

Abstract
It has long been recognized that the pathogenicity of a broad range of intracellular parasites depends on the availability of transition metal ions, especially iron. Nramp1 (natural resistance-associated macrophage protein 1), a proton-coupled divalent metal ion transporter, has been identified as a controlling factor in the resistance or susceptibility to infection with a diverse range of intracellular pathogens such as Toxoplasma, Salmonella, Mycobacterium, and Leishmania. The role of divalent metal ion transport is even more compelling given the existence of Nramp homologs in several intracellular parasites, such as mycobacteria. We have confirmed the functional homology of the Nramp homologue from Mycobacterium leprae by using a yeast complementation assay for divalent cation uptake. To facilitate a concerted biochemical and structural analysis of this important class of transporters, the M. leprae Nramp was expressed in Escherichia coli. Dual affinity tags were engineered at the N and C termini to allow for isolation of full-length protein at >95% purity. Site-directed spin labeling of Cys-299 reveals a flexible hinge-like domain. A weak dipolar interaction is detected between the nitroxide and paramagnetic transition ions, indicating this position is approximately 19 Å from the nearest high affinity binding site.
One of the most effective mechanisms of pathogen survival is the ability of some facultative and obligate intracellular parasites to reside, and hence multiply, within macrophages. Survival of a broad range of pathogens (protozoa such as Leishmania, Plasmodium, and Taxoplasma; bacteria such as Listeria monocytogenes; mycobacteria such as tuberculosis and leprae; and fluke worms such as Schistosoma) within macrophages involves a variety of mechanisms that allow the parasite to evade or inactivate the harmful effects of cytotoxic pathways, and particularly the effects of reactive oxygen and nitrogen intermediates.
After phagocytosis of a pathogen, the activated macrophage displays the “oxidative burst,” generating reactive oxygen and nitrogen intermediates. It has long been recognized that the innate ability of intracellular parasites, especially mycobacteria such as Mycobacterium leprae and Mycobacterium tuberculosis, to neutralize the toxic effects of reactive oxygen and nitrogen intermediates may be an important virulence factor (1). The neutralization of reactive intermediates generated by the activated macrophage is accomplished by enzymes containing iron and/or manganese cofactors (2–4).
Natural resistance to infection with unrelated intracellular parasites such as Toxoplasma, Mycobacteria, Salmonella, and Leishmania is controlled in the mouse by a single gene on chromosome 1, designated Bcg, Ity, or Lsh. The product for the Bcg gene, designated natural resistance-associated macrophage protein 1 (Nramp1), has been isolated and shown to encode a macrophage-specific membrane protein, which is altered in susceptible animals (5–7). Nramp1 plays an essential role only in the early part of the macrophage-parasite interaction, suggesting it may function during the initial cytocidal or cytostatic response.
An appreciation of the importance in limiting available iron to maintain immunity has existed for several decades (8). Strong evidence exists showing that the mammalian Nramps are divalent metal ion transporters that function by sequestering metal ions from the lumen into the macrophage cytoplasm by coupling the metal ion translocation to the downhill movement of protons from the acidified lumen (9, 10).
The identification of bacterial prokaryotic Nramp homologs has reinforced the central importance of metal ion homeostasis in immunity. Recently, the manganese transport functions of bacterial Nramp homologues (MntH) from Escherichia coli, Salmonella typhimurium, and M. tuberculosis have been demonstrated (11–13). Thus the resistance of the host against the pathogen, and the pathogenicity of the microorganism, may depend on the performance of the individual Nramp or homologous protein. To explore the structure-function relationships in this important class of proteins, we have initiated a biochemical and spectroscopic examination of the Nramp homologue from M. leprae (Ml MntH).
Materials and Methods
Materials.
(1-Oxyl-2,2,5,5-tetramethylpyrroline-3-methyl) methanethiosulfonate (methanethiosulfonate spin label) was purchased from Reanal Finechemical (Budapest, Hungary). Chromium oxalate (potassium trioxalatochromate) was obtained from G. Frederich Smith Chemicals (Powell, OH). n-Dodecyl β,d-maltopyranoside (DDM) and Triton X-100 were obtained from Anatrace (Maumee, OH). Complete protease inhibitor was purchased from Roche Diagnostics. StrepTactin Sepharose media was obtained from Genosys Biotechnologies (The Woodlands, Texas). The RPAS purification module was purchased from Amersham Pharmacia. Slyde-a-Lyzer dialysis cassettes [molecular weight cutoff (MWCO) of 10 kDa] were obtained from Pierce. Purified soybean polar lipid extract was purchased from Avanti Polar Lipids (Alabaster, AL). 54Mn (13.3 Ci/mmol) was purchased from Perkin–Elmer.
Cloning and Sequence Analysis.
