

Abstract
Bacterial σ factors combine with the catalytic core RNA polymerase to direct the process of transcription initiation through sequence-specific interactions with the −10 and −35 elements of promoter DNA. In the absence of core RNA polymerase, the DNA-binding function of σ is autoinhibited by its own N-terminal 90 amino acids (region 1.1), putatively by a direct interaction with conserved region 4.2, which binds the −35 promoter element. In the present work, this mechanism of autoinhibition was studied by using a combination of NMR spectroscopy and segmental isotopic labeling of a σ70-like subunit from Thermotoga maritima. Our data argue strongly against a high-affinity interaction between these two domains. Instead we suggest that autoinhibition of DNA binding occurs through an indirect steric and/or electrostatic mechanism. More generally, the present work illustrates the power of segmental isotopic labeling for probing molecular interactions in large proteins by NMR.
The 400-kDa bacterial core RNA polymerase (RNAP) is fully active in RNA polymerization but is incapable of promoter recognition and specific transcription initiation. Promoter recognition, promoter melting to form the open complex, and possibly other functions during transcription initiation, depend on the binding of a σ factor to the core RNAP (subunit composition α2ββ′ω) to form the RNAP holoenzyme (reviewed in ref. 1). The primary σ factor in Escherichia coli, responsible for the bulk of transcription during exponential growth, is σ70. Sequence comparisons reveal that σ70 belongs to a large homologous family of proteins with four regions of highly conserved amino acid sequence (2–5) (Fig. 1).
Figure 1.

Schematic illustrating the conserved regions within σA. The black bar represents the Thermotoga maritima σA primary sequence, with conserved regions within the σ70 family (3, 5) shown as colored boxes and labeled (color coding according to sequence identity with E. coli: green, 20–40%; yellow, 40–60%; orange, 60–80%; red, 80–100%). Sequence identity and similarity with E. coli σ70 are shown above the bar. Below the bar, the T. maritima sequence is shown in the region surrounding conserved region 4.2. Above the sequence is shown the secondary structure (α-helices indicated as rectangles, coils indicated as a line) from the x-ray structure of Thermus aquaticus σA region 4 (10). The colored bars below denote the boundaries of regions 4.1 (yellow) and 4.2 (red). To the left is shown a ribbons diagram of T. aquaticus region 4 (10), with region 4.1 yellow, region 4.2 red. The location of the ligation site is indicated in magenta for both the sequence and the structure.
The σ factors direct the process of transcription initiation by first locating the promoter through sequence-specific recognition of two hexamers of consensus DNA; the Pribnow box or −10 element, centered at about −10 with respect to the transcription start site (+1), and the −35 element (6, 7). Genetic and biophysical evidence strongly suggested that amino acid residues in region 4.2 recognize the −35 element (8, 9), and this was confirmed by structural studies (10). Nonetheless, most functions of σ, including sequence-specific DNA binding, manifest themselves only in the context of the RNAP holoenzyme. In the absence of core RNAP, σ70-like factors are unable to recognize promoter DNA in either double- or single-stranded form (11). Specific interactions between N-terminally truncated derivatives of σ70 and promoter DNA were detected by using competitive filter-binding assays (11, 12), leading to the hypothesis that the latent DNA-binding activity of σ is inhibited by the N-terminal region 1.1, and that this inhibition is relieved by conformational changes upon binding of σ to core RNAP. Studies of isolated σ70 fragments revealed that region 1.1 inhibited region 4.2 binding to the −35 element in trans, but not region 2 binding to the −10 element. These results led to a more detailed model in which region 1.1 directly masks the DNA-binding determinants of region 4.2 (11, 12). In the present work, we set out to characterize and quantify the effect of region 1.1 on σ factor interactions with promoter DNA. In particular, we have directly examined the putative intramolecular interaction between regions 1.1 and 4.2 of a σ70-like factor by using a combination of segmental isotopic labeling (13) and heteronuclear NMR spectroscopy.
Materials and Methods
Cloning and Protein Expression.
