

Abstract
An enhanced bioinformatics tool incorporating the participation of molecular structure as well as sequence in protein DNA recognition is proposed and tested. Boltzmann probability models of sequence-dependent DNA structure from all-atom molecular dynamics simulations were obtained and incorporated into hidden Markov models (HMMs) that can recognize molecular structural signals as well as sequence in protein–DNA binding sites on a genome. The binding of catabolite activator protein (CAP) to cognate DNA sequences was used as a prototype case for implementation and testing of the method. The results indicate that even HMMs based on probabilistic roll/tilt dinucleotide models of sequence-dependent DNA structure have some capability to discriminate between known CAP binding and nonbinding sites and to predict putative CAP binding sites in unknowns. Restricting HMMs to sequence only in regions of strong consensus in which the protein makes base specific contacts with the cognate DNA further improved the discriminatory capabilities of the HMMs. Comparison of results with controls based on sequence only indicates that extending the definition of consensus from sequence to structure improves the transferability of the HMMs, and provides further supportive evidence of a role for dynamical molecular structure as well as sequence in genomic regulatory mechanisms.
The idea that structure as well as sequence might serve as a useful bioinformatics screening criterion has considerable potential in genomics for elucidating similarities with low sequence consensus. The proposal that protein–DNA recognition involves molecular geometry as a supplement to sequence-based, nonbonded contacts dates back at least to the observation of structural irregularities in the first high-resolution x-ray crystal structure of B-DNA (1, 2). However, structural characteristics of DNA at the most fundamental level, Boltzmann statistical mechanics, is described in terms of the probability of achieving a particular conformational or helicoidal state at a given temperature, rather than a single time averaged form. We propose herein a methodology in which probability models of dynamical structure derived from molecular dynamics (MD) simulations on DNA including counterions and water (3, 4) are incorporated along with DNA sequence information into hidden Markov models (HMM) (5, 6) suitable for genomic analysis. HMMs already provide a statistical framework for protein and DNA sequence alignments (6–8), and the probabilistic nature of HMMs per se (9, 10) is ideally suited for incorporating Boltzmann probability models of molecular structural characteristics from MD into a bioinformatics tool. The binding of catabolite activator protein (CAP) to cognate DNA sequences (11–13) serves as a model protein–DNA binding system and basis for demonstration and testing of the methodology. The resulting HMMs are applied to scans of the Escherichia coli genome. The results provide further exploration of a role for molecular geometry as well as sequence in protein DNA recognition and specificity as a means of genomic searches.
Background
The hypothesis of a molecular structural component in protein–DNA recognition is the basis of a number of recent research studies aimed at using observable properties of DNA sequences as a basis for genomic searches. A base pair scale derived from the sequence dependence of propeller twist has been used in scans of aligned polymerase II promoters (14). A dinucleotide scale based on sequence-dependent melting propensities was shown to have diagnostic capabilities for prokaryotic promoter regions (15). Dinucleotide scales based on gel retardation have been developed by Bolshoy et al. (16) and used as a basis for a search for promoter sites by Ozoline et al. (17). Trinucleotide scales have been developed based on both DNase I digestion (18) and nucleosome positioning (19, 20), and used in scans of polymerase II promoters (14). Perez-Martin and de Lorenzo (21) have recently reviewed the literature on DNA bending and genomic transcription. Recently, Lavery and Lafontaine (22, 23) have proposed a method, ADAPT (24), which begins with the crystal structure of the DNA in a protein–DNA binding site and calculates the compatible sequences based on energy minimization. An integrative view of the structural hypothesis has been developed by Pedersen et al. (25) and presented in a novel color-coded computer graphic wheel called a DNA structural atlas for E. coli and 17 other prokaryotic genomes. Numerous empirical correlations between functional genomic sites and molecular properties of the DNA were noted, including regions in which all characteristics had extreme values simultaneously.
Experiments do not directly yield probability measures or models, but probabilistic description of dynamical structure can be obtained from MD simulation. MD simulation (26) involves numerical integration of Newtonian laws of motion based on intermolecular forces computed from empirical or semiempirical potential functions and results in a description of structure as a function of time. With the availability of increased computer power, MD has become widely used for computational modeling of biological macromolecules in solution (27–29). Although computationally quite intensive, MD can generate all-atom description of the dynamics of in vitro DNA oligonucleotides in solution including solvent water and counterions explicitly, and the structural characteristics of DNA suitable for this project can be obtained from an analysis of MD results. The field of MD applied to DNA and nucleic acids in general has been under serious development since the early 1980s (30). The assessments of current MD force fields and simulation protocols compared with experimental data show dramatic improvement in recent years (3, 4). Although some specific deficiencies remain, successful descriptions of DNA sequence effects on dynamical structure (31, 32), conformational transitions (33, 34), and salient features of DNA bending and bendability (35–37) have been reported. However, to our knowledge, MD on DNA has not heretofore served as the basis for a genomic search.
