

Abstract
Large-scale purification of recombinant human membrane proteins represents a rate-limiting step toward the understanding of their role in health and disease. There are only four mammalian membrane proteins of known structure, and these were isolated from natural sources (see http://www.mpibp-frankfurt.mpg.de/michel/public/memprotstruct.html). In addition, genetic diseases of membrane proteins are frequently caused by trafficking defects, and it is enigmatic whether these mutants are functional. Here, we report the employment of Escherichia coli for the functional expression, purification, and reconstitution of a human membrane protein, the human Na+/glucose cotransporter (hSGLT1). The use of an E. coli mutant defective in the outer membrane protease OmpT, incubation temperatures below 20°C, and transcriptional regulation from the lac promoter/operator are crucial to reduce proteolytic degradation. Purification of a recombinant hSGLT1 through affinity chromatography yields about 1 mg of purified recombinant hSGLT1 per 3 liters of cultured bacterial cells. Kinetic analysis of hSGLT1 in proteoliposomes reveals that a purified recombinant transporter, which is missorted in eukaryotic cells, retains full catalytic activity. These results indicate the power of bacteria to manufacture and isolate human membrane proteins implicated in genetic diseases.
Integral membrane proteins such as channels and transporters are essential for all living organisms by mediating the passage of ions and molecules across membranes. The understanding of how membrane proteins work in health and disease has been restricted largely because of the lack of purified protein. Most membrane proteins are present at low concentrations in their native tissue, and their overexpression is a prerequisite for purification. The use of eukaryotic expression systems has illuminated mechanistic features of human membrane proteins, but these are unsuitable for their large-scale purification (1, 2). In addition, genetic disorders of membrane proteins are frequently caused by improper trafficking in cells, and it is not known whether missorted proteins retain their catalytic activity (3). To obtain structural information on membrane proteins, prokaryotic homologues have been most amenable because they can often be expressed in bacteria in large quantities (4). However, the inexpensive and less time-consuming overexpression of animal and human integral proteins of the plasma membrane has not been applicable, and there are only a few examples in which eukaryote membrane proteins are functional when expressed in prokaryotic cells (5–11). Here, we report the employment of Escherichia coli for the expression, purification, and reconstitution of a fully functional recombinant human sodium/glucose transporter (hSGLT1)—a central protein for the homeostasis of glucose, salt, and water (12–14). Our strategy is based in part on our group's previous success in purifying functional transporters from bacteria (15, 16).
Experimental Procedures
Genetic Engineering.
A cassette version of the hSGLT1 cDNA has been generated to alleviate subcloning and site-directed amino acid replacements. In this construct, we have introduced unique restriction enzyme recognition sites about every 200 bps by introducing silent mutations in the hSGLT1 cDNA, and we have removed restriction sites in the 5′ and 3′ polylinker region. These modifications did not affect the kinetic properties of hSGLT1 expressed in Xenopus oocytes (not shown). The entire cDNA, flanked by unique NcoI and HindIII restriction sites, was cloned into similarly treated pTMH-6⋅FH expression vector (17), a derivative of pT7–5(lacY) (18).
For immunological detection and affinity chromatography, the FLAG epitope was introduced in hSGLT1 by changing the native peptide sequence D574AEEEN to D574YKDDDDK. Truncation of the N-terminal loop was performed by blunt-end religation of the EcoRV- and SacII-digested cDNA after removal of the SacII-generated 3′ overhang. The DNA sequence coding for the glycophorin A transmembrane span and the green fluorescence protein (GFP) reporter group was fused to the 3′ end of hSGLT1 by swapping the corresponding DNA fragment from pVNGFPH6 (15) into the hSGLT1 expression vector. Site-directed mutagenesis was performed by using standard PCR protocols with synthetic mutagenic oligonucleotides. All modifications were verified by DNA sequencing.
Expression, Purification, and Reconstitution.
E. coli strains CD41(DE3) and CD43(DE3) (19) were obtained from Avidis (Saint-Beauzire, France). All other strains were purchased from commercial sources. E. coli transformed with expression vectors carrying recombinant hSGLT1 was cultivated in LB (Miller; Difco) under selective conditions (100 μg/ml ampicillin). Cells were incubated at 16°C to mid-logarithmic phase before induction of the hSGLT1 expression by the addition of 0.3 mM isopropyl 1-thio-β-d-galactopyranoside for 5 h.