The sigA gene encoding σA from T. maritima was cloned as previously described (5). Wild-type σA[1–399] and Δ1.1-σA[137–399] were expressed in E. coli BL21(DE3)pLysS cells by using the vector pET15b (Novagen) and purified by Ni2+-charged Hi-Trap affinity chromatography (Pharmacia) followed by gel filtration chromatography on a Superdex 75 column (Pharmacia). Region 4.2 of σA (residues 349–399) was expressed in E. coli BL21(DE3) cells as a His-tagged fusion by using the vector pET28 (Novagen). The sequence -MIEGRCG-, which contains a factor Xa cleavage site, was inserted between the poly(His) tag and region 4.2. Note that introduction of a Gly residue immediately after the Cys was found to greatly improve the yield of the cleavage reaction (see below). Triple-labeled fusion protein was prepared by growing the cells in M9 minimal medium in 2H2O containing 0.2% [U-13C]glucose and 0.1% 15NH4Cl. His-tagged proteins were purified by affinity chromatography on Ni-NTA-beads (Qiagen, Chatsworth, CA; NTA, nitrilotriacetate) followed by preparative reversed-phase (RP)-HPLC (Vydac C18 column). The poly(His) tag was removed by factor Xa treatment and the desired protein, CG-σA[349–399], was further purified by preparative RP-HPLC. The intein fusion proteins σA[1–348]-GyrA-CBD and Δ1.1-σA[137–348]-GyrA-CBD (CBD, chitin-binding domain) were expressed in E. coli BL21(DE3)pLysS+ cells by using the vector pTXB1 (New England Biolabs). Both fusion proteins were purified by affinity chromatography on chitin-agarose beads (New England Biolabs). T4 AsiA was expressed in E. coli BL21(DE3) cells as an in-frame fusion protein to σA-[349–399] by using the vector pET28 (Novagen). A poly(His) tag and a thrombin cleavage site were introduced between the AsiA and σA coding sequences. After affinity purification on Ni-NTA beads, the fusion protein was cleaved with thrombin and the desired AsiA product was further purified by RP-HPLC and then refolded. Use of this fusion strategy was found to greatly improve the expression levels of AsiA.
Preparation of σA* and Δ1.1-σA*.
Segmental labeled proteins were obtained by chemically ligating the ethyl α-thioester derivatives of either σA[1–348] or Δ1.1-σA[137–348] with [U-2H,13C,15N]- CG-σA[349–399]. The protein thioesters were generated in situ in the ligation mixture by thiolysis of chitin beads containing either σA[1–348]-intein-CBD or σA[137–348]-intein-CBD. The ligation reaction was carried out in ligation buffer (1 mM EDTA/25 mM NaPi/200 mM guanidinium chloride/250 mM NaCl buffer at pH 7.2) containing 0.2% octyl glucoside and 3% (vol/vol) ethanethiol. Equimolar amounts of the two fragments were used at a concentration of ≈50 μM each. Reactions were allowed to proceed at room temperature overnight, after which the slurry was filtered and the beads were washed several times with ligation buffer. All washes were combined with the supernatant. The protein samples were then concentrated and exchanged into storage buffer (20 mM CHAPSO/20 mM DTT/30 mM Tris⋅HCl/100 mM NaCl buffer at pH 7.6; CHAPSO is 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate).
Abortive Initiation Assay.
Reactions were performed in 10 μl of standard transcription buffer (20 mM Tris⋅HCl, pH 8.0/50 mM NaCl/5 mM MgCl2); 0.1 μM E. coli core RNAP was incubated with 0.3 μM σ or σ derivatives for 15 min at 37°C. Reactions were initiated by the addition of 0.1 μM T7 A1 promoter fragment, 0.5 mM ApU initiating dinucleotide, and 1 μl of [α-32P]CTP. The reactions proceeded for 15 min at 37°C, then were terminated by the addition of an equal volume of gel loading buffer (8 M urea/90 mM Tris/64.6 mM boric acid/2.5 mM EDTA, pH 8.3). Reaction products were separated by polyacrylamide gel electrophoresis on a 23% gel in 7 M urea and visualized by PhosphorImager (Molecular Dynamics).
In Vitro DNA-Binding Assays.
The affinity of the σA constructs for −35 promoter duplex DNA were measured with a fluorescence-based titration assay using the 5′-fluorescein-labeled sequence (5′-AGGTATTGACAACATG-3′ = sense strand) derived from the T7 A1 promoter. Experiments were conducted at 25°C on a Spex Fluorolog-3 instrument using excitation and emission wavelengths of 488 nm and 525 nm, respectively. In all cases, the DNA was kept at 0.1 μM in assay buffer (20 mM CHAPSO/30 mM Tris⋅HCl, pH 7.2) containing either no NaCl or 100 mM NaCl. Control studies used the nonspecific 5′-fluorescein-labeled sequence, 5′-GATAGAAGTAGTAGTA-3′ The dissociation constants were determined by changes in the polarization of the fluorescence upon addition of the corresponding σA construct at defined concentrations; calculations were made assuming formation of 1:1 complex. All experiments were performed at least in duplicate.