HMM provide a natural way of incorporating both sequence information and probability models of structure into a form suitable for analyzing genomic DNA (6, 8). HMMs, following the notation of Baldi et al. (7), are a general statistical technique defined on a set of n states S = [S1, S2, . . . , Sn]. On moving from state to state consistent with a set of Markov transition probabilities T = [tij], each state emits, based on emission probabilities E = [eiα], a sequence of symbols αi from a well defined alphabet α = [α1, α2 . . . , αm]. The process is Markovian in that transitions depend only on the current step and that immediately preceding, and “hidden” because the path of the system from state to state is probabilistic and generally not an output of interest as long as the symbols emitted are consistent with the model. “Training” an HMM involves calculating numerically the transition and emission probabilities T and E based on a set of appropriate data. Extensive applications of HMM in computational biology are described in the recent literature (9). Notably, HMM studies of multiple sequence alignment (38), protein DNA binding sites (14), and gene finding (39) have already been developed on the basis of DNA sequence alone.
The binding of CAP to DNA, a well characterized example of a genomic regulatory system, was chosen as a demonstration case. CAP activates the transcription of many operons involved in the uptake and catabolism of various sugars and other carbon sources, and in addition functions as a repressor of its own gene. The crystal structure of CAP both uncomplexed (40) and complexed with a 30-bp oligonucleotide (41) have been reported (Fig. 1A). The CAP protein structure contains a helix–turn–helix (HTH) motif extending over some three turns of DNA helix (≈36 bp). The target site for CAP contains an interrupted inverted repeat with a highly conserved TGTGA cassette located one half turn away from the center of palindromic symmetry in one of the monomer units; the consensus of sequence in the binding region of the other monomer is not as strong. The HTH motif of CAP binds to the major groove of the cognate DNA sequence, which in the crystal form narrows the major groove and widens the minor groove compared with canonical B-form DNA structure. This occurs in conjunction with a ≈45° localized deformations produce by base pair roll at TpG steps, resulting in a ≈90° overall bend in the bound DNA. Recent studies of complexes involving DNA and CAP mutants (42, 43) provides additional perspective on structural issues as well direct and indirect readout in protein DNA complexation. Despite the low sequence homology in regions not involved in specific contact with the protein (Fig. 1B), certain combinations of mutations in this regions modulate the half-life of the bound species to on the order of 100-fold that of the biologically active site (44), further suggesting a role for indirect readout in the CAP–DNA system.
Figure 1.

(A) Crystal structure of the CAP DNA complex (41); (B) sequence logo indicating high information content in the half sites, constructed from set of known CAP binding sites considered in this article. The Logo figure was created from http://www.bio.cam.ac.uk/cgi-bin/seqlogo/logo.cgi. Arrows indicate mutations that, in various combinations, significantly affect binding affinity (44).
Methods
Molecular Dynamics Simulations.