Genomic DNA from M. leprae was cloned from genomic DNA kindly provided by Patrick Brennan, Colorado State University, Fort Collins, CO. The MnTH gene (U15184) from M. leprae (Ml MntH) was amplified by using PCR with primers complementary to the 5′ and 3′ ends of the gene and inserted into the pBAD-TOPO plasmid (Invitrogen). Silent mutagenesis was then performed to remove the native internal HindIII site in the Ml MntH coding sequence by using PCR. The topology of the protein was examined by the Wimley-White whole-residue hydropathy analysis, with Membrane Protein Explorer (http://blanco.biomol.uci.edu/mpex). Ambiguous regions were further evaluated by alignment analysis assuming the highest conservation among the membrane-spanning domains. Alignments were performed by using the clustal w algorithm within the commercial software vector nti.
Engineering the MntH Gene Bacterial Expression.
The gene Ml MntH in pBAD-TOPO was then reamplified to place a NcoI restriction site (at ATG) and insert the codons for the nine-residue NWSHPQFEK StrepII tag (14) at the 5′ end and insert the 13-residue GAPVPYPDPLEPR E tag (Amersham Pharmacia) sequence followed by a HindIII site at the 3′ end. The resulting fusion protein has the N-terminal StrepII sequence inserted after the ATG start and C-terminal E-tag sequence inserted just before the TGA stop codon. The PCR product was cloned into the NcoI and HindIII sites of the pBAD/Myc-HisB expression vector (Invitrogen) to generate the construct pBAD-ML-ST/ET. To generate the Cys-less control for EPR experiments, Cys-299 was substituted with an alanine by oligonucleotide-directed mutagenesis using PCR (15).
54Mn Uptake Measurement.
E. coli TOP 10 (Invitrogen) were transformed with pBAD-ML-ST/ET. Cells were grown aerobically overnight in 10 ml LB with 100 μg/ml ampicillin at 37°C and agitated at 250 rpm. The culture was transferred to 90 ml LB and grown aerobically at 30°C and 250 rpm in the presence of 100 μg/ml ampicillin and 1 mM MnCl2. Cells to be induced were grown to an OD600 of 0.63 and treated with 0.02% (wt/vol) arabinose. The cell cultures (induced and uninduced) were then incubated for an additional 3 h. Cells were pelleted at 7,277 g and washed with 100 ml of incubation medium (40 mM Mes/0.2% glucose, pH 5.8). Incubations contained induced or uninduced cells resuspended in incubation medium to an OD600 of 1.6 (final volume = 1 ml). Cells were preincubated at 37°C for 2 min before dosing with 54Mn (final concentration = 2.6 nM). At 0 and 10 min, metal transport was halted by dilution of the incubation with 4 ml of quench solution (5 mM Tris, pH 8.0, 0.2 mM EDTA) followed by immediate filtration through a 0.7-μm glass filter. The incubation tube was rinsed with another 4 ml of quench solution, which was then used to wash the filter. Radioactivity on each filter was measured by using liquid scintillation counting. For each culture, to eliminate the signal caused by nonspecific binding of 54Mn, the amount of radioactivity measured at T0 was subtracted from that measured after 10 min of incubation. The significance of difference between the amounts of radioactivity taken up by the uninduced and induced cells was determined by using the F test followed by the T test.
Insertion of the MntH Gene into the Yeast Expression Plasmid.
A DNA fragment encoding MntH was amplified by PCR using the primers TAT GAA TTC GTGGCAGCCAACGTCAG and TAT CTC GAG TTA ACT GGT CAC GGT CAG GTA that contain the EcoRI and XhoI sites, respectively. After digestion by these enzymes the fragment was cloned into the respective restriction sites of the yeast expression plasmid BFG1 (16). This way MntH was inserted in-frame at the EcoRI site and as a result contains 50 additional aa at the N terminus bearing three copies of the hemagglutinin epitope (YPYDVPDYA).
Yeast Mutant Complementation by MntH.
The yeast wild type that was used is S. cerevisiae W303 (MATaα trp1 ade2 his3 leu2 ura3). The other strains used in this work are: ΔSMF3 (MATα ade2 his3 trp1 leu2 SMF3∷URA3) and ΔSMF1 + 2+3 (MATα ade2 his3 leu2 ura3 SMF1∷URA3/FOA SMF2∷URA3/FOA SMF3∷URA3). The construction of these yeast strains has been described (17). The cells were grown in a YPD medium containing 1% yeast extract, 2% bactopeptone, and 2% dextrose. The yeast mutants in which the gene SMF3 was disrupted (ΔSMF3) or the three genes encoding Smf proteins were disrupted (ΔSMF1 + 2+3) were transformed with the BFG1 plasmid bearing the MntH gene. Yeast transformation was performed as described (18), and the transformed cells were grown on minimal plates containing a 0.67% yeast nitrogen base, 2% dextrose, 2% agar, and the appropriate nutritional requirements. For metal-ion limitation experiments, the cells were grown in a medium containing 0.25% yeast extract, 0.5% bactopeptone, 2% dextrose, 50 mM Mes, 3 mM EGTA, pH 6 (19, 20).
