

Abstract
Cyclooxygenase (COX)-1 or -2 and prostaglandin (PG) synthases catalyze the formation of various PGs and thromboxane (TX) A2. We have investigated the expression and activity of COX-1 and -2 during human megakaryocytopoiesis. We analyzed megakaryocytes from bone marrow biopsies and derived from thrombopoietin-treated CD34+ hemopoietic progenitor cells in culture. Platelets were obtained from healthy donors and patients with high platelet regeneration because of immune thrombocytopenia or peripheral blood stem cell transplantation. By immunocytochemistry, COX-1 was observed in CD34+ cells and in megakaryocytes at each stage of maturation, whereas COX-2 was induced after 6 days of culture, and remained detectable in mature megakaryocytes. CD34+ cells synthesized more PGE2 than TXB2 (214 ± 50 vs. 30 ± 10 pg/106 cells), whereas the reverse was true in mature megakaryocytes (TXB2 8,440 ± 2,500 vs. PGE2 906 ± 161 pg/106 cells). By immunostaining, COX-2 was observed in <10% of circulating platelets from healthy controls, whereas up to 60% of COX-2-positive platelets were found in patients. A selective COX-2 inhibitor reduced platelet production of both PGE2 and TXB2 to a significantly greater extent in patients than in healthy subjects. Finally, we found that COX-2 and the inducible PGE-synthase were coexpressed in mature megakaryocytes and in platelets. We conclude that both COX-isoforms contribute to prostanoid formation during human megakaryocytopoiesis and that COX-2-derived PGE2 and TXA2 may play an unrecognized role in inflammatory and hemostatic responses in clinical syndromes associated with high platelet turnover.
Prostanoids are biologically active metabolites of arachidonic acid (AA), formed through the sequential action of phospholipases, cyclooxygenase (COX)-1 or -2, and terminal prostaglandin (PG) synthases, which then generate PGE2, PGF2α, PGD2, PGI2, or thromboxane (TX)A2, known collectively as prostanoids (1). Despite the remarkable structural and functional homologies, COX-1 and -2 are encoded by different genes, subserve distinct functions, and have been shown to preferentially couple with different isoforms of PG synthases (1).
COX-1 generally serves physiologic, housekeeping functions, such as generation of proaggregatory TXA2 by platelets, and cytoprotective PGE2 by the gastric mucosa (1). In contrast, COX-2 is virtually undetectable under resting conditions and is induced by cytokines, endotoxins, growth factors, or tumor promoters (1). However, this simplified paradigm of constitutive COX-1 and inducible COX-2, has many exceptions: COX-1 can be regulated during development or by some hormones and growth factors (1–3), whereas COX-2 is constitutively expressed in the brain, reproductive tissues, kidney, and thymus (4).
Circulating blood cells are known to diversely express active COX isoforms: polymorphonuclear leukocytes and monocytes up-regulate COX-2 and produce PGs on cytokine, phagocytic or growth factor stimuli (3), which contribute to the local inflammatory/immunologic responses. Whereas the pathophysiologic significance of COX-1-dependent TXA2 production in human platelets is well established from biochemical measurements and aspirin trials (5), the presence of COX-2 in platelets and its biological significance are highly controversial (6, 7). Moreover, the expression and function of COX isozymes and prostanoids in hematopoietic progenitors and lineages remain largely unknown. Increasing evidence supports a role for PGs in the intrathymic development of mouse and human T cells (2, 3). In fact, murine CD4−CD8− lymphoid progenitors, which originate from hematopoietic tissues, require COX-1- and COX-2-dependent prostanoids at different stages to complete their maturation (2, 3). Fewer studies using human monocytic cell lines differentiated in vitro suggest a role for COX and PGs in the myeloid maturation (8, 9). Recent reports indicate the presence of COX-1 and PGs during differentiation of megakaryoblastic cell lines in vitro (10–14).
Physiologic megakaryocyte maturation occurs in the bone marrow from hematopoietic progenitors, and mature platelets originate from the fragmentation of cytoplasmic protrusions of megakaryocytes (15). The low number of megakaryocytes in normal bone marrow (≈0.05% of total mononuclear cells) hampers the direct study of megakaryocytopoiesis, only allowing the morphological examination of different stages of maturation. However, culture systems for hematopoietic progenitors and the discovery of thrombopoietin (TPO) have offered new insights into megakaryocytopoiesis (16, 17). In fact, TPO can drive full megakaryocyte development from CD34+ hematopoietic stem cells in culture (15–17).
Using these tools, together with the morphological characterization of megakaryocytes in bone marrow, we have studied the expression and activity of COX-1 and COX-2 during human megakaryocytopoiesis. We have also tried to clarify the controversial pattern of COX-2 expression in peripheral platelets and the contribution of COX-2 to prostanoid synthesis from activated platelets. Our study demonstrates that both COX isoforms contribute to prostanoid formation during megakaryopoiesis and suggests that COX-2-derived prostanoids may play unrecognized roles in clinical syndromes associated with accelerated platelet regeneration.