Seven MD simulations on DNA sequences were obtained from a recent study of sequence effects on DNA structure (37) and used to form probability models of structural variables. Full details of simulation protocols, force field (AMBER parm.94), and analysis of the results are reported elsewhere (37). This particular set of sequences was chosen because the MD simulations were all performed using the same protocol, environmental conditions, and temperature, and thus the results by base pair step are as comparable as possible. We use in this study only base pair roll and tilt by dinucleotide step. The roll/tilt pair (16, 45, 46), although not the only operational variables in this problem, is appropriate for a simple demonstration of our methodology. Polar “bending dials” (ref. 45; Fig. 2) were used to display the magnitude and direction of sequence-dependent deformations obtained from analysis of all of the MD by base pair step. Points on a bending dial carry magnitude and direction of stepwise deformation of the local dynamical structure from a reference state of canonical B DNA (47). To avoid artifacts from end effects, the first and last base pair steps from each sequence were not included. Each bending dial was digitized into directional quadrants k = {1, 2, 3, 4} associated with displacements of roll toward the major and minor grooves (k = 1, 3) and displacements of tilt toward the respective sugar phosphate backbone (k = 2, 4). The radial coordinate of each bending dial was digitized into rings of radii rαj, j = {1, 2, . . . , l) defined such that the area between each ring, specifying a range of conformational magnitudes, encompasses (100/l)% of the total points for the bending dial α. The average of these 10 radial coordinates r̄αr is just
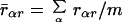 |
1 |
and taken to specify the reference B-form DNA base pair step behavior. This was used as the cutoff on all α dials and the point in each region (k, l) bounded by the annular rings 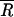 αr. Directional quadrant divisions k were counted and normalized to give values for the conformational probabilities Pα(k, l), where
αr. Directional quadrant divisions k were counted and normalized to give values for the conformational probabilities Pα(k, l), where
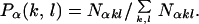 |
2 |
Here Nαkl is the number of MD data points for geometry (k, l) at base pair step α and the sum is over all discrete geometrical categories. Sample results for the optimal model in which l = 2 are presented in Table 2, which is published as supporting information on the PNAS web site, www.pnas.org. The MD results show essentially straight ApA steps (37) and a strong propensity for base pair roll toward the major groove at YpR steps (48), particularly CpA, and toward the minor groove at RpY steps. This behavior is generally consistent with that observed in oligonucleotide crystal structures (37, 46) and the general sequence DNA bending model of Dickerson and coworkers (49). The corresponding probability that a given base pair step α exhibits the geometry (k, l) is given by
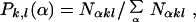 |
3 |
where the sum runs over all elements of the alphabet. The MD results obtained for Pk,l(α) are provided in Table 3, which is published as supporting information on the PNAS web site. The MD probabilities Pα(k, l) and Pk,l(α) in Tables 2 and 3 are used to incorporate dynamical structure into dinucleotide step HMMs as described below.
Figure 2.

Definition of roll/tilt bending dials for a DNA base pair step (45).
Hidden Markov Models.
All HMM calculations in this project were carried out with the program hmmpro, generously made available by NetID, Inc. Use of hmmpro in this project is based on an alphabet α consisting of the ten unique dinucleotide steps {ApA, ApT, . . . , CpC}. All HMM connectivities are based on the linear architecture shown in Fig. 3, in which the states Si are comprised of main states Mi, insert states Ii, and delete states Di, namely,
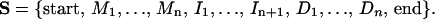 |
4 |
Based on the results of footprinting experiments (50), a site size with n = 36 encompasses the sequence length of CAP-related control elements. The HMM was trained on sixteen well characterized CAP binding sites observed to have regulatory functionality by using 350 cycles of the full gradient descent online option to obtain the emission and transition probabilities. Both transmission and emission probabilities were found to be well converged. The HMM at this point knows the probabilities for the observation of each of the unique base pair step in the known binding sites, and is referred to as a sequence HMM. These sequence-only-based HMMs (SEQ-HMMs) will score an unknown with respect to its probability of achieving the sequence characteristics learned from its training set.
Figure 3.

Connectivity of the linear architecture of the HMM used in this study. Our models have n = 36 instead of n = 3 as shown in the figure (see text).
Incorporating the MD description of sequence-dependent structure into HMMs (denoted MDS-HMMs) is accomplished here by a two-step process in which the emission probabilities eiα are transformed first to emission probabilities e′ikl and subsequently, by a second transformation, to emission probabilities e″iα. The e′iklare emission probabilities for a geometry (k, l) and the e″iαare emission probabilities conditional on simultaneously satisfying the probability model of structure from MD and probability model of sequence trained into the HMM. In step 1, the eiα are transformed by the Pα(k,l) of Table 2,
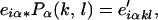 |
5 |
where the e′iαkl refer to state i, step α, and geometry (k, l). Because at this point we are not interested in the contribution from step α but in the geometry (k, l) corresponding to that step, we sum over all steps,
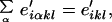 |
6 |
in which the e′ikl is the geometry emitted by the state i and the prime reminds us that the information on geometry was the result of an MD transformation subsequent to HMM training. However, e′ikl is indexed by geometry and not by step as in the original HMMs. To convert from structure to sequence, we apply a second transformation,
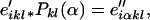 |
7 |
in which the e″iαkl are emission probabilities for symbol α, and geometry (k, l) in state i. Note that at this point the emission probability knows both step and geometry. Finally, we return this to the step level by summing over geometries,
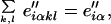 |
8 |
defining a matrix E" of transformed HMM emission probabilities e″iαthat incorporate the Boltzmann probability model of structure from MD by step. All probabilities are normalized at every opportunity, but this is omitted from the equations in order not to overly complicate the notation. The transition probabilities T remain unchanged.