Functional studies in Xenopus oocytes of each modified hSGLT1 variant were performed (20).
For purification, the membrane fraction of E. coli BL21 [F− ompT hsdSB(rB− mB−) gal dcm] harboring recombinant hSGLT1ΔN-GFP was collected by ultracentrifugation after disrupting the cells by sonication and the removal of unbroken cells and cell debris by low-speed centrifugation (21). The membrane fraction (7.5 mg of total membrane protein per ml) was solubilized with 1.2% FosCholine-12 (Anatrace, Maumee, OH) in buffer TG [20 mM Tris⋅Cl, pH 8.0/1 M NaCl/20% (vol/vol) glycerol and protease inhibitor mixture (Sigma)] at 4°C for 30 min. After removal of the insoluble fraction by ultracentrifugation, the supernatant was bound to preequilibrated anti-FLAG M2 (α-FLAG) affinity gel resin (Sigma) with a gel:supernatant ratio of 1:10 (vol/vol) at 4°C for 4 h by slight shaking. The unbound protein was removed by washing with 10 column volumes of buffer TG plus 0.2% FosCholine-12; recombinant hSGLT1ΔN-GFP was eluted by competition with 100 μg/ml FLAG peptide (Sigma). The fraction was concentrated to <0.2 ml by ultrafiltration in Centricon YM100 devices (Millipore) and separated by gel filtration on Superose R12 (Amersham Pharmacia).
Reconstitution of purified recombinant hSGLT1ΔN-GFP was performed into preformed Triton X-100-destabilized liposomes (22) composed of either E. coli lipids (Avanti Polar Lipids) or 90% (wt/vol) asolectin soy lecithin (Associated Concentrates, Woodside, NY) and 10% (wt/vol) cholesterol at a protein:lipid ratio of 1:400 (15). The detergent-mediated solubilization was followed by turbidity measurements (23).
Sugar Uptake.
Tracer uptake by intact E. coli cells and right-side-out membrane vesicles (17) was performed as described (15, 21). Before uptake assays, proteoliposomes (preloaded with 100 mM potassium phosphate, pH 7.5/2 mM β-mercaptoethanol) were subjected to three sonication/freeze/thaw cycles, and uptake of methyl α-d-[14C]glucopyranoside ([14C]α-MDG) was performed in 100 mM NaCl or choline chloride buffer at a final protein concentration of 0.125 μg/ml (22). Sugar-transport kinetics were determined from 1 min α-MDG uptakes by varying the α-MDG concentration from 0.01 to 25 mM. All experiments were performed at least in triplicate, and errors indicate the SEM or the SE of the fit (see Fig. 5C only).
Figure 5.

Sugar uptake kinetics of proteoliposomes containing purified recombinant hSGLT1ΔN-GFP. (A) The time course of 10 μM α-MDG uptake in the presence of 100 mM sodium (■) shows an ≈30-fold increase in the initial rate of sugar uptake before reaching concentration equilibrium (>5 h). After addition of 100 μM phlorizin (▾) or removal of Na+ (Δ), the initial rate was identical to control liposomes, and sugar accumulation above the equilibrium is not observed. (B) Uptake of 100 μM α-MDG is inhibited by high-affinity substrates or inhibitors of SGLT1 (10 mM d-galactose, Gal; 10 mM d-glucose, Glc; 100 μM phlorizin, Phlz) but not by low-affinity substrates (e.g., 10 mM 2-deoxy-d-glucose, 2-doGlc). (C) Concentration dependence of the initial-rate sugar uptake shows a half-saturation constant (K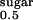 ) of 0.3 ± 0.05 mM and a maximum velocity (Vmax) of 2.2 ± 0.06 μmol × mg hSGLT1ΔN-GFP−1 × min−1.
) of 0.3 ± 0.05 mM and a maximum velocity (Vmax) of 2.2 ± 0.06 μmol × mg hSGLT1ΔN-GFP−1 × min−1.