Materials and Methods
Subjects.
Bone samples were obtained from the iliac crests of three subjects undergoing orthopedic surgery (one male (M) aged 18, two females (F) aged 25 and 50), with normal hematological parameters. For CD34+ purification, umbilical cord blood specimens collected according to institutional guidelines were obtained during normal deliveries from 22 healthy females. Patients (Table 1) and healthy subjects (n = 10, five M, five F, age range 29–51 yr) recruited for whole blood studies did not take any aspirin, nonsteroidal antiinflammatory drugs (NSAIDs), corticosteroids, or COX-2 inhibitors (Coxibs) in the 10 days before blood withdrawal. Informed consent was obtained from all subjects participating in the studies, and the protocol was approved by the Institutional Review Board of the Catholic University of Rome.
Table 1.
Patient characteristics
| Patient ID | Sex | Age | Diagnosis | Platelet count, ×103/μl | Reticulated platelets, %* |
|---|---|---|---|---|---|
| 1 | M | 36 | ITP | 52.000 | 15 |
| 2 | F | 39 | MM/allo | 38.000 | 10 |
| 3 | F | 36 | MM/allo | 38.000 | 6 |
| 4 | M | 24 | MM/auto | 25.000 | 5 |
| 5 | M | 42 | NHL/auto | 19.000 | 6 |
| 6 | M | 40 | CML/allo | 49.000 | 8 |
Normal values: 1.2 ± 0.6% (20). MM, multiple myeloma; CML, chronic myeloid Leukemia; NHL, non Hodgkin's lymphoma; ITP, immune thrombocytopenia; allo, allogeneic peripheral blood stem cell transplant; auto, autologous peripheral blood stem cell transplant.
CD34+ Cell Purification and Cultures.
For CD34+ cells, mononuclear cells were isolated from cord blood with Ficoll-Paque gradient as previously described (16), by using the MiniMACS magnetic cell sorting program (Miltenyi Biotec, Auburn, CA). Purity of CD34+ cells averaged 85–98% of the cell population. CD34+ cells were differentiated in TPO-treated liquid suspension cultures as previously described (17). Briefly, CD34+ cells were resuspended in a serum-free Iscove's modified Dulbecco's medium (IMDM; GIBCO) with 10−4 M BSA-adsorbed cholesterol, nucleosides (10 μg/ml), 0.5% BSA (fraction V Cohn), 10 μg/ml insulin, 200 μg/ml iron-saturated transferrin, 5 × 10−5 M β-mercaptoethanol (all from Sigma), and 100 ng/ml human recombinant TPO (Genzyme). Every 3 days, cultures were amplified by removing half medium, substituted with fresh medium. Viable cells were scored by Trypan blue dye exclusion. Each well was supplemented with 100 ng/ml of fresh TPO.
Phenotypic Analyses.
Phenotype of purified or cultured cells was analyzed by FACScan (Becton Dickinson). Staining was performed with 10 μg of CD61, CD42b, or CD34 fluorescein-conjugated mAbs (FITC, Serotec) as already described (17).
Reticulated platelets were determined by the thiazole orange method in platelet-rich plasma according to the method of Ault with minor modifications (18).
Immunohistochemistry.
For immunocytochemistry on platelets, blood was withdrawn in tubes containing 0.38% sodium citrate (final concentration) and 10 μM PGE1 (Cayman Chemicals, Ann Arbor, MI). Platelet-rich plasma was obtained by centrifuging at 300 × g for 15 min. Platelets or cells, as indicated, were washed twice with PBS, layered on slides, fixed in ice cold 70% methanol/30% acetone, and kept at −80°C until use. Bone marrow biopsies were fixed 12 h in 4% formalin, decalcified in formic acid for 4 h, embedded in paraffin, cut at 3–5 mm, mounted on aminosilane-coated slides, and dewaxed. Slides were rehydrated, treated with 0.3% H2O2/methanol for 10 min to block endogenous peroxidase, and incubated for 1 h with one of the following primary Abs: COX-1 polyclonal Ab at 1/100 dilution (19), COX-2 polyclonal Ab at 1/200 dilution (20), COX-2 mAb at 1/100 dilution (19), CD34 at 1/100 dilution, CD61 and CD42b mAbs at 1/50 dilution (YLEM, Rome, Italy), and inducible PGE2 synthase (PGES) polyclonal Ab (Cayman Chemicals; 1/50 dilution). Labeling specificity was checked by including single staining, omitting specific Ab or with an irrelevant Ab. For immunoperoxidase, ABC-peroxidase technique (Vector Laboratories) was used. Endogenous biotin was saturated by biotin blocking kit. Peroxidase was developed with the DAB substrate kit (Vector Laboratories). For immunofluorescence, Abs were detected by using goat anti-mouse FITC and/or goat antirabbit tetramethylrhodamine B isothiocyanate Abs (Dako). Specimens were observed and digitalized by a fluorescence Zeiss Axioskop (Zeiss, Jena, Germany) equipped with an intensified charge-coupled device (CCD) camera system (Photometrics, Tucson, AZ).