When a protein–DNA binding site shows a tract of strong sequence consensus making physical contacts, an HMM model in which the SEQ condition is applied locally is desired, with no variation permitted in this region based on structure. Emissions for steps outside the contact region are subjected to transformation. For the 36-bp CAP DNA system, bases 10–15 are the highly conserved TGTGA sequence motif known from the crystal structure (41) to be involved in specific intermolecular contacts between the CAP protein and DNA. The result is a hybrid HMM of sequence only in the consensus region and sequence plus dynamical structure in the remainder of the site, referred to as a consensus sequence–dynamical structure hybrid (CSQ/MDS) HMM. This HMM scores unknowns based on probability for consensus sequence in positions 10–15 plus a probability of consensus sequence and structure in the remainder of the query site.
With all of the above in place, hmmpro with any one of the specified HMMs loaded as the active model can be used to score how well a given “new” (i.e., unknown to the model) or query sequence fits the established profile. All scores were generated using the Viterbi algorithm in global mode. The scoring system was calibrated by scoring the “test set” consisting of 25 known CAP binding sites [refs. 11, 50, and 51; including the 16 from the original HMM training set (black bars) plus 9 not included before (gray bars)]. plus 110 eukaryotic nonbinding sequences (white bars). A range of scores was obtained in this process, from which a threshold value that provides optimal discrimination between sequences that do and do not exhibit the pattern displayed by the training set. Because the optimal cutoff for each HMM is particular to the model, a standard way to compare the models was devised based on the success rate of finding 92% (missing two) and 88% (missing three) of the known binding sites.
In comparing and assessing results from the various HMMs described above, it is important to determine how well an HMM distinguishes between binding and nonbinding sites with respect to some threshold value in scoring (discriminatory ability), and how successfully an HMM can locate binding sites not in the training set (transferability). A well known problem arises with HMMs when the training set is biased in favor of a particular feature. The resulting HMM has strong discriminatory ability (with respect to this feature) but weak transferability—i.e., its ability to recognize unknown binding sites will be compromised by overtraining (6, 10). Examining scores on a test set that consists of both binding sites not in the original training set and nonbinding sites can recognize this problem.
Results
A total of 23 HMMs employing various choices of disposable parameters were tested so far in this project, and fall into the general categories sequence only (SEQ), molecular dynamical structure (MDS), and consensus sequence–dynamical structure hybrids (CSQ/MDS), as defined in the preceding section. The SEQ-HMM serves as a control. The test set was constructed as described above and scored with the MDS-HMM. Details of the HMMs are provided in Table 1.
Table 1.
Designations and details of hidden Markov models referred to in this study
| Model
|
MDS-HMM
|
CSQ/MDS-HMM
|
||||
|---|---|---|---|---|---|---|
| #k | #r | 92% cut | 88% cut | 92% cut | 88% cut | |
| 4 | × | 0 | 1.8 | 0.0 | 9.1 | 9.6 |
| 4 | × | 1 | 37.3 | 30.0 | 12.7 | 6.4 |
| 4 | × | 2* | 35.5 | 32.7 | 12.7 | 6.4 |
| 4 | × | 3 | 60.0 | 60.0 | 14.5 | 9.1 |
| 4 | × | 4 | 66.4 | 66.4 | 16.4 | 13.6 |
| 4 | × | 5 | 74.5 | 61.8 | 21.8 | 20.9 |
| 4 | × | 6 | 70.0 | 68.2 | 17.3 | 10.0 |
| 4 | × | 7 | 75.5 | 70.9 | 17.3 | 10.0 |
| 4 | × | 8 | 79.1 | 76.4 | 16.4 | 13.6 |
| 4 | × | 9 | 80.9 | 78.2 | 18.2 | 18.2 |
| 4 | × | 10 | 80.9 | 77.3 | 22.7 | 22.7 |
| D | N | C | * | * | 2.7 | 2.7 |
Model selected for further study.
The results are presented in Figs. 4–6. For clarity only 20 of the 110 nonbinding sites considered are included, but these are well representative of the entire set. Two thresholds, which scored correctly 92% and 88% of the known binding sequences, respectively, were considered. The scoring based on the MDS-HMM 4 × 2 is shown in Fig. 4. This model, based on four divisions of roll/tilt orientations and three divisions of magnitude (small, medium, and large), scored the test set with 35.5% and 32.7% error under the respective thresholds.
Figure 4.