NMR Spectroscopy.
NMR samples were prepared by exchanging the corresponding pure protein (final concentration 100–200 μM) into a buffer containing 30 mM Tris⋅HCl at pH 7.6, 100 mM NaCl, 20 mM CHAPSO, 20 mM [2H10]DTT, 0.1% NaN3, and 10% (vol/vol) 2H2O. Unlabeled −35 element promoter DNA or T4 AsiA was added to the σA* and Δ1.1-σA* NMR samples to a final molar ratio of 1.2:1. 1H{15N} HSQC-TROSY (ref. 14; HSQC is heteronuclear single-quantum correlation and TROSY is transverse relaxation optimized spectroscopy) and 1H{13C} constant-time HSQC (15) spectra were collected at 35°C on a Bruker DMX spectrometer operating at a 1H frequency of 600 MHz with 1,000 scans per transient. We collected 512 complex points in 1H, 15N, and 13C dimensions and multiplied by a cosine-bell window function and zero-filled to 1,000 points before Fourier transformation by using xwinnmr (Bruker Instruments). The corresponding sweep-widths were 12.5 ppm, 30 ppm, and 70 ppm in 1H, 15N, and 13C dimensions, respectively. The uniform 13C,15N,2H-labeling procedure used in this work afforded low proton density in region 4.2 within otherwise protonated σA* and Δ1.1-σA* samples. Protonation of methyl and methylene groups of region 4.2 is highly selective and amino acid specific (A.S. and D.C., unpublished results).
1H{15N} HSQC-TROSY and 1H{13C} HSQC NMR spectra of all σA samples prepared as part of this study are shown in Figs. 6–23, which are published as supporting information on the PNAS web site, www.pnas.org.
Results
Design and Biosynthesis of Segmental Isotopic Labeled σA Constructs.
All of our studies were carried out with σA (or derivatives thereof) from T. maritima (5, 16). This σ factor has a high degree of sequence similarity to σ70 from E. coli (Fig. 1), but it is considerably smaller (47 kDa vs. 70 kDa) and more thermostable (the optimal growth temperature for T. maritima is 80°C), making it more suitable for NMR studies. Two σA constructs were prepared for NMR analysis; one corresponding to the full-length sequence (σA*) and the other lacking region 1.1 (Δ1.1-σA*). The site of truncation within Δ1.1-σA* was based on alignment of the T. maritima σA and E. coli σ70 sequences. In both cases, region 4.2 was uniformly labeled with 15N, 13C, and 2H. This segmental isotopic labeling pattern was accomplished by using expressed protein ligation (EPL) (17). The only requirements for this reaction are an N-terminal Cys residue (α-Cys) in the C-terminal fragment and an α-thioester moiety in the N-terminal fragment (18). Because there are no Cys residues within σA, it was necessary to introduce a Cys between residues Gly-348 and Lys-349 in the sequence (Fig. 1). The ligation junction is in a loop between subregions 4.1 and 4.2 (10), an area that is poorly conserved among σ70-like factors (3, 5). Furthermore, we have shown previously that a Cys residue is functionally tolerated at this position in E. coli σ70 (19).
Triple-labeled region 4.2, containing the required α-Cys residue for ligation, was prepared by using a proteolysis strategy (Fig. 2A). The N-terminal fragments (either residues 1–348 for σA* or residues 137–348 for Δ1.1-σA*) were both expressed as C-terminal fusions to a modified GyrA intein (Fig. 2A). Treatment of the purified fusion proteins with ethanethiol yielded the desired ethyl α-thioester derivatives for expressed protein ligation. The σA* and Δ1.1-σA* ligation reactions were initiated by mixing equimolar amounts of the appropriate fragments. Both ligation reactions were found to be extremely rapid and efficient. Remarkably, both obeyed simple first-order kinetics (Fig. 2B), whereas a second-order process was expected. Inclusion of 6 M guanidinium chloride in the ligation buffer dramatically inhibited the reactions (data not shown), suggesting that the rapid, unimolecular kinetics observed under physiological conditions required the native folded states of the two fragments. This finding strongly suggests that in isolated σ, region 4.2 interacts with structural elements in the rest of σ (residues 1–348). Based on the crystal structure of region 4 from Thermus aquaticus (10), it is likely that at least part of this interaction surface involves region 4.1 (Fig. 1). It is interesting to note that the presence or absence of region 1.1 had very little effect on the ligation kinetics (Fig. 2B). The extraordinary efficiency of the two reactions meant that a simple solvent exchange and concentration step yielded the desired high-purity NMR samples (Fig. 3A).