Arachidonate Stimulation and Inhibitor Studies.
Cells were harvested from cultures, and viable cells were scored by Trypan blue, washed twice with Hanks' balanced salt solution (HBSS) with 1 mg/ml BSA, and incubated for 40 min in 1 ml of the same buffer with 10 μM AA (Cayman Chemicals). Supernatants were collected and stored at −80°C until use.
In inhibitor experiments, cells were preincubated for 40 min with HBSS/BSA containing different concentrations of SC-560, NS-398, valeryl salicylate, or with indomethacin (all from Cayman Chemicals). Indomethacin was solubilized in ethanol; NS-398 and SC-560 stocks were prepared in DMSO. Aliquots from stocks were added to fresh HBSS/BSA, and each preparation contained ≤0.1% ethanol or DMSO (vol/vol, final concentration), as did the control medium. Cells were washed with HBSS/BSA and incubated for 40 min in the same buffer containing 10μM AA and various concentrations of inhibitors. Supernatants were frozen for prostanoid measurements.
Measurement of Prostanoids.
PGE2 and TXB2 were determined in the supernatants of cell cultures or in sera by using previously validated radioimmunoassays (21, 22).
Whole Blood Studies.
Blood was collected and immediately placed in glass tubes containing vehicle, 50 μM aspirin or 10 μM NS-398. Serum TXB2 and PGE2 production during whole blood clotting was measured as a reflection of thrombin-stimulated COX activity, as previously described (22).
Statistical Analysis.
Results were evaluated by using ANOVA with subsequent comparisons by Student's t test for paired or non-paired data, as appropriate. Statistical significance was defined as P < 0.05. Values are reported as means ± 1 SD. The IC50s were calculated by using grafit Software.
Results
Megakaryocyte Studies.
Immunohistochemistry of bone marrow sections showed that megakaryocytes express COX-1 (Fig. 1), which is consistent with the current notion that platelets express only COX-1. Quite unexpectedly, megakaryocytes stained positively also for COX-2 (Fig. 1), indicating that both COX isoforms are present in human megakaryocytes.
Figure 1.
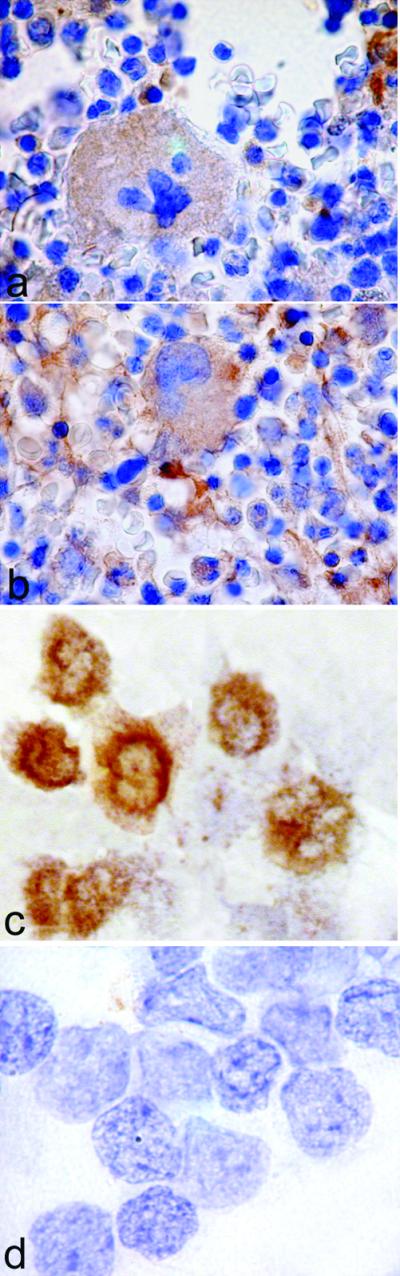
(a and b) Bone marrow biopsies were reacted with anti-COX-1 (a) or anti-COX-2 (b) Ab, and peroxidase-labeled secondary Ab. Megakaryocytes were positive for both COX-1 (a) and COX-2 (b); original magnifications: ×400. (c and d) Cytospins of purified CD34+ cells were stained with anti-COX-1 Ab (c) or anti-COX-2 (d) and peroxidase-labeled secondary Ab. The purity of this preparation was 94%. Original magnifications: ×1,000.
We next investigated the expression of COX-1 and -2 during megakaryocyte-oriented maturation of CD34+ progenitors, cultured with TPO. Cells were collected for immunohistochemistry at day +6, +14, and +16 of culture to study different stages of differentiation. The surface antigens CD61 (GpIIIa complex) and/or CD42b (GpIb complex) were used as markers of megakaryocyte-oriented maturation. In fact, GpIIIa is known to appear on the cell surface earlier, whereas GpIb is expressed on more mature megakaryocytes (23).