Log odds scoring of the MDS-HMM with four categories of direction and three categories of magnitude on the 25 CAP binding and 110 nonbinding sites. Of the 25 CAP binding sites, 16 are used for training. For the clarity of the figure, only results from 20 nonbinding sites are explicitly included, but all are well representative of the full complement.
Figure 6.

Log odds scoring of the SEQ-HMM on the CAP binding test set.
We proceeded to improve the HMM by restricting to sequence consensus only at positions 10–15 as described above, producing a CSQ/MDS model. With this information added, the scoring error was reduced to 12.7% and 6.4% for the 92% and 88% thresholds, respectively (Fig. 5). Thus, as expected, restricting the region where specific contacts are required between the CAP protein and its DNA binding site to a probability model of sequence information improved the discriminatory abilities of the HMM.
Figure 5.

Log odds scoring of the CSQ/MDS-HMM with four categories of direction and three categories of magnitude in the MDS region on the CAP binding test set. Sites 11–15 are restricted to an HMM model of sequence only.
The level of resolution to be applied to the magnitude of deviations in the DNA structures was explored by sensitivity analysis. The CSQ/MDS models with small and large (4 × 1) and with small, medium, and large magnitude (4 × 2) performed with 6.4% error at the 88% threshold. At a resolution level of 4%, the percentage points attributed to a single known binding site, exploratory models with four, seven, and eight magnitude components show comparable discriminatory ability in scoring protein binding and nonbinding sites in the test set as well. The digitization of magnitude into three divisions seems the most reasonable compromise at this point.
Results on scoring the test set based on sequence only (SEQ-HMM) were obtained as a control. It scored known binding sites in the training set significantly better than those in the test set (Fig. 6), clear indication of overtraining. The corresponding MDS-HMMs score binding sites in the training and in the test set at a similar level (for example, Fig. 5), presumably as a consequence of a broader definition of consensus (sequence plus structure). To elucidate this point, a purposely overtrained model problem was constructed. Seven of the original sixteen sequences were arbitrarily chosen and used to train a SEQ-HMM. Applied to the test set, the resulting HMMs exhibited errors of 35% and 30% for 92% and 88% of the binding sites found, respectively. This SEQ-HMM was transformed using the MD results into an MDS-HMM and tested (Fig. 7). The scoring improved markedly, to 8% and 4% false positives at the two levels, indicating that the MD transformation results in substantive difference in the ability of an HMM to successfully locate binding sites.
Figure 7.

Log odds scoring results on (A) a purposely overtrained SEQ-HMM compared to (B) a corresponding MDS-HMM. See text for details.
Discussion
The results of the preceding section indicate that HMMs based on probabilistic roll/tilt dinucleotide models of sequence-dependent DNA structure have a capability to discriminate between known CAP binding and nonbinding sites and to predict putative CAP binding sites in unknowns. Restricting HMMs to sequence in regions of high consensus in which the protein makes base-specific contacts further improved the discriminatory capabilities of the HMMs. The incorporation of dynamical structure in HMMs and thereby introducing a broader definition of consensus was shown to improve the transferability of the HMMs for a case in which sequence-only HMMs were overtrained. In the following, we discuss the approximations and sources of uncertainty in the method as implemented, and also further implications and assessment of the results.
The key operational quantities in MDS-HMM methodology are the transformed emission probabilities E′ and E". The transformations per se, Eqs. 5 and 7, involve a product of probabilities, and implicit in this step is that sequence and dynamical structure are independent events. After the first transformation process is complete (Eq. 6), the alphabet corresponding to the emission probabilities is that of dynamical structure (k, l) for each base pair in the site. This is unwieldy for scoring per se, and resolution of this requires the second transformation step, Eqs. 7 and 8. The summations by which the transformed emission probabilities are reduced, Eqs. 6 and 8, are averaging procedures—i.e., effectively integrations over aspects of structure. MDS-HMMs thus seek consensus in a way that combines the known sequence binding characteristics with intrinsic dynamical structure characteristics of the uncomplexed DNA in whatever way this might contribute to binding.
Our training set in this study is of course relatively small, but we feel the results obtained are such that at least provisional conclusions are justified. The value added from incorporating dynamical structure is evident most clearly in the improved transferability of the transformed models, which we attribute to the expanded definition of consensus to include dynamical structure as well as sequence. Because any training set of finite size engenders some degree of overtraining, it is especially encouraging to find from our results on the model problem described in the previous section that incorporating dynamical structure appears to provide some compensation. The transformation procedure thus results in an improved HMM for identification of any DNA element characterized by its dynamic properties.