Figure 2.

Segmental isotopic labeling of region 4.2 from T. maritima σA. (A) Expressed protein ligation strategy used to prepare segmental labeled σA* and Δ1.1-σA* constructs. (B) Kinetics of σA* and Δ1.1-σA* reactions. Ligation reactions were followed by SDS/PAGE, and reaction progress was quantified from the SDS/PAGE bands; curves were fit to a first-order kinetic function.
Figure 3.

Characterization of σA* and Δ1.1-σA*. (A) Electrospray mass spectra of purified σA* and Δ1.1-σA* (predicted masses are in parenthesis). (B) Abortive transcription initiation reactions on a T7 A1 promoter in the presence of E. coli RNAP core and the indicated σ factor. The reaction products were resolved by denaturing PAGE and visualized by autoradiography. (C) In vitro DNA-binding assays at low and high ionic strength. Shown is the change in fluorescence-polarization of 5′-fluorescein-labeled −35 promoter DNA as a function of added σA* or Δ1.1-σA*.
Purified σA* and Δ1.1-σA* were both active in an in vitro abortive initiation assay using E. coli core RNAP and a −10/−35 promoter, T7 A1 (Fig. 3B). Moreover, the level of activity for the two ligated proteins was indistinguishable from that of the control molecules σA and Δ1.1-σA that were prepared by standard expression techniques and lacked the two mutations between regions 4.1 and 4.2 (Fig. 3B, compare lanes 4 and 5 with 6 and 7). It is interesting to note that the presence or absence of region 1.1 in σA had no effect on the level of transcription in this assay. All σA constructs tested were only slightly less active than E. coli σ70 (Fig. 3B, lane 3). This result almost certainly stemmed from the use of E. coli core RNAP in the in vitro assays. Presumably, this polymerase slightly prefers σ70 over σA.
Binding of σA* Constructs to Promoter DNA.
The ability of the σA* constructs to bind promoter DNA was tested by using fluorescence spectroscopy. Binding isotherms were obtained by measuring the change in polarization of a solution of a 5′-fluorescein-labeled double-stranded DNA containing the −35 element from the T7 A1 promoter upon adding increasing amounts of the corresponding σA* construct (Fig. 3C). In the absence of salt in the buffer, both σA constructs were able to bind the −35 promoter DNA; however Δ1.1-σA* had a dissociation constant around 1/10 that of σA* (Kd = 0.6 ± 0.06 μM vs. 6.8 ± 0.07 μM). In contrast, at higher ionic strength (100 mM NaCl), full-length σA* bound the −35 promoter DNA at least two orders of magnitude more weakly than Δ1.1-σA* (Kd = 0.7 ± 0.07 μM for Δ1.1-σA*). These equilibrium-binding measurements suggest that the interaction between full-length σA* and −35 promoter DNA is weak and rather nonspecific. Indeed, σA* bound a nonspecific DNA sequence with a Kd of ≈11 μM under low-salt conditions (data not shown). Importantly, deletion of region 1.1 allows the truncated σA factor, Δ1.1-σA*, to make tight and specific interactions with the −35 promoter DNA. These results are in good agreement with those found previously by Gross and coworkers with E. coli σ70 (11, 12).
Solution NMR Studies.
Segmental isotopic labeling allows a specific domain within a protein to be examined by NMR spectroscopy; signals from unlabeled regions of the protein can be filtered out by using heteronuclear correlation experiments, leaving signals from only the labeled part of the protein (13). Thus, this technique significantly reduces the spectral complexity for large proteins and allows a variety of solution-based NMR strategies to be applied. In the present study, we were interested in simply comparing the spectra of the segmentally labeled σA* constructs. Changes in the structure around region 4.2 would be reflected in chemical shift perturbations in the HSQC spectra, which essentially provide structural fingerprints. Thus, it should be possible to determine whether or not regions 1.1 and 4.2 are in direct contact.