Purified CD34+ cells expressed COX-1, whereas COX-2 was undetectable (Fig. 1). Megakaryocyte-committed cells isolated from cultures at any time point uniformly expressed COX-1, as shown by double immunostainings with CD61 or CD42b and COX-1 (Fig. 2). At variance with COX-1, COX-2 was almost absent in CD61+ cells at day +6, whereas by day +14 the entire CD61+/CD42b+ population expressed COX-2 (Fig. 2), consistently with the data of bone marrow. Therefore, COX-1 is present in undifferentiated CD34+ cells and remains expressed throughout the entire megakaryocytopoiesis, whereas COX-2 is induced later.
Figure 2.

(a and b) Double immunofluorescence of cytospins from CD34+ cells at day +6 of culture, stained for CD61 revealed by FITC-conjugated secondary Abs (a) and COX-1 revealed by phycoerythrin (PE)-conjugated secondary Abs (b). (c and d) Double immunofluorescence of cytospins from CD34+ cells at day +6 of culture, stained for CD61 revealed by PE-conjugated secondary Abs (c) and COX-2 revealed by FITC-conjugated secondary Abs (d). (e and f) double immunofluorescence of cytospins from CD34+ cells at day +14 of culture, stained for CD42b revealed by FITC-conjugated secondary Abs (e) and COX-2 revealed by PE-conjugated secondary Abs (f).
The presence of COX isoforms in stem and megakaryocyte-committed cells prompted us to investigate COX activity as reflected by prostanoid production. Cells were harvested at the same time points used for the immunocytochemistry, and PGE2 and TXB2 were measured in the cell supernatants. PGE2 was the main product of purified CD34+ cells, whereas TXB2 production was considerably lower (Fig. 3). By day +6, there was a shift in the profile of prostanoid synthesis, with a prevalence of TXB2 (≈500-fold increase vs. basal), whereas PGE2 did not change significantly compared with untreated CD34+ cells (Fig. 3). Finally, both TXB2 and PGE2 showed a parallel increase from day +6 to day +14/+16, and TXB2 remained the most abundant prostanoid (Fig. 3), with a ratio of approx. 10 to 1 vs. PGE2.
Figure 3.

TPO-treated CD34+ cells were harvested at different days of culture, counted, and incubated with AA. PGE2 or TXB2 were measured in the supernatants. Results are means of two to six determinations for each time point.
Because of the presence of both COX isoforms in mature megakaryocytes, we studied the relative contribution of each isoenzyme to the synthesis of TXB2 and PGE2. Cells harvested at day +16, when both isoforms are observed, were treated with the following: (i) selective COX-1 inhibitors, i.e., SC-560 and valeryl salicylate; (ii) the COX-2 inhibitor NS-398; or (iii) the COX-1/COX-2 nonselective inhibitor indomethacin. Selective inhibitors suppressed PGE2 and TXB2 synthesis with significantly different IC50s (Fig. 4), whereas indomethacin inhibited both with equal potency (100 ± 26 nM for PGE2 and 90 ± 5 nM for TXB2). These data suggest a preferential, although not exclusive, coupling of COX-1 with TXA2 production, and of COX-2 with PGE2 synthesis, in mature megakaryocytes.
Figure 4.

Cells from day +14 of culture were incubated with selective COX inhibitors before AA incubation. PGE2 (Right) and TXA2 (Left) production were measured, and the dose-response curves for each compound are shown. The respective IC50s are indicated on each plot. Results are means of two determinations for each concentration.
Platelet Studies.
Because the data illustrated above indicated that COX-2 is present and metabolically active in mature megakaryocytes, we investigated COX-2 expression and activity in circulating platelets, which are the end-product of megakaryocytopoiesis. Immunostaining for COX-2 performed on platelets from healthy volunteers showed that very few platelets (between 3 and 8%, based on the fields of observation) expressed COX-2 (Fig. 5). Interestingly, the fraction of COX-2-expressing platelets is reasonably compatible with the rate of platelet turnover, as calculated on platelet lifespan, i.e., approximately 10 days (24), with an estimated 10% renewal of the platelet population per day. We then hypothesized that younger platelets would bear COX-2, directly exported from the cytoplasm of parent megakaryocytes. To verify this hypothesis, we isolated platelets from patients with either immune thrombocytopenia or recovering from peripheral stem cell transplantation (Table 1), as paradigm of high platelet regeneration, anticipating that a higher percentage of platelets would express COX-2 as compared with healthy controls. To verify that patients had an increased thrombocytopoiesis, we measured reticulated platelets, i.e., the youngest platelets with residual mRNA (18, 24), as an index of high platelet turnover. Indeed, reticulated platelets were significantly higher in patients (Table 1) than in controls from our own lab (8 ± 3% vs. 1.2 ± 0.6%, respectively). Consistently with our hypothesis, a much higher percentage of platelets from patients expressed COX-2, i.e., between 30 and 60%, based on immunoperoxidase (Fig. 5).