One advantage of this approach is that the need for any explicit consideration of phenomena such as intrinsic curvature, bending, flexibility, bendability, or induced fit is avoided. Each of these “modelistic” terms is not well defined in an operational sense (D.L.B., unpublished observation), and as a consequence there is some confusion in the literature about what each of the terms really signifies at the level of dynamical structure. For example, the trinucleotide DNase and nucleosome positioning scales used to derive DNA bendability do not correlate well with each other (D.L.B., unpublished data). In this project, sequence-dependent dynamical structure is simply defined in probability form with respect to B-form DNA reference state, based not on phenomenological definitions but on the calculated dynamical structure and Boltzmann statistical mechanics of the system.
As for a higher-order demonstration case, the E. coli genome was obtained from PubMed (http://www.ncbi.nlm.nih.gov/PubMed/) and converted from base pairs to base pair steps. The converted genome (4.6 Mb) was scanned with the various HMMs on a sliding window of 36 steps moved in single-base-pair increments sequentially through the genome. Each fragment was scored and compared based on the previously established scoring scale. The results are shown in Fig. 8. The results identify a plausible number of putative binding sites. However, one seeks sequences that not only bind protein, but also have a regulatory function. False regulatory sites could arise by chance or be remnants of evolutionarily extinct genes. To be useful, an expanded protocol should include information about how far the regulatory element is located in sequence space with respect to the beginning of the ORF. In some cases, genes are already annotated, but ORF-predicting programs are also currently available (52), some of which are tailored to specific organisms. System-specific searching could be achieved by the use of additional MDS-HMMs with complex architectures allowing for the different positions of the binding site with respect to the transcriptional initiation site, and in conjunction with in vivo expression data in the form of microarray analysis, could provide a method for predicting groups of genes that are regulated by the same transcription factor.
Figure 8.

CSQ/MDS-HMM log odds scorings of the E. coli genome.
Summary and Conclusions
Boltzmann probability models of DNA sequence-dependent structure from MD simulations on a set of oligonucleotides have been incorporated into HMMs, resulting in a bioinformatics tool that can recognize molecular structural signals as well as sequence in protein DNA binding sites on a genome. The binding of CAP to cognate DNA sequences served as a well characterized model system for demonstrating and testing of the method, and HMMs based on MD were used in an analysis of the E. coli genome. The results indicate that HMMs based on probabilistic roll/tilt dinucleotide models of sequence-dependent DNA structure have a capability to discriminate between known CAP binding and nonbinding sites and to predict putative CAP binding sites in unknowns. Restricting HMMs to sequence only in regions of high consensus in which the protein makes base specific contacts further improved the discriminatory capabilities of the HMMs. The incorporation of dynamical structure in HMMs and thereby the introduction of a broader definition of consensus was shown to improve the transferability of the HMMs. Collectively, these results provide supportive evidence of a role for molecular geometry as well as sequence in regulatory mechanisms. The method described is readily extended to definitions of sequence-dependent DNA structure involving additional helicoidal parameters, and to include sequence context effects with trinucleotide or higher-order models. In conclusion, we note that however encouraging one finds the results of this study, this is not an unequivocal proof of concept because agreement or plausible behavior compared with experiment does not prove a model per se. The proposed methodology has considerable potential applications in bioinformatics beyond those described herein, but it is not yet possible to say how general these results are. In particular, the issues we have raised about overtraining (i.e., developing a proper training set with all potentially interesting characteristics included) and robustness of the method will require subsequent detailed and critical study.
Supplementary Material
Acknowledgments
Discussions with Dr. Richard Lavery are gratefully acknowledged. This research was supported by National Institutes of Health Grant GM 37909 (to D.L.B.). K.M.T. is the recipient of a National Institutes of Health Traineeship in Molecular Biophysics from National Institutes of Health Grant 08271.