Fig. 4 shows the 1H{15N} HSQC-TROSY and 1H{13C} HSQC spectra of region 4.2 in the context of Δ1.1-σA* and in σA*. Both pairs of spectra have well-dispersed signals indicating that region 4.2 has assumed a defined tertiary fold in Δ1.1-σA* and σA* (Fig. 4 A, B and D, E). In contrast, the 1H{15N} and 1H{13C} fingerprints obtained for the isolated region 4.2 indicate that it lacks a defined fold in solution; many peaks are overlapping, characteristic of residues of the same amino acid type having virtually indistinguishable structural environments (see supporting information). This conclusion is also supported by far-UV circular dichroism studies that indicate an absence of secondary structure in the isolated domain (data not shown). Presumably, region 4.2 makes contacts in Δ1.1-σA* and in σA that stabilize its tertiary fold, and it is likely that the interaction with region 4.1 provides some of this stabilization energy (Fig. 1) (10).
Figure 4.

Effect of context on the solution structure of σA region 4.2. (A and B) 1H{15N} HSQC-TROSY spectra of σA* (A) and Δ1.1-σA* (B). (D and E) 1H{13C} HSQC spectra of σA* (D) and Δ1.1-σA* (E). (C and F) Comparison of the reconstructed 1H{15N} HSQC-TROSY (C) and 1H{13C} HSQC (F) spectra of Δ1.1-σA* (circles) and σA* (crosses), using chemical shifts extracted from the individual spectra (A, B and D, E).
Surprisingly, we find that the 1H{15N} HSQC-TROSY and 1H{13C} HSQC spectra for Δ1.1-σA* and in σA* are nearly superimposable (Fig. 4 C and F). The extent of this similarity is reflected in the very low spectral similarity factors (SSFs) calculated for the two sets of spectra (see Table 1). This finding is inconsistent with a strong interaction over a significant interface between region 1.1 and region 4.2. As a control for the sensitivity for the chemical shift perturbation method, we measured the effect on the NMR spectra of adding ligands known to bind to region 4.2. Addition of T4 AsiA, a ≈90-residue anti-σ factor known to bind to E. coli σ70 region 4.2 (19, 20), resulted in significant changes in the NMR spectra of Δ1.1-σA* (Fig. 5 A and B and Table 1). Binding of AsiA to region 4.2 of σA was expected because region 4.2 is highly homologous in σA and σ70 (Fig. 1). Interestingly, addition of AsiA to σA* did not result in changes in the HSQC spectra, and hence the calculated SSFs are small (Table 1). This finding indicates that the presence of region 1.1 inhibits binding of AsiA to region 4.2 of σA. A similar pattern of results was obtained when −35 element promoter DNA was added to the NMR samples—i.e., the NMR spectra of Δ1.1-σA* were significantly perturbed, whereas those of σA* were not (Fig. 5 C and D and Table 1). Collectively, these NMR data demonstrate that region 1.1 and region 4.2 do not directly interact in σA.
Table 1.
Spectral similarity factors (SSFs) for comparisons of two-dimensional spectra of segmentally labeled σA* constructs and their liganded forms
| a | b | Comparison between NMR spectra a and b
|
|||
|---|---|---|---|---|---|
|
1H{15N} HSQC-TROSY
|
1H{13C} HSQC
|
||||
| SSF,† Hz | Total no. of peaks in spectra a and b‡ | SSF,† Hz | Total no. of peaks in spectra a and b‡ | ||
| Δ1.1-σA* | σA* | 3 | 48, 48 | 1.1 | 102, 102 |
| Δ1.1-σA* | Complex of Δ1.1-σA* with AsiA | 13 | 48, 48 | 23 | 102, 92 |
| Δ1.1-σA* | Complex of Δ1.1-σA* with promoter DNA | 62 | 48, 29 | 49 | 102, 102 |
| σA* | Complex of σA* with AsiA | 0.5 | 48, 48 | 0.9 | 102, 102 |
| σA* | Complex of σA* with promoter DNA | 0.3 | 48, 48 | 0.7 | 102, 102 |
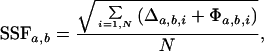 |
where for peaks within a 300-Hz radius of each other Δa,b,i = (ωH,a,i − ωH,b,i)2 + (ωX,a,i − ωX,b,i)2 and Φa,b,i = 0 and for unmatched peaks outside the 300-Hz radius Δa,b,i = 0 and Φa,b,i = (300 Hz)2, N = max(Na, Nb), ωH(X),a(b),i is the resonant proton (heteronuclear) frequency (Hz) of the ith peak of the NMR spectrum a(b), and Na(b) is a total number of peaks in the compared NMR spectra a and b.