Figure 5.

(a and b) Cytospins of washed platelets were reacted with anti-COX-2 Ab, and peroxidase-labeled secondary Ab. (a) Platelets from a control subject. Note that few platelets display a clear positivity (in brown color, one positive platelet is indicated by the arrowhead), the majority are negative (one example of negative platelet is indicated by the asterisk). (b) Platelets from a patient who underwent peripheral stem cell transplantation. Several positive platelets are present in the field (indicated by the arrowhead); a negative platelet is indicated by the asterisk. The increased platelet size of patient compared with control is typical of high platelet regeneration status. (c and d) Double immunofluorescence of platelets stained for COX-2 revealed by PE-conjugated secondary Abs (c) and iPGES revealed by FITC-conjugated secondary Abs (d). (e and f) Double immunofluorescence of cytospins from CD34+ cells at day +14 of culture stained for iPGES revealed by FITC-conjugated secondary Abs (e) and COX-2 revealed by PE-conjugated secondary Abs (f). Original magnifications ×1,000. (c and d Insets) Digital magnification ×10.
Next, we investigated the contribution of COX-2 to prostanoid release in vitro from activated platelets under normal and pathological conditions. We measured TXB2 and PGE2 produced by platelets during whole blood clotting in the presence of vehicle, aspirin (50 μM), or the selective COX-2 inhibitor, NS-398 (10 μM). Prostanoids released under these conditions reflect the maximal biosynthetic capacity of circulating platelets in response to thrombin generated during clotting (22), a process highly sensitive to inhibition by low-dose aspirin ex vivo (25). In vehicle-treated samples, we observed a significantly higher PGE2 synthesis in patients vs. controls (101 ± 53 vs. 32 ± 15 pg/103 platelets, respectively, P < 0.001), whereas TXA2 production was not significantly higher in patients than healthy subjects (1,444 ± 679 vs. 1,060 ± 186 pg/103 platelets, respectively). TXB2 and PGE2 synthesis was differentially sensitive to NS-398 and aspirin inhibition in patients vs. controls (Fig. 6). Thus, the selective COX-2 inhibitor reduced platelet production of both prostanoids to a significantly greater extent in patients than controls. These data are consistent with the histological observations of a greater fraction of COX-2-positive platelets in patients and may indicate a preferential association of platelet COX-2 with PGE2 synthesis, similarly to what observed in megakaryocytes. Indeed, functional and morphological associations between COX-2 and the inducible PGES (iPGES) have been recently reported (26–28), and we examined whether they where coexpressed in platelets as well. By double immunostains, iPGES and COX-2 were present in the same platelets (Fig. 5). Conversely, platelets without COX-2 did not express iPGES. We finally checked whether iPGES was also present in mature megakaryocytes, and found that it was largely expressed at culture day +16 (Fig. 5).
Figure 6.

TXB2 (Upper) and PGE2 (Lower) production during whole blood clotting. Peripheral venous samples were obtained from patients with high platelet regeneration and healthy subjects and clotted in the presence of vehicle, the COX-2 inhibitor NS-398 (10 μM), or aspirin (ASA: 50 μM). Values are expressed as percentage of the respective vehicle-treated sample. *, P < 0.01 vs. vehicle-treated samples.
Discussion
In the present study, we investigated the pattern and timing of expression and activity of COX-1 and -2 during human megakaryocytopoiesis, starting from the undifferentiated CD34+ stem/progenitor cell down to the final product of maturation, i.e., circulating platelets. To study megakaryocytopoiesis, cord blood CD34+ cells were cultured in the presence of TPO, and we analyzed intermediate (day +6) and late (day +14/+16) time points of maturation. This model is considered to reflect the development occurring in the bone marrow more physiologically than differentiation of leukemic cell lines (16).
The precursor CD34+ cells and megakaryocytes at any given time point expressed COX-1. The observation of COX-1 protein in human CD34+ stem cells suggests a role for COX-1 in the function of hematopoietic progenitors. Indeed, mRNA for COX-1, but not for COX-2, has been found in the CD4−CD8− lymphoid precursors, originated from hematopoietic organs (3). Consistently, COX-1 has also been reported in human megakaryoblastic cell lines differentiated in vitro (11–15).
At variance with COX-1, COX-2 appears to be selectively induced along megakaryocytic differentiation, as demonstrated by CD34+ cultures and immunohistochemistry of bone marrow. Interestingly, the COX-2 promoter contains a recognition site for GATA-1 (29), a well known hemopoietic transcription factor playing a major role in megakaryocytopoiesis (30). Therefore, COX-2 expression may be specifically related to the megakaryocytopoiesis, whereas the myeloid differentiation appears to up-regulate COX-1 instead (8, 9).
