control T and B lymphocytes toward gradients of CXCL12 was tested by transwell assays. There were no statistically significant differences in migration between GRK5−/− and control lymphocytes, but there was a significant difference in chemotaxis of GRK6−/− compared to control T cells. Thus, migration of GRK6-deficient lymphocytes was further evaluated by transendothelial migration assays (C). Shown are the means and SEs of at least three independent experiments for A–C. Statistically significant differences are indicated.
Discussion
Leukocyte recruitment to sites of inflammation involves a complex series of events including the diapedesis (transendothelial migration) and directed cell movement in response to a chemotactic gradient within the tissue. Recognition of, and response to, chemotactic ligands are mediated by seven membrane-spanning receptors that generally activate members of the Gαi family of heterotrimeric G proteins (1, 2, 25). In cellular systems, such chemokine receptors have been shown to be regulated by classical GRK and β-arrestin mechanisms which “desensitize” second messenger signaling and promote internalization of the receptors (8, 26).
Our migration assays provide direct evidence that lymphocytes genetically deficient in either β-arrestin2 or GRK6 are impaired in chemotactic responses to CXCL12, despite the enhanced sensitivity of G protein stimulation assessed by ligand activated GTPase. This chemotactic defect is more pronounced in T cells than B cells, possibly because of the higher levels of β-arrestin2 in T cells. Moreover, the migration defect is not limited to lymphocytes or CXCL12-mediated chemotaxis because neutrophil chemotactic responses to the IL-8 homolog KC are also defective (data not shown). Neither can β-arrestin1 substitute for β-arrestin2, nor can GRK2 or GRK5 substitute for GRK6, in supporting chemotaxis.
How does the GRK-β-arrestin system function to positively regulate chemotactic responses while dampening GPCR–G protein coupling? At this point, one can only speculate, although recent advances in understanding the functions of β-arrestins suggest several possibilities. Originally discovered as molecules that bind to GRK phosphorylated receptors, thereby uncoupling them from G proteins, β-arrestins are also known to function as multifunctional adapter and scaffold proteins (27, 28). By targeting a wide variety of signaling molecules and components of the endocytic machinery to the GPCRs in an agonist-dependent manner, the β-arrestins coordinate a wide range of receptor activities as well as receptor trafficking and degradation. A partial list of such molecules that interact with β-arrestins includes clathrin, clathrin adapter protein complex 2, N-ethylmaleimide-sensitive fusion factor, the ubiquitin ligase MDM2, the small G protein ADP-ribosylation factor and its guanine nucleotide exchange factor ADP-ribosylation factor nucleotide binding site opener, several Src family nonreceptor tyrosine kinases, and components of at least two mitogen-activated protein kinase cascades, c-jun terminal kinase 3, and extracellular receptor kinase1/2 (9, 11, 12, 29–33). Thus, β-arrestins appear to play roles in both termination and initiation of various seven membrane-spanning receptor-signaling pathways. It is possible that either or both of these functions might be required for efficient chemotaxis. For example, sensing and responding to a ligand gradient must involve different levels of receptor activation at various points along the advancing edge of the migrating cell. Moreover, these levels are continuously changing as the cell moves through the gradient. Thus, the dynamic temporal and spatial control of receptor signaling required to achieve effective chemotaxis would almost certainly require a means not only for rapidly turning on receptors, but for turning them off as well. Thus, a deficiency of β-arrestin might impair the coordinated process of receptor activation and deactivation required for chemotaxis.
β-arrestin2 and GRK6 might positively regulate chemotaxis by preventing cell paralysis from GPCR overstimulation, as thought to occur at high chemokine concentrations. Previous in vitro studies, however, have given conflicting results regarding the possible involvement of chemokine receptor phosphorylation (and presumably β-arrestin binding) in chemotaxis. CXCR1, CXCR2, and CCR2B truncation or point mutants that cannot be phosphorylated and have enhanced G protein-signaling activity are defective in chemotaxis (refs. 34–36, and R. M. Richardson, R. J. Marjoram, L. S. Barak & R. Snyderman, unpublished data) On the other hand, similar CCR5 mutants are not defective in chemotaxis (37).
The β-arrestins might directly influence chemotaxis by their ability to serve as signaling adapters or scaffolds. The remarkably complex signaling networks involved in chemotaxis are just beginning to be elucidated. It is clear that in addition to GPCRs and heterotrimeric G proteins, the networks involve small G proteins, and their exchange factors, and extracellular receptor kinase mitogen-activated protein kinases. Because these families of proteins are known to interact with β-arrestins, it seems plausible that β-arrestins might exert positive regulatory effects on chemotaxis, in part, by coordinating the activities of such signaling molecules. Further, because β-arrestins translocate to GPCRs in proportion to the level of receptor stimulation, it is also possible that β-arrestins participate in forming the intracellular gradient leading to cell polarization.