We used the xeasy program (24) to create a peak list for the spectrum a. This peak list was used for the spectrum b and the peak positions were visually inspected and manually adjusted.
Figure 5.

Binding of T4 AsiA and promoter DNA to Δ1.1-σA*. (A and C) 1H{15N} HSQC-TROSY spectra of Δ1.1-σA* with 1.2 molar equivalents of purified AsiA (A) and promoter DNA (C). (B and D) Reconstructed comparisons of the 1H{15N} HSQC spectra of Δ1.1-σA* with (crosses) and without (circles) AsiA (B) and DNA (D).
Discussion
Upon association with core RNAP, many bacterial σ factors are converted from an autoinhibited state, which cannot interact with promoter DNA, to an active form that can. Biochemical and genetic studies on E. coli σ70 have implicated the most N-terminal 90 amino acids, region 1.1, in this autoinhibitory process (11, 12). Consistent with this idea, fluorescence-based measurements performed in the current study indicate that removal of region 1.1 from a closely related σ factor, σA from T. maritima, increases the binding affinity for −35 element promoter DNA by at least 100-fold. The prevailing view in the field is that region 1.1 elicits its inhibitory effect by masking the DNA-binding residues within region 4.2 (11, 12).
The NMR data presented in the current study do not support a direct high-affinity interaction between region 1.1 and region 4.2. We were unable to detect any significant chemical shift perturbations within region 4.2 when region 1.1 was removed. In contrast, dramatic perturbations were observed when known ligands to region 4.2 were added to Δ1.1-σA*. There are additional biochemical observations that further argue against a direct region 1.1/region 4.2 association. Hydroxy-radical-footprinting studies have shown that region 4.2 of σ70 is only weakly protected from cleavage in the autoinhibited state compared with the active core-RNAP-bound state (21). Moreover, mutation of Arg-588 within the recognition helix of σ70 region 4.2 does not relieve region 1.1-mediated inhibition (12) and, as shown here, the apparent first-order kinetics of ligation are essentially unaffected by removal of region 1.1.
If region 1.1 does not interact directly with region 4.2, then how does it inhibit DNA binding? Conceivably, region 1.1 could protect the ligand-binding surface of region 4.2 by some indirect steric mechanism. Our observation that region 1.1 reduces the affinity of region 4.2 for AsiA is also consistent with this idea. Analysis of region 1.1 from σA reveals a high content of Asp and Glu residues (calculated pI = 4.4). Thus, it is possible that region 1.1 also inhibits promoter binding through an electrostatic mechanism based on mutual charge repulsion between itself and DNA. Such steric and/or electrostatic inhibition requires that region 1.1 and region 4.2 are in close proximity in the autoinhibited state, but not necessarily in direct contact. The location of the primary binding site for region 1.1 with σ70-like factors remains unknown. It is worth noting that the competition binding experiments performed by Gross and coworkers (11) used a fragment of σ70 that comprised all of region 4 and part of region 3. The NMR data presented herein, thus, focus attention on regions 3 and 4.1 as potential binding sites for region 1.1. This possibility awaits further structural and biochemical investigation.
Segmental isotopic labeling permits the observation of domain–domain interactions, without the absolute requirement for assignments, because the labeled residues are known from the molecular synthesis. While lack of full sequential assignment lowers the resolution of any mapping (cf. ref. 22), the ability to observe directly the effects of ligands on a domain is dependent solely on its segmental labeling and appropriate NMR observation. The development of TROSY for 15N labeling (23) and the spin dilute 13C labeling method illustrated here and to be described more fully elsewhere (A.S. and D.C., unpublished work) provide such NMR windows into molecular masses of 100 kDa and more. This ability to localize molecular interactions provides a unique tool for the production of domain-specific probes in large proteins.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants GM59908 (T.W.M.), GM53759 (S.A.D.), GM 47021 (D.C.), and GM31625 (D.A.B.). A.S. is a recipient of National Cancer Institute Fellowship F037244.