The catalytic turnover number for the reconstituted transporter in proteoliposomes was determined by the ratio of Vmax:number of transporters, whereas that for the transporter in oocytes was based on electrophysiological experiments using the maximum current:maximum charge (Imax:Qmax) ratio (24).
Analytical Methods.
Equal amounts of protein (1 μg per lane) were subjected to SDS/10% PAGE. Protein was assayed according to a modified Lowry method (25) with BSA as standard. Silver staining of SDS/PAGE gels and immunological detection of the recombinant gene product were performed (21).
Results and Discussion
Expression of hSGLT1 in E. coli.
E. coli BL21 harboring a plasmid with the hSGLT1 cDNA under control of the lac promoter/operator (17, 18) proves to be most suitable for the high-level expression and stability of recombinant hSGLT1. The transporter is detected in the bacterial cells by using a FLAG antibody against the engineered FLAG epitope (Fig. 1), and it is active as judged by sodium-stimulated sugar (d-glucose) uptake (not shown). However, employing the strong trc or T7 promoter, incubation above 20°C, or using OmpT+ E. coli strains as expression hosts, results in proteolytic degradation of hSGLT1 (Fig. 2). Another E. coli host strain (UT5600) lacking the outer membrane protease OmpT is also crucial for the functional expression of the mouse multidrug-resistance protein (mdr-1) (5). Although derivatives of E. coli CD41(DE3) or CD43(DE3) (19) have been successfully used for the production of mitochondrial membrane proteins (8, 11), FLAG-tagged hSGLT1 is not detectable in either strain employing the T7 system. Full-length hSGLT1 (plus FLAG epitope) is detected in whole-cell lysates of BL21 harboring the hSGLT1 expression vector when incubated at 20°C. However, incubation of the same cells at 37°C results in the proteolytic degradation of hSGLT1 as indicated by immuno-reaction of the monoclonal FLAG M2 antibody with hSGLT1 fragments at about 30 kDa and smaller (Fig. 2).
Figure 1.

Secondary structure model of hSGLT1. hSGLT1 consists of 14 α-helical transmembrane domains (shaded columns) with the N and C termini located on the extracellular phase of the membrane (26, 28). The N-glycosylation site (Asn-248; N) and the position of the engineered FLAG epitope are indicated. After deletion of the DNA sequence encoding amino acid residues 12–28 (white circles), amino acid residue Thr-11 was fused to residue Ile-29 (black circles). Asp-28 is highlighted (D). “Real-time” detection of the recombinant gene product (hSGLT1ΔN-GFP) during expression/purification was facilitated by use of the GFP as a reporter group fused to the C terminus of hSGLT1 in combination with the single glycophorin A transmembrane span (GpA, white column; connecting spacer regions are shown as white lines; ref. 15).
Figure 2.

Immunological detection of hSGLT1 in different expression hosts. Equal amounts (10 μg) of lysates of whole Xenopus oocytes or E. coli BL21 cells were subjected to SDS/10% PAGE and electroblotted onto nitrocellulose. hSGLT1 was immunologically detected by incubating the blot with an SGLT1 (α-SGLT1) or the α-FLAG antibody. Although the SGLT1 antibody crossreacts with intrinsic E. coli proteins (BL21-co, E. coli BL21 harboring plasmid pT7–5 as control), no unspecific signals in E. coli are detectable by using the α-FLAG monoclonal antibody. Incubation of E. coli BL21 harboring hSGLT1-FLAG (BL21-hS) at 37°C results in the proteolytic degradation of the transporter (degradation products are detectable at ≈30 kDa and ≈18 kDa; not shown). Reduction of the incubation temperature to 20°C yields the full-length transporter (≈55 kDa, arrowhead), which corresponds to the apparent molecular mass of a nonglycosylated hSGLT1 mutant expressed in oocytes (oocytes).
Because bacterial cotransporters possess significantly shorter N-terminal hydrophilic extensions than those in eukaryotes (26), we adapted this region in hSGLT1 by deleting amino acid residues 12–28 (Fig. 1). This deletion increases the amount of hSGLT1 inserted into the membrane of E. coli by about fivefold as judged by Western blot analysis (data not shown) to comparable levels with a bacterial cotransport protein [the Na+/proline transporter (PutP)] overexpressed in E. coli (21). Accumulation of α-MDG, an SGLT1-specific, nonmetabolized glucose analog, in E. coli cells producing truncated hSGLT1 (subsequently referred to as hSGLT1ΔN) is Na+-dependent and completely blocked by phlorizin, the classical SGLT1-inhibitor (Fig. 3A). The affinity of the transporter for sugar transport (K ) is 0.42 mM—a value comparable to that for native and FLAG-tagged hSGLT1 determined in oocytes (0.35 mM; Table 1).