In summary, our results indicate a previously unsuspected positive regulatory role for β-arrestin2 and GRK6 in lymphocyte chemotaxis. These roles appear to be fairly specific because other GRKs and β-arrestins present in the cells are not able to rescue the chemotactic defect. Elucidating the mechanisms by which the β-arrestin-GRK system participates in chemotactic signaling should enhance understanding of the molecular basis for this extraordinarily important and complex cellular process. It also may shed light on the pathophysiology of certain inflammatory conditions such as rheumatoid arthritis in which lymphocyte GRK6 is dramatically down regulated (38, 39).
Acknowledgments
We thank Kristina Riebe, Aron Boney, and Leona Whichard for their technical assistance. We thank Drs. Julia Walker and David Zidar for helpful discussions and a critical review of the manuscript. Dr. Lefkowitz is an Investigator of the Howard Hughes Medical Institute. These studies were supported by National Institutes of Health Grants AR39162, AI38910, and HL16037.
Abbreviations
- GRK
G protein-coupled receptor kinase
- GPCR
G protein-coupled receptor
- GTPase
guanosine triphosphatase
References
- 1.Neptune E R, Bourne H R. Proc Natl Acad Sci USA. 1997;94:14489–14494. doi: 10.1073/pnas.94.26.14489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neptune E R, Iiri T, Bourne H R. J Biol Chem. 1999;274:2824–2828. doi: 10.1074/jbc.274.5.2824. [DOI] [PubMed] [Google Scholar]
- 3.Benard V, Bohl B P, Bokoch G M. J Biol Chem. 1999;274:13198–13204. doi: 10.1074/jbc.274.19.13198. [DOI] [PubMed] [Google Scholar]
- 4.Akasaki T, Koga H, Sumimoto H. J Biol Chem. 1999;274:18055–18059. doi: 10.1074/jbc.274.25.18055. [DOI] [PubMed] [Google Scholar]
- 5.Belisle B, Abo A. J Biol Chem. 2000;275:26225–26232. doi: 10.1074/jbc.M002743200. [DOI] [PubMed] [Google Scholar]
- 6.Vanhaesebroeck B, Jones G E, Allen W E, Zicha D, Hooshmand-Rad R, Sawyer C, Wells C, Waterfield M D, Ridley A J. Nat Cell Biol. 1999;1:69–71. doi: 10.1038/9045. [DOI] [PubMed] [Google Scholar]
- 7.Ma A D, Metjian A, Bagrodia S, Taylor S, Abrams C S. Mol Cell Biol. 1998;18:4744–4751. doi: 10.1128/mcb.18.8.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lefkowitz R J. J Biol Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- 9.Luttrell L M, Roudabush F L, Choy E W, Miller W E, Field M E, Pierce K L, Lefkowitz R J. Proc Natl Acad Sci USA. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daaka Y, Luttrell L M, Ahn S, Della Rocca G J, Ferguson S S, Caron M G, Lefkowitz R J. J Biol Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- 11.Miller W E, Lefkowitz R J. Curr Opin Cell Biol. 2001;13:139–145. doi: 10.1016/s0955-0674(00)00190-3. [DOI] [PubMed] [Google Scholar]
- 12.McDonald P H, Chow C W, Miller W E, Laporte S A, Field M E, Lin F T, Davis R J, Lefkowitz R J. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 13.Bohn L M, Lefkowitz R J, Gainetdinov R R, Peppel K, Caron M G, Lin F T. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 14.Gainetdinov R R, Bohn L M, Walker J K, Laporte S A, Macrae A D, Caron M G, Lefkowitz R J, Premont R T. Neuron. 1999;24:1029–1036. doi: 10.1016/s0896-6273(00)81048-x. [DOI] [PubMed] [Google Scholar]
- 15.Bohn L M, Gainetdinov R R, Lin F T, Lefkowitz R J, Caron M G. Nature (London) 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 16.Attramadal H, Arriza J L, Aoki C, Dawson T M, Codina J, Kwatra M M, Snyder S H, Caron M G, Lefkowitz R J. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- 17.Stoffel R H, Inglese J, Macrae A D, Lefkowitz R J, Premont R T. Biochemistry. 1998;37:16053–16059. doi: 10.1021/bi981432d. [DOI] [PubMed] [Google Scholar]