Growth of Cells for Recombinant Protein Production.
E. coli TOP 10 (Invitrogen) was transformed with pBAD-ML-ST/ET. Cultures were grown aerobically at 30°C in LB broth with ampicillin (100 μg/ml). A 1-liter cell culture was grown overnight and then diluted 11-fold in a 12-liter fermentor. The cells were stirred at 400 rpm and aerated with a flow rate of 12 ml/min. Cells were grown to an OD600 of 0.90–0.95 and then treated with 1 mM Fe, Mn, and Zn followed by 0.02% l-arabinose 5 min later. Cells were fermented for an additional 3.5 h.
Enrichment of Ml MntH.
Cells were harvested by centrifugation at 4,657 g for 15 min at 4°C. The pelleted cells were resuspended in 5 ml of buffer A (100 mM Tris⋅HCL/1 mM EDTA buffer, pH 7.4) containing 1 pill (crushed) of protease inhibitor. Cells were disrupted by passage through a French pressure cell. Cell fragments were sedimented at 27,000 g for 20 min at 4°C. The supernatant was centrifuged at 205,076 g for 70 min at 4°C to pellet the cell membranes. The pellet was resuspended in buffer A, 1% sodium cholate, and 1 mM DTT and stirred for 60 min at 4°C to solubilize peripheral membrane proteins. The mixture was centrifuged for 60 min at 205,076 g and 4°C. The pellet was resuspended in 1% DDM, 4% Triton, and 1 mM DTT in buffer A (total volume of 24 ml) and stirred for 60 min at 4°C to solubilize integral membrane proteins. The mixture was centrifuged at 205,076 g for 60 min at 4°C to pellet unsolubilized material. The supernatant was diluted to a final volume of 1 liter in buffer A and applied to a 20-ml open column containing 4 ml of StrepTactin media equilibrated with EQ buffer [0.096% (vol/vol) Triton X-100 and 0.024% (wt/vol) DDM in buffer A]. The column was washed with 100 ml of the EQ buffer, and the target protein was eluted with 40 ml of EQ buffer containing 2.5 mM desthiobiotin. This eluent was then applied four times to an RPAS purification module column containing the anti-E tag media, which was equilibrated in the kit's binding buffer containing 0.096% (vol/vol) Triton X-100 and 0.024% (wt/vol) DDM. The column was then washed and the protein was eluted as described in the manufacturer's instructions with the exception that all buffers used contained 0.096% (vol/vol) Triton X-100 and 0.024% (wt/vol) DDM. Protein concentrations in the presence of lipid and detergent were performed by using the Non Interacting protein assay from GenoTech (St. Louis, MO).
The anti-E tag column eluent was concentrated to a volume of 0.3 ml by using a 30-kDa MWCO concentrator. The retentate was injected into a Slyde-a-Lyzer (MWCO of 10 kDa) along with 100 μM of the sulfhydryl-specific methanethiosulfonate spin label to generate the spin-labeled protein C299R1. The mixture was incubated for 60 min at 4°C. The sample was then dialyzed at 4°C against 500 ml of 10 mM Hepes, pH 7.4 containing 0.1 mg/ml BSA, and containing 0.02% (wt/vol) DDM or the detergent mixture of 0.096% (vol/vol) Triton X-100 and 0.024% (wt/vol) DDM for proteins to be reconstituted. After 1–2 h, a second dialysis was carried out overnight at 4°C against 500 ml of the same buffer. The solution containing the dialyzed and spin-labeled Ml MntH was removed from the Slyde-a-Lyzer and concentrated to ≈50 μl on a 10-kDa MWCO concentrator for EPR analysis or reconstitution.
SDS/PAGE and Immunoblots.
Protein electrophoresis was carried out on 12% acrylamide gels as described (21). Gels were stained either for total protein with Coomassie blue or proteins were electroblotted from the gels onto 0.2 μm nitrocellulose or poly(vinylidene difluoride) membranes by using a semidry transfer apparatus. For immunoblots, membrane fractions were isolated after French press disruption and the equivalent to 0.5 OD600 units of cells were run on a 12% SDS/PAGE gel. After transfer to a blotting membrane, the blot was probed with an anti-V5 mAb (Invitrogen) specific for the C-terminal V5 epitope tag on the fusion protein or the anti-E-tag mAb (Amersham Pharmacia), followed by an horseradish peroxidase-conjugated goat-anti-mouse secondary Ab (BioRad). Bands were visualized by enhanced chemiluminescence (SuperSignal, Pierce). Slot blots were performed by using a 0.45 μm nitrocellulose membrane on the Bio-Dot SF manifold both from BioRad. Detergent-extracted membrane samples for slot blotting were prepared as described above. Three hundred micrograms of protein was loaded into each slot and blotted according to the manufacturer's instructions. The blot was subsequently probed with the anti-E-tag mAb.