) is 0.42 mM—a value comparable to that for native and FLAG-tagged hSGLT1 determined in oocytes (0.35 mM; Table 1).
Figure 3.

Expression of recombinant hSGLT1 in E. coli. (A) Sugar uptake (50 μM α-MDG) by right-side-out vesicles of E. coli BL21 producing hSGLT1ΔN is stimulated by sodium (Na+, 25 mM) and inhibited by phlorizin (Phlz, 10 μM). No Na+-dependent, phlorizin-sensitive uptakes were observed with vesicles of BL21 harboring plasmid pT7–5 as a control (Co). For the control vesicles, there was no significant difference in sugar uptake in the presence or absence of Na+ (39 ± 12 nmol α-MDG × mg protein−1 × min−1), which was not affected by the addition of phlorizin. (B) Western blot analysis of the membrane fraction of E. coli B21 producing hSGLT1ΔN. No signal was detected in other cell fractions. The apparent molecular mass of hSGLT1 corresponds to that of the nonglycosylated transporter in Xenopus oocytes (compare Fig. 2).
Table 1.
Kinetics of hSGLT1 in different systems
|
Xenopus oocytes
|
Proteoliposomes
|
|||
|---|---|---|---|---|
| hSGLT1* | hSGLT1-FLAG | hSGLT1-FLAG-GFP | hSGLT1ΔN-GFP | |
K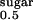 , mM† , mM†
|
0.41 ± 0.03 | 0.33 ± 0.02 | 0.38 ± 0.1 | 0.35 ± 0.05 |
| Turnover number, s−1‡ | 50 | 48 | 10 | 4 |
The K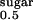 and the turnover number for hSGLT1 wild type and the cassette version of hSGLT1 (see Experimental Procedures) are indistinguishable.
and the turnover number for hSGLT1 wild type and the cassette version of hSGLT1 (see Experimental Procedures) are indistinguishable.
The K in oocytes was determined at −50 mV. Data are obtained from representative measurements in single oocytes (repeated in triplicate with oocytes from different batches) and the error described the SE of the fit. The K
in oocytes was determined at −50 mV. Data are obtained from representative measurements in single oocytes (repeated in triplicate with oocytes from different batches) and the error described the SE of the fit. The K for the transporter reconstituted into proteoliposomes represents the mean of at least three independent measurements with errors indicating the SEM.
for the transporter reconstituted into proteoliposomes represents the mean of at least three independent measurements with errors indicating the SEM.
The catalytic turnover number for the reconstituted transporter in proteoliposomes was determined by the ratio of Vmax:number of transporters, whereas that for the transporter in oocytes was based on electrophysiological experiments using the maximum current (Imax):maximum charge (Qmax) ratio (24).
However, although Western blot analysis of whole-oocyte lysates indicates that hSGLT1ΔN is expressed at similar levels as hSGLT1 wild type, the protein is not inserted into the oocyte plasma membrane as determined by steady-state and presteady-state measurements (20, 24). This finding demonstrates that this variation causes improper trafficking or missorting in the eukaryotic host cell. On the other hand, Western blot analysis of fractionated E. coli cells reveals that recombinant hSGLT1ΔN is inserted into the bacterial plasma membrane and not into inclusion bodies in the cytoplasm (Fig. 3B).
Purification, Reconstitution, and Functional Analysis of Recombinant hSGLT1.
To monitor the purification progress, we adopted a strategy used by Turk et al. (15) and fused a GFP reporter group to the C terminus of hSGLT1 (see Fig. 1). To determine the functional effect of this modification, we analyzed the kinetics of this construct by means of electrophysiological methods. Because the K of native hSGLT1 and full-length hSGLT1 with the GFP fusion in oocytes are indistinguishable (Table 1), we used an hSGLT1-GFP fusion protein for the subsequent purification procedure.
of native hSGLT1 and full-length hSGLT1 with the GFP fusion in oocytes are indistinguishable (Table 1), we used an hSGLT1-GFP fusion protein for the subsequent purification procedure.