Abbreviations
- CAP
catabolite activator protein
- MD
molecular dynamics
- HMM
hidden Markov model
- MDS
molecular dynamical structure
- CSQ/MDS
consensus sequence–dynamical structure hybrid
- SEQ-HMM
sequence-only-based HMM
- MDS-HMM
MD and sequence-based HMM
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Wing R M, Drew H R, Takano T, Broka C, Tanaka S, Itakura I, Dickerson R E. Nature (London) 1980;287:755–758. doi: 10.1038/287755a0. [DOI] [PubMed] [Google Scholar]
- 2.Dickerson R E. Sci Am. 1983;249:94–111. [Google Scholar]
- 3.Beveridge D L, McConnell K J. Curr Opin Struct Biol. 2000;10:182–196. doi: 10.1016/s0959-440x(00)00076-2. [DOI] [PubMed] [Google Scholar]
- 4.Cheatham T E, III, Young M A. Biopolymers. 2001;56:232–256. doi: 10.1002/1097-0282(2000)56:4<232::AID-BIP10037>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 5.Rabiner L R. Proc IEEE. 1989;77:257–286. [Google Scholar]
- 6.Durbin R, Eddy S, Krogh A, Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, U.K.: Cambridge Univ. Press; 1998. [Google Scholar]
- 7.Baldi P, Chauvin Y, Hunkapiller T, McClure M A. Proc Natl Acad Sci USA. 1994;91:1059–1063. doi: 10.1073/pnas.91.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldi P, Brunak S. Bioinformatics: The Machine Learning Approach. Cambridge, MA: MIT Press; 1998. [Google Scholar]
- 9.Eddy S R. Curr Opin Struct Biol. 1996;6:361–365. doi: 10.1016/s0959-440x(96)80056-x. [DOI] [PubMed] [Google Scholar]
- 10.Krogh A. In: Computational Methods in Molecular Biology. Salzberg S L, Searls D B, Kasif F, editors. New York: Elsevier; 1998. pp. 45–63. [Google Scholar]
- 11.Berg O G, von Hippel P H. J Mol Biol. 1988;200:709–723. doi: 10.1016/0022-2836(88)90482-2. [DOI] [PubMed] [Google Scholar]
- 12.Ebright R H, Ebright Y W, Gunasekera A. Nucleic Acids Res. 1989;17:10295–10305. doi: 10.1093/nar/17.24.10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gunasekera A, Ebright Y W, Ebright R H. J Biol Chem. 1992;267:14713–14720. [PubMed] [Google Scholar]
- 14.Pedersen A G, Baldi P, Brunak S, Chauvin Y. Proc Int Conf Intell Syst Mol Biol. 1996;4:182–191. [PubMed] [Google Scholar]
- 15.Margalit H, Shapiro B A, Nussinov R, Owens J, Jernigan R L. Biochemistry. 1988;27:5179–5188. doi: 10.1021/bi00414a035. [DOI] [PubMed] [Google Scholar]
- 16.Bolshoy A, McNamara P, Harrington R E, Trifonov E N. Proc Natl Acad Sci USA. 1991;88:2312–2316. doi: 10.1073/pnas.88.6.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ozoline O N, Deev A A, Trifonov E N. J Biomol Struct Dyn. 1999;16:825–831. doi: 10.1080/07391102.1999.10508295. [DOI] [PubMed] [Google Scholar]
- 18.Brukner I, Sanchez R, Suck D, Pongor S. EMBO J. 1995;14:1812–1818. doi: 10.1002/j.1460-2075.1995.tb07169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baldi P, Brunak S, Chauvin Y, Krogh A. J Mol Biol. 1996;263:503–510. doi: 10.1006/jmbi.1996.0592. [DOI] [PubMed] [Google Scholar]
- 20.Brukner I, Sanchez R, Suck D, Pongor S. J Biomol Struct Dyn. 1995;13:309–317. doi: 10.1080/07391102.1995.10508842. [DOI] [PubMed] [Google Scholar]
- 21.Perez-Martin J, de Lorenzo V. Annu Rev Microbiol. 1997;51:593–628. doi: 10.1146/annurev.micro.51.1.593. [DOI] [PubMed] [Google Scholar]
- 22.Lafontaine I, Lavery R. Comb Chem High Throughput Screening. 2001;4:707–717. doi: 10.2174/1386207013330698. [DOI] [PubMed] [Google Scholar]
- 23.Lafontaine I, Lavery R. Biophys J. 2000;79:680–685. doi: 10.1016/S0006-3495(00)76326-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lafontaine I, Lavery R. Biopolymers. 2000;56:292–310. doi: 10.1002/1097-0282(2000)56:4<292::AID-BIP10028>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 25.Pedersen A G, Jensen L J, Brunak S, Staerfeldt H H, Ussery D W. J Mol Biol. 2000;299:907–930. doi: 10.1006/jmbi.2000.3787. [DOI] [PubMed] [Google Scholar]