In-Gel Digestion and Peptide Sequencing.
Protein digestion, peptide purification, and amino acid analysis were carried out at the Molecular Structure Facility at the University of California at Davis. Briefly, the Coomassie-stained protein band on the gel containing the Ml MntH protein was excised from the gel. The protein within the band was reduced with 10 mM DTT in 100 mM NH4HCO3, alkylated with 55 mM iodoacetamide in 100 mM NH4HCO3, and digested overnight at 37°C in 50 mM NH4HCO3, pH 7.8, using modified porcine trypsin. The resulting peptides were extracted from the gel with 0.1% trifluoroacetic acid followed by 5% formic acid in 50% acetonitrile. The extracts were sequenced with mass spectrometry.
Reconstitution of the Ml MntH Protein.
Reconstitution of ≈0.7 mg of purified and spin-labeled protein was carried out as described (22). EPR analysis was carried out on the sample dialyzed against 10 mM Hepes, pH 7.4.
EPR Spectroscopy.
EPR measurements were carried out in a JEOL X-band spectrometer fitted with a loop-gap resonator (23, 24). An aliquot of purified, spin-labeled protein (5 μl) at a final concentration of about 100 μM protein in 10 mM Hepes, pH 7.4 containing ≈0.14% (wt/vol) DDM was placed in a sealed quartz capillary contained in the resonator. Spectra of samples at room temperature (20–22°C) were obtained by a single 60-s scan over 100 G at a microwave power of 2 mW, a receiver gain of 125, and a modulation amplitude optimized to the natural line width of the individual spectrum. Because the Cys-less Cys-299→Ala mutant gave no detectable signal at this receiver gain, subtraction for background label was unnecessary. Relaxer accessibilities were obtained from power saturation measurements as described (25, 26).
For the reconstituted protein (about 10 μM), signal averaging was carried out for five scans because of the reduced signal strength. This was also carried out for the buffer control. The resulting spectrum was subtracted from that of the reconstituted protein to remove background interference. EPR instrument conditions were as stated earlier except for a scan length of 120 s, a microwave power of 4 mW, and a receiver gain of 630.
Data Analysis.
Power saturation curves were analyzed as described (27). Spectral broadening caused by dipolar interaction between a paramagnetic metal ion and a nitroxide as described by Leigh (28) has been described (15, 29, 30). Briefly, the line width (δH) of the relaxed spin is influenced by the strength of the interaction and the interspin vector orientation (θ′R) relative to the applied magnetic field according to:
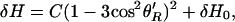 |
1 |
where δH0 is the line width in the absence of interaction. C, the dipolar broadening coefficient, is related to the interspin distance r by:
 |
2 |
where g is the electronic g factor of the nitroxide, β is the Bohr magneton, and μ is the magnetic moment of the metal. This treatment assumes that τ is equal to T1e, the electronic relaxation time of the metal and is less than the inverse of the frequency of the dipolar interaction. The T1e of Cu(II) at room temperature is on the order of 3 × 10−9 s (29).
Results
The Ml MntH Complements Divalent Metal Transport in Null Mutant Yeast.
To confirm that the sequence homology of the Ml MntH extends to functional homology as well, we expressed the protein in a yeast strain deficient in three SMF genes, which code for divalent metal transport proteins (17). This yeast stain (ΔSMF3) is unable to grow under low (+EGTA) metal ion conditions, unless complemented by the expression of a functional divalent cation transport protein (31). As shown in Fig. 1, heterologous expression of the mycobacterial MntH proteins in the ΔSMF3 strain promotes growth in the presence of EGTA, thereby demonstrating its ability to function as a divalent transition ion transporter.
Figure 1.

Functional complementation of the yeast SMF null mutant (ΔSMF3) by the Ml MntH gene. The various yeast strains were grown on YPD-agar plates as described in Materials and Methods. The medium in the right plate was supplemented by 3 mM EGTA. (A) Wild-type strain W 303. (B) ΔSMF3 strain. (C) ΔSMF3 strain transformed with the yeast expression vector pBFG1. (D) ΔSMF3 strain transformed with pBFG1 containing the cloned Ml MntH.
Overexpression and Purification of the MntH Protein in E. coli.
The sequence for the putative Nramp-related metal ion transporter from M. leprae encodes for a basic, hydrophobic 40-kDa protein of 377 residues that shares approximately 40% similarity with the human macrophage Nramp1. Like the hNramp1, hydropathy analysis of Ml MntH predicts 12 transmembrane (TM) domains (Fig. 2), a common topography for secondary active transport proteins. This topography conforms to the general features associated with membrane proteins, namely, a preponderance of positive charges on the cytoplasmic side of the membrane (32), and a high frequency of aromatic residues positioned near the membrane interface (33).