Recombinant hSGLT1ΔN-GFP was purified by α-FLAG immuno-affinity chromatography, followed by gel filtration (Fig. 4). The presence of the GFP reporter group greatly facilitated following the purification progress. This method confirms the observation that the recombinant transporter is inserted into the bacterial plasma membrane as opposed to inclusion bodies. In addition, measurement of the GFP fluorescence after each purification step indicates some proteolytic degradation of the recombinant transporter, even though all purification steps were performed at 0–4°C. These degradation products (see Fig. 4, lane F) also are detectable by their immuno-reactivity with the FLAG M2 antibody (not shown). The subsequent gel filtration step (Fig. 4, lane GF) removes these smaller protein fragments. Overall, about 1 mg of purified protein can be isolated from 3 liters of cell culture (about 10 g of cell wet weight). The apparent molecular mass (≈100 kDa) of the single protein band detectable on a silver-stained SDS/PAGE gel (Fig. 4, lane GF) corresponds to the mass of recombinant hSGLT1ΔN-GFP (103,131 Da). Western blot analysis (using the FLAG M2 and SGLT1 antibodies), GFP fluorescence, and activity measurements (Fig. 5) unequivocally identify this band as recombinant hSGLT1.
Figure 4.

Purification of recombinant hSGLT1. Recombinant hSGLT1ΔN-GFP was solubilized from the membrane fraction of E. coli BL21 (S) and purified by using α-FLAG affinity chromatography (F) followed by gel filtration (GF). Purified recombinant hSGLT1ΔN-GFP was reconstituted into preformed detergent-destabilized liposomes (L) to obtain hSGLT1ΔN-GFP-containing proteoliposomes (PL). Samples of the purification procedure were subjected to SDS/10% PAGE followed by silver staining of the gel and Western blot analysis using the FLAG M2 monoclonal antibody.
Purified recombinant hSGLT1ΔN-GFP was reconstituted into preformed detergent-destabilized liposomes with a detergent concentration adjusted to the onset of solubilization (22, 23). Pilot uptake assays with 100 μM α-MDG reveal that the initial rates of sugar accumulation in cholesterol-containing proteoliposomes (15) are 38% higher (230 ± 14 nmol × mg hSGLT1ΔN-GFP−1 × min−1) than those measured in proteoliposomes made of E. coli lipids (144 ± 7 nmol × mg hSGLT1ΔN-GFP−1 × min−1). Therefore, we performed the subsequent kinetic analysis in cholesterol-containing proteoliposomes (Fig. 5). Sodium-stimulated α-MDG uptake is completely blocked by phlorizin (Fig. 5 A and B) and by other high-affinity substrates (Fig. 5B). The K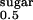 for sugar uptake is 0.3 mM (Fig. 5C, Table 1). Because giant excised patch-clamp measurements reveal a more than 10-fold higher K
for sugar uptake is 0.3 mM (Fig. 5C, Table 1). Because giant excised patch-clamp measurements reveal a more than 10-fold higher K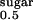 on the cytoplasmic phase of SGLT1 (27), our data suggests a unidirectional right-side-out orientation of the recombinant transporter in proteoliposomes. However, we cannot exclude the possibility of randomly oriented hSGLT1-GFP in proteoliposomes. The catalytic turnover number of hSGLT1ΔN-GFP based on the Vmax of 2.2 μmol × mg hSGLT1ΔN-GFP−1 × min−1 is ≈4 s−1, which is in good agreement with that obtained for full-length hSGLT1-GFP determined in oocytes (≈10 s−1) and other Na+-dependent transporters [e.g., the Na+/glucose transporter of Vibrio parahaemolyticus (vSGLT) (15), and PutP (22)].
on the cytoplasmic phase of SGLT1 (27), our data suggests a unidirectional right-side-out orientation of the recombinant transporter in proteoliposomes. However, we cannot exclude the possibility of randomly oriented hSGLT1-GFP in proteoliposomes. The catalytic turnover number of hSGLT1ΔN-GFP based on the Vmax of 2.2 μmol × mg hSGLT1ΔN-GFP−1 × min−1 is ≈4 s−1, which is in good agreement with that obtained for full-length hSGLT1-GFP determined in oocytes (≈10 s−1) and other Na+-dependent transporters [e.g., the Na+/glucose transporter of Vibrio parahaemolyticus (vSGLT) (15), and PutP (22)].