Based on prostanoid measurements in the cell supernatants, COX isoforms appear functionally active along megakaryocyte maturation. Interestingly, PGE2 is the main product of CD34+ cells, which express COX-1 only. As megakaryocyte commitment proceeds, TXB2 becomes the main product, and by culture day +6, TXB2 level is 10-fold higher than PGE2. This pattern of differentiation-dependent shift in the profile of prostanoids is consistent with data reporting only traces of TX-synthase mRNA in CD34+ cells, whereas a higher expression was observed in purified megakaryocytes (31). In addition, TX-synthase has been shown to be controlled by the p45 NF-E2 transcription factor, which specifically regulates megakaryocytopoiesis (31).
Because of the presence of both COX-1 and -2 in mature megakaryocytes, we used selective inhibitors to study the activity of each isoform. Based on the different IC50s for each prostanoid, it seems reasonable to hypothesize that, in these cells, PGE2 synthesis is preferentially coupled to COX-2, whereas TXB2 depends more on COX-1 activity. It is becoming increasingly apparent that downstream PG isomerases are coordinately regulated with COX isoforms (26–28). In the case of PGES, the inducible and the constitutive isoforms have been described to be functionally coupled with the corresponding COX (26–28). Enzymatic and/or morphologic associations similar to the one here reported (COX-1/TXA2, COX-2/PGE2, COX-2/iPGES) have been described in tissues and overexpressing cells (26–28). Our data extend these observations and demonstrate that these coordinate metabolic routes of AA may also operate in megakaryocytopoiesis.
Because of the presence of both COX isoforms in megakaryocytes, we explored whether platelets transiently express COX-2. Reports on COX-2 mRNA or protein in platelets are conflicting (6, 7), mainly based on platelet extracts, and do not describe the distribution of COX-2 within the platelet population. Our data show that COX-2 is detectable only in a minor fraction (<10%) of normal platelets whereas, in clinical conditions associated with high platelet regeneration, the COX-2-positive fraction can be substantially increased, up to 60%. Altogether, our data are consistent with the hypothesis that only newly released platelets express COX-2, which are likely to originate from the cytoplasm of parent megakaryocytes. At variance with COX-1, the high mRNA turnover and translational inhibition reported for COX-2 (32) may account for rapid protein disappearance in peripheral platelets, shortly after release from megakaryocytes. Such a low frequency of COX-2 expression in normal platelets may explain the conflicting data based on immunoblottings (6, 7), where differences in experimental conditions may contribute to different sensitivity and results. Our data may also explain the weak COX-2 mRNA signal reported in platelets (10). Immunohistochemical observations are consistent with functional data on prostanoids released by platelets during whole blood clotting in patients and controls. In fact, COX-2-dependent TXB2 released from platelets appears negligible in controls, in agreement with previous reports (7). On the other hand, in patients with high platelet regeneration, where a larger fraction of peripheral platelets express COX-2, we observed a more profound inhibitory effect of NS-398 on prostanoid synthesis and release. The disproportionate increase of PGE2 vs. TXB2 production in patients as compared with healthy subjects, further supports a preferential functional coupling of COX-2 with PGE2 synthesis in circulating platelets as well.
Recently, the term aspirin “resistance” has been used to indicate an ill-defined phenomenon in which platelets are not fully inhibited by low-dose aspirin (33). Low-dose aspirin is known to profoundly inhibit platelet COX-1 activity (22, 25), but low-dose, aspirin-insensitive TXA2 production has been reported in patients with unstable coronary syndromes (34, 35). Thus, in clinical conditions with enhanced platelet regeneration, COX-2-dependent TXA2 biosynthesis may be more prevalent and therefore a potential source of aspirin-insensitive platelet activation. Interestingly, baseline urinary excretion of 11-dehydro-TXB2 was an independent predictor of major vascular events during follow-up of aspirin-treated patients with vascular disease recruited in the Heart Outcome Prevention Evaluation (HOPE) trial (36).
Overall, a small amount of PGE2 is released from platelets during blood clotting compared with TXA2 (1:30 ratio in controls). Whereas the role of TXA2 in the regulation of platelet function is well known, the hemostatic significance, if any, of platelet-released PGE2 is controversial. In vitro studies have reported that high (μM) or low (nM) PGE2 concentrations inhibit or potentiate platelet aggregation, respectively (37–39). On the other hand, platelet-derived PGE2 in concert with inflammatory cytokines released from activated platelets (40) may participate in the local inflammatory response to vascular injury.
In conclusion, we have demonstrated that COX-1 and COX-2 are differentially expressed in hematopoietic progenitors and during megakaryocytopoiesis, and functionally coupled to various bioactive prostanoids starting from the CD34+ stem cells, ending to mature circulating platelets. The transient nature of COX-2 expression and activity in newly formed platelets is likely to have negligible functional consequences under physiologic circumstances. However, accelerated platelet turnover may represent a novel clinical paradigm through which to explore the relevance of COX-2-derived prostanoids in hemostatic and/or inflammatory responses and to reassess the dose- and time-requirements for the antiplatelet effect of aspirin.