Figure 2.

A topology model for the Ml MntH protein. The approximate sequence positions for residues at the bilayer interface are numerically indicated. The relative positions of the conserved charged residues (+ or −) and histidine residues (H) are also indicated. The location of Strep II (ST) and E tag (ET) affinity sequences in the fusion protein are noted; however, the residue numbering is given according to the native sequence. The prediction places three acidic and three basic residues within membrane spanning regions, suggesting the presence of ion pairs within the bilayer. The approximate position of Gly-95, the conserved glycine residue of TM IV found mutated in Nramp1 and Nramp2-responsible defects, is indicated.
Initial expression of the M. leprae gene was detected by using the V5 epitope, fused at the C terminus (Fig. 3A). Similar to most hydrophobic proteins, the Ml MntH protein migrates faster (≈33 kDa) than its size would predict relative to standard SDS/PAGE molecular mass markers. Peptide sequencing of the 33-kDa band confirms the sequence of the engineered Ml MntH protein (not shown).
Figure 3.

(A) Inducible expression of Ml MntH in the membrane fraction of E. coli. Shown is the Western blot analysis of expression from the gene cloned into the pBAD-TOPO plasmid containing the C-terminal V5 peptide epitope. The positions of protein molecular mass markers are indicated to the left in kDa. Lane 1, membranes from uninduced cells; lane 2, membranes from cells induced with l-arabinose. (B) SDS/PAGE gel of purified Ml MntH protein after affinity chromatography with the StrepTag and then E tag columns. The purified sample was loaded into lane 1. Lane 2 shows the molecular size standards in kDa. (C) Ml MntH-dependent 54Mn uptake. Uptake of radioactivity in uninduced (U) and induced (I) cells after 10 min of incubation. Levels of accumulation were determined from four individual samples measured for each time point. Each value represents the mean ± SE of three experiments performed in separate cultures.
Based on immunoblots, protein turnover of overexpressed Ml MntH is significant in E. coli isolated membranes. This degradation problem results in fragments that coisolate with full-length protein when a single-phase affinity step is used. To facilitate the isolation of recombinant protein, the V5 epitope was replaced with the 13-aa E tag peptide GAPVPYPDPLEPR (Amersham Pharmacia) at the C terminus, and the 9-aa Strep II tag (34) sequence NWSHPQFEK was inserted at the N terminus. By engineering affinity tags at each end of the molecule, a convenient two-pass affinity purification was achieved, resulting in the isolation of only full-length protein. Using this approach to purify the Ml MntH protein from E. coli, the yield is ≈1 mg per 10 liters of culture, and SDS/PAGE gels (Fig. 3B) indicate a purity of >95%.
The functionality of the Ml MntH protein from E. coli was confirmed by comparing the 54Mn uptake of E. coli carrying the pBAD-Ml MntH plasmid with and without l-arabinose induction (Fig. 3C). Induction of MntH expression results in a significantly (P < 0.05) higher amount of radioactivity accumulated (491 ± 28 cpm) compared with the background transport pathways of uninduced cells (290 ± 22 cpm).
Interestingly, yields of Ml MntH overexpressed in E. coli are enhanced by 90–120% with the inclusion of 1 mM divalent transition ions (Mn, Zn, and Fe) in the culture (Fig. 4). This finding suggests that substrate stabilization of the protein structure may account for the diminished degradation.
Figure 4.

Slot blot expression analysis of the Ml MntH protein as a function of metal ion presence in the growth media during induction. (A) Detergent-solubilized membranes from cells transformed with pBAD-mycHis plasmid (control). (B) Detergent-solubilized membranes from cells transformed with pBAD-mycHis plasmid containing the Ml MntH gene. (C) Same as B, but with 1 mM Mn2+, Zn2+, and Fe2+ in the growth media. Detection was performed by using the anti-E tag antibody. Each slot was loaded with 300 μg total membrane protein.
Site-Directed EPR Spectroscopy of Wild-Type Ml MntH.
The Ml MntH contains a single Cys at position 299 (position 309 in the Strep II tag fusion), which is not conserved among Nramps. Reaction of the protein with the methanethiosulfonate spin label results in specific attachment of the nitroxide spin label at position 299, resulting in the protein C299R1. Based on the hydropathy prediction, the label will report information from the approximate center of TM domain X (TM X). Examination of the labeled protein in DDM micelles (≈0.1%) by EPR spectroscopy reveals the label is attached to a flexible domain (Fig. 5A).
Figure 5.