This report describes the unequivocal successful use of bacteria for the isolation of a functional human membrane protein. We conclude that E. coli may be used for the functional manufacture, purification, and reconstitution of medically important human membrane proteins. The kinetics of recombinant hSGLT1 in intact cells or membrane vesicles, and reconstituted into proteoliposomes, mirror those determined for hSGLT1 in native tissue and Xenopus oocytes. Furthermore, these studies indicate that eukaryotic membrane proteins do not require cholesterol for activity—a steroid is missing among the E. coli membrane lipids—although the presence of cholesterol in the proteoliposomes increases the initial rates of sugar uptake by about 40%. This report also supports the observation that N-glycosylation is not required for hSGLT1 activity (28), because bacteria do not possess glycosylation machinery.
Finally, by taking advantage of the bacterial machinery for the cotranslational insertion of membrane proteins into the plasma membrane (29), we demonstrate that a missorted mutant protein retains full activity when purified and reconstituted into proteoliposomes. Note that engineered hSGLT1ΔN lacks Asp-28, a residue that, when mutated to Asn, causes glucose galactose malabsorption syndrome through a defect in trafficking (30). The employment of the E. coli expression system now allows the expression of known hSGLT1 mutants (3), which could permit functional analyses of these mutants.
Acknowledgments
We thank J. Tomasevic for technical assistance, E. Turk for providing plasmid pVNGFPH6, and B. Hirayama, D. Loo, E. Turk, and G. Sachs for critical reading of the manuscript. This work was supported by National Institutes of Health Grants DK19567 and DK44582 (to E.M.W.) and Center for Ulcer Research and Education Grant DK41301-13 (to M.Q.).
Abbreviations
- GFP
green fluorescence protein
- α-MDG
methyl α-d-glucopyranoside
References
- 1.Fleming K G. Curr Opin Biotechnol. 2000;11:67–71. doi: 10.1016/s0958-1669(99)00056-7. [DOI] [PubMed] [Google Scholar]
- 2.Tate C G. FEBS Lett. 2001;504:94–98. doi: 10.1016/s0014-5793(01)02711-9. [DOI] [PubMed] [Google Scholar]
- 3.Martín M G, Turk E, Lostao M P, Kerner C, Wright E M. Nat Genet. 1996;12:216–220. doi: 10.1038/ng0296-216. [DOI] [PubMed] [Google Scholar]
- 4.Doyle D A, Cabral J M, Pfuetzner R A, Kuo A, Gulbis J M, Cohen S L, Chait B T, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 5.Bibi E, Gros P, Kaback H R. Proc Natl Acad Sci USA. 1993;90:9209–9213. doi: 10.1073/pnas.90.19.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuhaus H, Thom E, Möhlmann T, Steup M, Kampfenkel K. Plant J. 1997;11:73–82. doi: 10.1046/j.1365-313x.1997.11010073.x. [DOI] [PubMed] [Google Scholar]
- 7.Tjaden J, Schwöppe C, Möhlmann T, Quick P, Neuhaus H. J Biol Chem. 1998;273:9630–9636. doi: 10.1074/jbc.273.16.9630. [DOI] [PubMed] [Google Scholar]
- 8.Fiermonte G, Palmieri L, Dolce V, Larorsa F M, Palmieri F, Runswick M J, Walker J E. J Biol Chem. 1998;273:24754–24759. doi: 10.1074/jbc.273.38.24754. [DOI] [PubMed] [Google Scholar]