Acknowledgments
We thank Dr. A. Montanaro for bone biopsies, and Dr. A. Habib for generously providing anti-COX antibodies. These studies were supported by a grant from the Ministry of Research and Education to the Center of Excellence on Aging of the University of Chieti.
Abbreviations
- PG
prostaglandins
- COX
cyclooxygenases
- TX
thromboxane
- PGES
PGE2 synthase
- iPGES
inducible PGES
- AA
arachidonic acid
- TPO
thrombopoietin
- PE
phycoerythrin
References
- 1.Smith W-L, Langenbach R. J Clin Invest. 2001;107:1491–1495. doi: 10.1172/JCI13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rocca B, Spain L-M, Pure E, Langenbach R, Patrono C, FitzGerald G-A. J Clin Invest. 1999;103:1469–1477. doi: 10.1172/JCI6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rocca B, FitzGerald G-A. Int Immunopharmacol. 2002;2:603–630. doi: 10.1016/s1567-5769(01)00204-1. [DOI] [PubMed] [Google Scholar]
- 4.FitzGerald G-A, Patrono C. N Engl J Med. 2001;345:433–442. doi: 10.1056/NEJM200108093450607. [DOI] [PubMed] [Google Scholar]
- 5.Patrono C. N Engl J Med. 1994;330:1287–1294. doi: 10.1056/NEJM199405053301808. [DOI] [PubMed] [Google Scholar]
- 6.Weber A-A, Zimmermann K-C, Meyer-Kirchrath J, Schror K. Lancet. 1999;353:900. doi: 10.1016/S0140-6736(99)00498-5. [DOI] [PubMed] [Google Scholar]
- 7.Patrignani P, Sciulli M-G, Manarini S, Santini G, Cerletti C, Evangelista V. J Physiol Pharmacol. 1999;50:661–667. [PubMed] [Google Scholar]
- 8.Hoff T, DeWitt D, Kaever V, Resch K, Goppelt-Struebe M. FEBS Lett. 1993;20:38–42. doi: 10.1016/0014-5793(93)81653-h. [DOI] [PubMed] [Google Scholar]
- 9.Smith C-J, Morrow J-D, Roberts L-J, Marnett L-J. Biochem Biophys Res Commun. 1993;192:787–793. doi: 10.1006/bbrc.1993.1483. [DOI] [PubMed] [Google Scholar]
- 10.Matijevic-Aleksic N, Sanduja S-K, Wang L-H, Wu K-K. Biochim Biophys Acta. 1995;1269:167–175. doi: 10.1016/0167-4889(95)00116-a. [DOI] [PubMed] [Google Scholar]
- 11.Mroske C, Plant M-H, Franks D-J, Laneuville O. Exp Hematol. 2000;28:411–421. doi: 10.1016/s0301-472x(00)00125-9. [DOI] [PubMed] [Google Scholar]
- 12.Vitrat N, Letestu R, Masse A, Lazar V, Vainchenker W, Debili N. Thromb Haemost. 2000;83:759–768. [PubMed] [Google Scholar]
- 13.Mahmud I, Ueda N, Yamaguchi H, Yamashita R, Yamamoto S, Kanaoka Y, Urade Y, Hayaishi O. J Biol Chem. 1997;272:28263–28266. doi: 10.1074/jbc.272.45.28263. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe T, Yatomi Y, Sunaga S, Miki I, Ishii A, Nakao A, Higashihara M, Seyama Y, Ogura M, Saito H. Blood. 1991;78:2328–2336. [PubMed] [Google Scholar]
- 15.Zauli G, Catani L. Crit Rev Oncol Hematol. 1995;21:135–157. doi: 10.1016/1040-8428(94)00171-5. [DOI] [PubMed] [Google Scholar]
- 16.Zucker-Franklin D, Kaushansky K. Blood. 1996;88:1632–1638. [PubMed] [Google Scholar]
- 17.Zauli G, Vitale M, Falcieri E, Gibellini D, Bassini A, Celeghini C, Columbaro M, Capitani S. Blood. 1997;90:2234–2243. [PubMed] [Google Scholar]
- 18.Catani L, Vianelli N, Luatti S, Rizzi S, Castellani S, Valdre L, Lemoli R-M, Tura S. Bone Marrow Transplant. 1999;24:1191–1194. doi: 10.1038/sj.bmt.1702062. [DOI] [PubMed] [Google Scholar]
- 19.Creminon C, Frobert Y, Habib A, Maclouf J, Pradelles P, Grassi J. Biochim Biophys Acta. 1995;1254:333–340. doi: 10.1016/0005-2760(94)00196-6. [DOI] [PubMed] [Google Scholar]
- 20.Habib A, Creminon C, Frobert Y, Grassi J, Pradelles P, Maclouf J. Biol Chem. 1993;268:23448–23454. [PubMed] [Google Scholar]