The EPR line shape of position 299 in the Ml MntH protein in DDM micelles (A) and after reconstitution into a lipid bilayer (B). In A, the distance of position 299 to the nearest metal binding site was measured by adding 0.5 mM CuCl2 (solid dark line), which will broaden the spectrum in a distance-dependent manner. The control spectrum (broken gray line) was obtained in the presence of 0.5 mM ZnCl2.
To probe the proximity of position 299 to the nearest high affinity divalent metal ion binding site, the spectrum was acquired in either the presence (0.5 mM) of paramagnetic CuCl2 or diamagnetic ZnCl2. A magnetic dipolar interaction between the bound Cu(II) ion and the spin-labeled side chain produces a broadened nitroxide spectrum, which simply appears as a reduction in signal amplitude (29). The strength of the interaction depends on the distance separating the nitroxide and Cu(II) centers, such that the closer the bound metal ion, the greater the reduction in observed signal amplitude. The +Cu(II) spectrum shown in Fig. 5A displays a 10% drop in amplitude relative to the control. This finding indicates the nitroxide at position 299 is somewhat removed from the nearest metal site, on the order of 19 Å. Increasing the added Cu(II) to 0.8 mM did not induce further broadening (not shown).
We also examined the protein after reconstitution into soybean lecithin liposomes. The resulting EPR spectrum of reconstituted Ml MntH C299R1 is shown in Fig. 5B. Remarkably, the line shape of reconstituted C299R1 also reflects a flexible environment, although as predicted, the motions are more limited when the protein is in a lipid bilayer.
The chemical environment surrounding the spin-labeled side chain R1 can be directly assessed by comparing the collision frequencies of nonpolar, polar, and relaxation agents (27). Thus, the accessibility of C299R1 in detergent micelles to oxygen and chromium oxalate (CrOx) was measured by using power saturation EPR. The position labeled displays relatively high accessibility to oxygen (Π(O2) = 0.46), and low accessibility to 20 mM CrOx [Π(CrOx) = 0.32]. The resulting Φ value (Φ = ln [(Π(nonpolar)/Π(polar)] = 0.35) reveals the spin label is in a hydrophobic environment, perhaps exposed to the detergent hydrocarbon chains.
Discussion
Nramps are increasingly recognized for their central importance in mineral homeostasis and host resistance to intracellular microbes in humans. The striking similarity of prokaryotic members of this family, including genes in obligate intracellular parasites M. tuberculosis and M. leprae, to the Nramp1 macrophage protein has illuminated the importance for a concerted structure-function analysis of this family of proteins. Here, we provide purification and spectroscopic studies on a Nramp.
Based on immunoblots, protein turnover of overexpressed Ml MntH is significant in E. coli isolated membranes. This degradation problem results in fragments that coisolate with full-length protein when a single-phase affinity step is used. We have circumvented this problem by designing a dual-affinity tag protein that facilitates a convenient, two-step isolation leading to >95% purity. An attractive feature of both the StrepTag and the E tag is their small size, which minimizes the probability that the fusions will interfere with native structure or trafficking. Because of the protein's role in binding and translocating divalent transition ions, the more common polyHis affinity tag was not explored.
Adding to the difficulty in obtaining sufficient amounts of recombinant protein is the low expression level of Ml MntH in E. coli, a common problem for secondary active transport proteins. Thus, our finding that divalent metals increase levels of protein in the membrane by 90–120% is significant. Recent studies on prokaryotic Nramp homologues from S. typhimurium and E. coli have shown that transcription of these proteins is diminished when the cells are exposed to divalent transition ions (11, 12), presumably caused by promoter elements that recognize divalent metals (12). In our case, we suspect that the metals serve a substrate-protective role, through structural stabilization, which may benefit insertion and/or shield the protein from proteases. Presumably the efficacy of any individual ion to increase yields is a function of its solubility, oxidation rate, and binding affinity to the protein.