- 18.Tomhave E D, Richardson R M, Didsbury J R, Menard L, Snyderman R, Ali H. J Immunol. 1994;153:3267–3275. [PubMed] [Google Scholar]
- 19.Haribabu B, Richardson R M, Fisher I, Sozzani S, Peiper S C, Horuk R, Ali H, Snyderman R. J Biol Chem. 1997;272:28726–28731. doi: 10.1074/jbc.272.45.28726. [DOI] [PubMed] [Google Scholar]
- 20.Harrison J K, Fong A M, Swain P A, Chen S, Yu Y R, Salafranca M N, Greenleaf W B, Imai T, Patel D D. J Biol Chem. 2001;276:21632–21641. doi: 10.1074/jbc.M010261200. [DOI] [PubMed] [Google Scholar]
- 21.Edgell C J, McDonald C C, Graham J B. Proc Natl Acad Sci USA. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lidington E, Nohammer C, Dominguez M, Ferry B, Rose M L. Clin Exp Immunol. 1996;104:66–71. doi: 10.1046/j.1365-2249.1996.d01-660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palczewski K, Saari J C. Curr Opin Neurobiol. 1997;7:500–504. doi: 10.1016/s0959-4388(97)80029-3. [DOI] [PubMed] [Google Scholar]
- 24.Orsini M J, Parent J L, Mundell S J, Benovic J L, Marchese A. J Biol Chem. 1999;274:31076–31086. doi: 10.1074/jbc.274.43.31076. [DOI] [PubMed] [Google Scholar]
- 25.Arai H, Tsou C L, Charo I F. Proc Natl Acad Sci USA. 1997;94:14495–14499. doi: 10.1073/pnas.94.26.14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferguson S S, Downey W E, III, Colapietro A M, Barak L S, Menard L, Caron M G. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 27.Luttrell L M, Lefkowitz R J. J Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 28.Perry S J, Lefkowitz R J. Trends Cell Biol. 2002;12:130–138. doi: 10.1016/s0962-8924(01)02239-5. [DOI] [PubMed] [Google Scholar]
- 29.Pierce K L, Maudsley S, Daaka Y, Luttrell L M, Lefkowitz R J. Proc Natl Acad Sci USA. 2000;97:1489–1494. doi: 10.1073/pnas.97.4.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laporte S A, Oakley R H, Zhang J, Holt J A, Ferguson S S, Caron M G, Barak L S. Proc Natl Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonald P H, Cote N L, Lin F T, Premont R T, Pitcher J A, Lefkowitz R J. J Biol Chem. 1999;274:10677–10680. doi: 10.1074/jbc.274.16.10677. [DOI] [PubMed] [Google Scholar]
- 32.Shenoy S K, McDonald P H, Kohout T A, Lefkowitz R J. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 33.Claing A, Chen W, Miller W E, Vitale N, Moss J, Premont R T, Lefkowitz R J. J Biol Chem. 2001;276:42509–42513. doi: 10.1074/jbc.M108399200. [DOI] [PubMed] [Google Scholar]
- 34.Richardson R M, Ali H, Pridgen B C, Haribabu B, Snyderman R. J Biol Chem. 1998;273:10690–10695. doi: 10.1074/jbc.273.17.10690. [DOI] [PubMed] [Google Scholar]
- 35.Arai H, Monteclaro F S, Tsou C L, Franci C, Charo I F. J Biol Chem. 1997;272:25037–25042. doi: 10.1074/jbc.272.40.25037. [DOI] [PubMed] [Google Scholar]
- 36.Richardson R M, Pridgen B C, Haribabu B, Ali H, Snyderman R. J Biol Chem. 1998;273:23830–23836. doi: 10.1074/jbc.273.37.23830. [DOI] [PubMed] [Google Scholar]
- 37.Kraft K, Olbrich H, Majoul I, Mack M, Proudfoot A, Oppermann M. J Biol Chem. 2001;276:34408–34418. doi: 10.1074/jbc.M102782200. [DOI] [PubMed] [Google Scholar]
- 38.Lombardi M S, Kavelaars A, Schedlowski M, Bijlsma J W, Okihara K L, Van de P M, Ochsmann S, Pawlak C, Schmidt R E, Heijnen C J. FASEB J. 1999;13:715–725. doi: 10.1096/fasebj.13.6.715. [DOI] [PubMed] [Google Scholar]
- 39.Lombardi M S, Kavelaars A, Cobelens P M, Schmidt R E, Schedlowski M, Heijnen C J. J Immunol. 2001;166:1635–1640. doi: 10.4049/jimmunol.166.3.1635. [DOI] [PubMed] [Google Scholar]