- 21.Ciabattoni G, Pugliese F, Spaldi M, Cinotti G-A, Patrono C. J Endocrinol Invest. 1979;2:173–182. doi: 10.1007/BF03349310. [DOI] [PubMed] [Google Scholar]
- 22.Patrono C, Ciabattoni G, Pinca E, Pugliese F, Castrucci G, De Salvo A, Satta M-A, Peskar B-A. Thromb Res. 1980;17:317–327. doi: 10.1016/0049-3848(80)90066-3. [DOI] [PubMed] [Google Scholar]
- 23.Debili N, Issaad C, Masse J-M, Guichard J, Katz A, Breton-Gorius J, Vainchenker W. Blood. 1992;80:3022–3035. [PubMed] [Google Scholar]
- 24.Dale G L. Curr Opin Hematol. 1997;4:330–334. doi: 10.1097/00062752-199704050-00006. [DOI] [PubMed] [Google Scholar]
- 25.Patrignani P, Filabozzi P, Patrono C. J Clin Invest. 1982;69:1366–1372. doi: 10.1172/JCI110576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jakobsson P-J, Thoren S, Morgenstern R, Samuelsson B. Proc Natl Acad Sci USA. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cipollone F, Prontera C, Pini B, Marini M, Fazia M, De Cesare D, Iezzi A, Ucchino S, Boccoli G, Saba V, et al. Circulation. 2001;104:921–927. doi: 10.1161/hc3401.093152. [DOI] [PubMed] [Google Scholar]
- 28.Ueno N, Murakami M, Tanioka T, Fujimori K, Tanabe T, Urade Y, Kudo I. J Biol Chem. 2001;276:34918–34927. doi: 10.1074/jbc.M100429200. [DOI] [PubMed] [Google Scholar]
- 29.Kosaka T, Miyata A, Ihara H, Hara S, Sugimoto T, Takeda O, Takahashi E, Tanabe T. Eur J Biochem. 1994;221:889–897. doi: 10.1111/j.1432-1033.1994.tb18804.x. [DOI] [PubMed] [Google Scholar]
- 30.Shivdasani R-A. Stem Cells. 2001;19:397–407. doi: 10.1634/stemcells.19-5-397. [DOI] [PubMed] [Google Scholar]
- 31.Deveaux S, Cohen-Kaminsky S, Shivdasani R-A, Andrews N-C, Filipe A, Kuzniak I, Orkin S-H, Romeo P-H, Mignotte V. EMBO J. 1997;16:5654–5661. doi: 10.1093/emboj/16.18.5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixon D-A, Kaplan C-D, McIntyre T-M, Zimmerman G-A, Prescott S-M. J Biol Chem. 2000;275:11750–11757. doi: 10.1074/jbc.275.16.11750. [DOI] [PubMed] [Google Scholar]
- 33.Patrono C, Coller B, Dalen J-E, FitzGerald G-A, Fuster V, Gent M, Hirsh J, Roth G. Chest. 2001;119:39S–63S. doi: 10.1378/chest.119.1_suppl.39s. [DOI] [PubMed] [Google Scholar]
- 34.Vejar M, Fragasso G, Hackett D, Lipkin D-P, Maseri A, Born G-V, Ciabattoni G, Patrono C. Thromb Haemost. 1990;63:163–168. [PubMed] [Google Scholar]
- 35.Cipollone F, Ciabattoni G, Patrignani P, Pasquale M, Di Gregorio D, Bucciarelli T, Davi G, Cuccurullo F, Patrono C. Circulation. 2000;102:1007–10013. doi: 10.1161/01.cir.102.9.1007. [DOI] [PubMed] [Google Scholar]
- 36.Eikelboom J-W, Hirsh J, Weitz J-I, Johnston M, Yi Q, Yusuf S. Circulation. 2002;105:1650–1655. doi: 10.1161/01.cir.0000013777.21160.07. [DOI] [PubMed] [Google Scholar]
- 37.MacIntyre D-E, Gordon J-L. Nature (London) 1975;258:337–339. doi: 10.1038/258337a0. [DOI] [PubMed] [Google Scholar]
- 38.Gray S-J, Heptinstall S. Eur J Pharmacol. 1991;194:63–70. doi: 10.1016/0014-2999(91)90124-9. [DOI] [PubMed] [Google Scholar]
- 39.Fabre J-E, Nguyen M, Athirakul K, Coggins K, McNeish J-D, Austin S, Parise L-K, FitzGerald G-A, Coffman T-M, Koller B-H. J Clin Invest. 2001;107:603–610. doi: 10.1172/JCI10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindemann S, Tolley N-D, Dixon D-A, McIntyre T-M, Prescott S-M, Zimmerman G-A, Weyrich A-S. J Cell Biol. 2001;154:485–490. doi: 10.1083/jcb.200105058. [DOI] [PMC free article] [PubMed] [Google Scholar]