Cys-299 in the native Ml MntH immediately precedes a Pro-Ala sequence, which is predicted to introduce a hinge-like feature in α-helixes. Analysis of the thermofactors around proline hinges has shown the residues N terminal to the proline display the highest backbone dynamics (35). The EPR line shape of C299R1 in DDM detergent micelles is consistent with such a lack of order, as reflected by the narrow lines arising from motional averaging of the hyperfine anisotropy (36). Reconstitution of the protein into liposomes moderately restricts the dynamics reported by the spin label, suggesting the flexibility of this domain is maintained in the native membrane. In addition, the accessibility ratio Π of the spin-labeled side chain at position 299 confirms the topological prediction (based on hydropathy analysis) of a membrane-imbedded location. These findings are consistent with what can be ascertained from a helical wheel representation of TM X. High-resolution structures of membrane proteins have revealed that—like soluble proteins—the interior side chains are mostly hydrophobic, but the lipid-exposed residues are even more hydrophobic (37). The lack of H-bonding possibilities for residues exposed to the lipid acyl chains results in polar residues being distributed along the interior-oriented faces of membrane-spanning segments. In the protein interior, polar side chains can form H-bonding and salt bridges with side chains on neighboring membrane-spanning segments. Thus helical wheel projections of TM domains can help direct the targeting of spin labels and the construction of models. Considering our initial EPR of the Ml MntH protein, we have labeled position Cys-299 within TM X, which is not conserved among MntH proteins. Within TM X, the polar arginines, theronine, and glycine (presents exposed amide bond) cluster to the face opposite of Cys-299 when this region is projected as a helix. In contrast, Cys-299 projects along a hydrophobic face of TM X. Thus by coupling the propensity for high conservation on the interior-facing residues (38), and the propensity for nonpolar side chains being exposed to the lipid bilayer, models for the orientation of TM domains can be constructed and tested experimentally. In the case of TM X, the high mobility of the spin label attached to position 299, excludes the possibility that this residue faces the protein interior. But rather, the data reveal the position is in a fluid, hydrophobic environment and attached to a flexible backbone. Upon reconstitution, the mobility is significantly attenuated, giving rise to a spectrum resembling a surface α-helix position (39). Finally, we probed the proximity of Cys-299 to the nearest divalent transition metal binding site by observing the magnitude of C299R1 broadening caused by exogenous Cu(II). The low broadening suggests the C-terminal portion of the protein lacks a high affinity metal binding site.
It is noteworthy that the Ml MntH contains a GGV sequence in the fourth membrane-spanning domain (Fig. 2). This motif is conserved in the eukaryotic Nramp family, and mutations at the second Gly position to a charged residue are responsible for mice and rat phenotypes associated with abnormal iron transport (40), resulting in either susceptibility to intracellular pathogens such as mycobacteria, leishmania, and Salmonella (Nramp1 mutation) or anemia (Nramp2 mutation) that cannot be corrected by parenteral administration of iron (40). It is also noteworthy that a single histidine (predicted within TM VI, see Fig. 2) is conserved across the eukaryotic and mycobacterial transporters. Thus, this position is a good candidate for participating in substrate binding and translocation (see below). Interestingly, the MntHs of mycobacteria contain a HXHXHX (HX3, where X is G, P, A, or S) motif within the predicted cytoplasmic loop connecting TMs VI and VII. This motif, which is expected to form a strong binding site for divalent transition metals (41), constitutes the signature sequence for the ZIP and ZnT zinc transporters (42). Thus, it will be important to explore the functional and structural consequences of mutations to sites conserved across all members, as well as elucidating the significance of features unique to the prokaryotic or eukaryotic members.
Acknowledgments
We thank Dr. P. Brennan for providing genomic M. leprae DNA. We are grateful to Angela Jones for excellent technical assistance. This work was supported by The March of Dimes Basil O'Connor Award.
Abbreviations
- Nramp
natural resistance-associated macrophage protein
- DDM
n-dodecyl β,d-maltopyranoside
- TM
transmembrane
References
- 1.Krahenbuhl J L, Adams L B. Immunol Ser. 1994;60:281–302. [PubMed] [Google Scholar]
- 2.Weinberg J, Hibbs J. Nature (London) 1977;269:245–247. doi: 10.1038/269245a0. [DOI] [PubMed] [Google Scholar]