- 9.Fiermonte G, Dolce V, Palmieri F. J Biol Chem. 1998;273:22782–22787. doi: 10.1074/jbc.273.35.22782. [DOI] [PubMed] [Google Scholar]
- 10.Fiermonte G, Dolce V, Palmieri L, Ventura M, Runswick M J, Palmieri F, Walker J E. J Biol Chem. 1998;276:8225–8230. doi: 10.1074/jbc.M009607200. [DOI] [PubMed] [Google Scholar]
- 11. Fiermonte, G., Palmieri, L., Todisco, S., Agrimi, G., Palmieri, F. & Walker, J. E. (2002) J. Biol. Chem., in press. [DOI] [PubMed]
- 12.Wright E M. Am J Physiol. 2001;280:F10–F18. doi: 10.1152/ajprenal.2001.280.1.F10. [DOI] [PubMed] [Google Scholar]
- 13.Wright E M, Hirayama B A, Loo D D F, Turk E, Hager K. In: Physiology of the Gastrointestinal Tract. Johnson L R, Alpers D H, Christensen J, Jacobson E D, Walsh J H, editors. New York: Raven; 1994. pp. 1751–1772. [Google Scholar]
- 14.Wright E M, Loo D D F. Ann NY Acad Sci. 2000;915:54–66. doi: 10.1111/j.1749-6632.2000.tb05223.x. [DOI] [PubMed] [Google Scholar]
- 15.Turk E, Kim O, le Coutre J, Whitelegge J P, Eskandari S, Lam J T, Kreman M, Zampighi G A, Faull K F, Wright E M. J Biol Chem. 2000;275:25711–25716. doi: 10.1074/jbc.M003127200. [DOI] [PubMed] [Google Scholar]
- 16.Panayotova-Heiermann M, Leung D W, Hirayama B A, Wright E M. FEBS Lett. 2000;459:386–390. doi: 10.1016/s0014-5793(99)01292-2. [DOI] [PubMed] [Google Scholar]
- 17.Quick M, Jung H. Biochemistry. 1998;37:13800–13806. doi: 10.1021/bi980562j. [DOI] [PubMed] [Google Scholar]
- 18.Bibi E, Kaback H R. Proc Natl Acad Sci USA. 1990;87:4325–4329. doi: 10.1073/pnas.87.11.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miroux B, Walker J E. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 20.Quick M, Loo D D F, Wright E M. J Biol Chem. 2001;276:1728–1734. doi: 10.1074/jbc.M005521200. [DOI] [PubMed] [Google Scholar]
- 21.Quick M, Tebbe S, Jung H. Eur J Biochem. 1996;239:732–736. doi: 10.1111/j.1432-1033.1996.0732u.x. [DOI] [PubMed] [Google Scholar]
- 22.Jung H, Tebbe S, Schmid R, Jung K. Biochemistry. 1998;37:11083–11088. doi: 10.1021/bi980684b. [DOI] [PubMed] [Google Scholar]
- 23.Rigaud J-L, Pitard B, Levy D. Biochim Biophys Acta. 1995;1231:223–246. doi: 10.1016/0005-2728(95)00091-v. [DOI] [PubMed] [Google Scholar]
- 24.Zampighi G A, Kreman M, Boorer K J, Loo D D F, Bezanilla F, Chandy G, Hall J E, Wright E M. J Membr Biol. 1995;148:65–78. doi: 10.1007/BF00234157. [DOI] [PubMed] [Google Scholar]
- 25.Peterson G L. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 26.Turk E, Wright E M. J Membr Biol. 1997;159:1–20. doi: 10.1007/s002329900264. [DOI] [PubMed] [Google Scholar]
- 27.Sauer G A, Nagel H, Koepsell H, Bamberg E, Hartung K. FEBS Lett. 2000;469:98–100. doi: 10.1016/s0014-5793(00)01255-2. [DOI] [PubMed] [Google Scholar]
- 28.Turk E, Kerner C J, Lostao M P, Wright E M. J Biol Chem. 1996;271:1925–1934. doi: 10.1074/jbc.271.4.1925. [DOI] [PubMed] [Google Scholar]
- 29.Bibi E. Trends Biochem Sci. 1998;23:51–55. doi: 10.1016/s0968-0004(97)01134-1. [DOI] [PubMed] [Google Scholar]
- 30.Turk E, Zabel B, Mundlos S, Dyer J, Wright E M. Nature (London) 1991;350:354–356. doi: 10.1038/350354a0. [DOI] [PubMed] [Google Scholar]