Abbreviations
- CBD
chitin-binding domain
- CHAPSO
3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate
- HSQC
heteronuclear single-quantum correlation
- RNAP
RNA polymerase
- SSF
spectral similarity factor
- TROSY
transverse relaxation optimized spectroscopy
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Gross C A, Chan C, Dombroski A, Gruber T, Sharp M, Tupy J, Young B. Cold Spring Harbor Symp Quant Biol. 1998;63:141–155. doi: 10.1101/sqb.1998.63.141. [DOI] [PubMed] [Google Scholar]
- 2.Gribskov M, Burgess R R. Nucleic Acids Res. 1986;14:6745–6763. doi: 10.1093/nar/14.16.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lonetto M, Gribskov M, Gross C A. J Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stragier P, Parsot C, Bouvier J. FEBS Lett. 1985;187:11–15. doi: 10.1016/0014-5793(85)81203-5. [DOI] [PubMed] [Google Scholar]
- 5.Gruber T M, Bryant D A. J Bacteriol. 1997;179:1734–1747. doi: 10.1128/jb.179.5.1734-1747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harley C B, Reynolds R P. Nucleic Acids Res. 1987;15:2343–2361. doi: 10.1093/nar/15.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawley D K, McClure W R. Nucleic Acids Res. 1983;11:2237–2255. doi: 10.1093/nar/11.8.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siegele D A, Hu J C, Walter W A, Gross C A. J Mol Biol. 1989;206:591–603. doi: 10.1016/0022-2836(89)90568-8. [DOI] [PubMed] [Google Scholar]
- 9.Gardella T, Moyle H, Susskind M M. J Mol Biol. 1989;206:579–590. doi: 10.1016/0022-2836(89)90567-6. [DOI] [PubMed] [Google Scholar]
- 10.Campbell E A, Muzzin O, Chlenov M, Sun J L, Olson C A, Weinman O, Trester-Zedlitz M L, Darst S A. Mol Cell. 2002;9:527–539. doi: 10.1016/s1097-2765(02)00470-7. [DOI] [PubMed] [Google Scholar]
- 11.Dombroski A J, Walter W A, Record M T, Jr, Siegele D A, Gross C A. Cell. 1992;70:501–512. doi: 10.1016/0092-8674(92)90174-b. [DOI] [PubMed] [Google Scholar]
- 12.Dombroski A J, Walter W A, Gross C A. Genes Dev. 1993;7:2446–2455. doi: 10.1101/gad.7.12a.2446. [DOI] [PubMed] [Google Scholar]
- 13.Xu R, Ayers B, Cowburn D, Muir T W. Proc Natl Acad Sci USA. 1999;96:388–393. doi: 10.1073/pnas.96.2.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pervushin K, Wider G, Wüthrich K. J Biomol NMR. 1998;12:345–348. doi: 10.1023/A:1008268930690. [DOI] [PubMed] [Google Scholar]
- 15.Cavanagh J, Fairbrother W J, Palmer A G, Skelton N J. Protein NMR Spectroscopy. San Diego: Academic; 1996. [Google Scholar]
- 16.Huber R, Stetter K O. In: The Prokaryotes. Balows A, editor. Berlin: Springer; 1992. pp. 3809–3815. [Google Scholar]
- 17.Muir T W. Synlett. 2001;6:733–740. [Google Scholar]
- 18.Dawson P E, Kent S B H. Annu Rev Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 19.Severinov K, Muir T W. J Biol Chem. 1998;273:16205–16209. doi: 10.1074/jbc.273.26.16205. [DOI] [PubMed] [Google Scholar]
- 20.Severinova E, Severinov K, Darst S A. J Mol Biol. 1998;279:9–18. doi: 10.1006/jmbi.1998.1742. [DOI] [PubMed] [Google Scholar]
- 21.Nagai H, Shimamoto N. Genes Cells. 1997;2:725–734. doi: 10.1046/j.1365-2443.1997.1600357.x. [DOI] [PubMed] [Google Scholar]
- 22.Ayed A, Mulder F A A, Yi G-S, Lu Y, Kay L E, Arrowsmith C H. Nat Struct Biol. 2001;8:756–760. doi: 10.1038/nsb0901-756. [DOI] [PubMed] [Google Scholar]
- 23.Riek R, Pervushin K, Wüthrich K. Trends Biochem Sci. 2000;25:462–468. doi: 10.1016/s0968-0004(00)01665-0. [DOI] [PubMed] [Google Scholar]
- 24.Bartels C, Xia T, Gunter P, Billeter M, Wüthrich K. J Biomol NMR. 1995;5:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.