- 3.Drapier J, Hibbs J. J Clin Invest. 1986;78:790–797. doi: 10.1172/JCI112642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pryor W, Squadrito G. Am J Physiol. 1995;268–722:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 5.Cellier M, Prive G, Belouchi A, Kwan T, Rodrigues V, Chia W, Gros P. Proc Natl Acad Sci USA. 1995;92:10089–10093. doi: 10.1073/pnas.92.22.10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Govoni G, Vidal S, Cellier M, Lepage P, Malo D, Gros P. Genomics. 1995;27:9–19. doi: 10.1006/geno.1995.1002. [DOI] [PubMed] [Google Scholar]
- 7.Vidal S, Gros P, Skamene E. J Leukocyte Biol. 1995;58:382–390. doi: 10.1002/jlb.58.4.382. [DOI] [PubMed] [Google Scholar]
- 8.Bullen J J, Griffiths E. Iron and Infection: Molecular, Physiological, and Clinical Aspects. New York: Wiley; 1987. [Google Scholar]
- 9.Gunshin H, MacKenzie B, Berger U V, Gunshin Y, Romero M F, Boron W F, Nussberger S, Gollan J L, Hediger M A. Nature (London) 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 10.Sacher A, Cohen A, Nelson N. J Exp Biol. 2001;204:1053–1061. doi: 10.1242/jeb.204.6.1053. [DOI] [PubMed] [Google Scholar]
- 11.Makui H, Roig E, Cole S T, Helmann J D, Gros P, Cellier M F. Mol Microbiol. 2000;35:1065–1078. doi: 10.1046/j.1365-2958.2000.01774.x. [DOI] [PubMed] [Google Scholar]
- 12.Kehres D G, Zaharik M L, Finlay B B, Maguire M E. Mol Microbiol. 2000;36:1085–1100. doi: 10.1046/j.1365-2958.2000.01922.x. [DOI] [PubMed] [Google Scholar]
- 13.Agranoff D, Monahan I M, Mangan J A, Butcher P D, Krishna S. J Exp Med. 1999;190:717–724. doi: 10.1084/jem.190.5.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt T G M, Koepke J, Frank R, Skerra A. J Mol Biol. 1996;255:753–766. doi: 10.1006/jmbi.1996.0061. [DOI] [PubMed] [Google Scholar]
- 15.Voss J, Hubbell W L, Kaback H R. Biochemistry. 1998;37:211–216. doi: 10.1021/bi972152l. [DOI] [PubMed] [Google Scholar]
- 16.Yelin R, Rotem D, Schuldiner S. J Bacteriol. 1999;181:949–956. doi: 10.1128/jb.181.3.949-956.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen A, Nelson H, Nelson N. J Biol Chem. 2000;275:33388–33394. doi: 10.1074/jbc.M004611200. [DOI] [PubMed] [Google Scholar]
- 18.Ito H, Fukuda Y, Murata K, Kimura A. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson H, Nelson N. Proc Natl Acad Sci USA. 1990;87:3503–3507. doi: 10.1073/pnas.87.9.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noumi T, Beltrán C, Nelson H, Nelson N. Proc Natl Acad Sci USA. 1991;88:1938–1942. doi: 10.1073/pnas.88.5.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Wu J, Voss J, Hubbell W L, Kaback H R. Proc Natl Acad Sci USA. 1996;93:10123–10127. doi: 10.1073/pnas.93.19.10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Froncisz W, Hyde J S. J Magn Reson. 1982;47:515–521. [Google Scholar]
- 24.Hubbell W L, Froncisz W, Hyde J S. Rev Sci Instrum. 1987;58:1879–1886. [Google Scholar]
- 25.Altenbach C, Flitsch S L, Khorana H G, Hubbell W L. Biochemistry. 1989;28:7806–7812. doi: 10.1021/bi00445a042. [DOI] [PubMed] [Google Scholar]
- 26.Voss J, Hubbell W L, Hernandez-Borrell J, Kaback H R. Biochemistry. 1997;36:15055–15061. doi: 10.1021/bi971726j. [DOI] [PubMed] [Google Scholar]
- 27.Altenbach C, Greenhalgh D A, Khorana H G, Hubbell W L. Proc Natl Acad Sci USA. 1994;91:1667–1671. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leigh J S. J Chem Phys. 1970;52:2608–2612. [Google Scholar]
- 29.Voss J, Salwinski L, Kaback H R, Hubbell W L. Proc Natl Acad Sci USA. 1995;92:12295–12299. doi: 10.1073/pnas.92.26.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voss J, Wu J, Hubbell W L, Jacques V, Meares C F, Kaback H R. Biochemistry. 2001;40:3184–3188. doi: 10.1021/bi002333e. [DOI] [PubMed] [Google Scholar]
- 31.Nelson N. EMBO J. 1999;18:4361–4371. doi: 10.1093/emboj/18.16.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von Heijne G. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 33.Yau W M, Wimley W C, Gawrisch I K, White S H. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 34.Voss S, Skerra A. Protein Eng. 1997;10:975–982. doi: 10.1093/protein/10.8.975. [DOI] [PubMed] [Google Scholar]
- 35.Sansom M S, Weinstein H. Trends Pharmacol Sci. 2000;21:445–451. doi: 10.1016/s0165-6147(00)01553-4. [DOI] [PubMed] [Google Scholar]
- 36.Hubbell W L, Cafiso D S, Altenbach C. Nat Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 37.Stowell M H, Rees D C. Adv Protein Chem. 1995;46:279–311. doi: 10.1016/s0065-3233(08)60338-1. [DOI] [PubMed] [Google Scholar]
- 38.Baldwin J M. EMBO J. 1993;12:1693–1703. doi: 10.1002/j.1460-2075.1993.tb05814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mchaourab H, Lietzow M A, Hideg K, Hubbell W L. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 40.Fleming M D, Andrews N C. J Lab Clin Med. 1998;132:464–468. doi: 10.1016/s0022-2143(98)90123-8. [DOI] [PubMed] [Google Scholar]
- 41.Regan L. Annu Rev Biophys Biomol Struct. 1993;22:257–287. doi: 10.1146/annurev.bb.22.060193.001353. [DOI] [PubMed] [Google Scholar]
- 42.Eng B H, Guerinot M L, Eide D, Saier M H., Jr J Membr Biol. 1998;166:1–7. doi: 10.1007/s002329900442. [DOI] [PubMed] [Google Scholar]