- 26.Haile J M. Molecular Dynamics Simulation: Elementary Methods. New York: Wiley; 1992. [Google Scholar]
- 27.McCammon J A, Harvey S C. Dynamics of Proteins and Nucleic Acids. Cambridge, U.K.: Cambridge Univ. Press; 1987. [Google Scholar]
- 28.Brooks C L, III, Karplus M, Pettitt B M. Proteins: A Theoretical Perspective of Dynamics, Structure and Thermodynamics. New York: Wiley; 1988. [Google Scholar]
- 29.van Gunsteren W F, Berendsen H J C. Angew Chem Int Ed Engl. 1990;29:992–1023. [Google Scholar]
- 30.Beveridge D L, Swaminathan S, Ravishanker G, Withka J M, Srinivasan J, Prevost C, Louise-May S, Langley D R, DiCapua F M, Bolton P H. In: Water and Biological Molecules. Westhof E, editor. London: Macmillan Press; 1993. pp. 165–225. [Google Scholar]
- 31.Young M A, Ravishanker G, Beveridge D L. Biophys J. 1997;73:2313–2336. doi: 10.1016/S0006-3495(97)78263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McConnell K J, Beveridge D L. J Mol Biol. 2000;304:803–820. doi: 10.1006/jmbi.2000.4167. [DOI] [PubMed] [Google Scholar]
- 33.Cheatham T E, III, Kollman P A. J Mol Biol. 1996;259:434–444. doi: 10.1006/jmbi.1996.0330. [DOI] [PubMed] [Google Scholar]
- 34.Sprous D, Young M A, Beveridge D L. J Phys Chem. 1998;102:4658–4667. [Google Scholar]
- 35.Young M A, Beveridge D L. J Mol Biol. 1998;281:675–687. doi: 10.1006/jmbi.1998.1962. [DOI] [PubMed] [Google Scholar]
- 36.Sprous D, Young M A, Beveridge D L. J Mol Biol. 1999;285:1623–1632. doi: 10.1006/jmbi.1998.2241. [DOI] [PubMed] [Google Scholar]
- 37.McConnell K J, Beveridge D L. J Mol Biol. 2001;314:23–40. doi: 10.1006/jmbi.2001.4926. [DOI] [PubMed] [Google Scholar]
- 38.Eddy S R. Proc Int Conf Intell Syst Mol Biol. 1995;3:114–120. [PubMed] [Google Scholar]
- 39.Krogh A, Mian I S, Haussler D. Nucleic Acids Res. 1994;22:4768–4778. doi: 10.1093/nar/22.22.4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weber I T, Steitz T A. J Mol Biol. 1987;198:311–326. doi: 10.1016/0022-2836(87)90315-9. [DOI] [PubMed] [Google Scholar]
- 41.Schultz S C, Shields G C, Steitz T A. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- 42.Chen S, Gunasekera A, Zhang X, Kunkel T A, Ebright R H, Berman H M. J Mol Biol. 2001;314:75–82. doi: 10.1006/jmbi.2001.5090. [DOI] [PubMed] [Google Scholar]
- 43.Chen S, Vojtechovsky J, Parkinson G N, Ebright R H, Berman H M. J Mol Biol. 2001;314:63–74. doi: 10.1006/jmbi.2001.5089. [DOI] [PubMed] [Google Scholar]
- 44.Gaston K, Kolb A, Busby S. Biochem J. 1989;261:649–653. doi: 10.1042/bj2610649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young M A, Ravishanker G, Beveridge D L, Berman H M. Biophys J. 1995;68:2454–2468. doi: 10.1016/S0006-3495(95)80427-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Beveridge D L. J Biomol Struct Dyn. 2001;18:505–526. doi: 10.1080/07391102.2001.10506684. [DOI] [PubMed] [Google Scholar]
- 47.Arnott S, Campbell-Smith P J, Chandrasekaran R. In: CRC Handbook of Biochemistry and Molecular Biology. Fasman G, editor. Vol. 2. Cleveland: CRC; 1976. pp. 411–422. [Google Scholar]
- 48.Barber A M, Zhurkin V B. J Biomol Struct Dyn. 1990;8:213–232. doi: 10.1080/07391102.1990.10507803. [DOI] [PubMed] [Google Scholar]
- 49.Goodsell D S, Dickerson R E. Nucleic Acids Res. 1994;22:5497–5503. doi: 10.1093/nar/22.24.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Crombrugghe B, Busby S, Buc H. Science. 1984;224:831–838. doi: 10.1126/science.6372090. [DOI] [PubMed] [Google Scholar]
- 51.Ebright R H, Cossart P, Gicquel-Sanzey B, Beckwith J. Nature (London) 1984;311:232–235. doi: 10.1038/311232a0. [DOI] [PubMed] [Google Scholar]
- 52.Mount D W. Bioinformatics: Sequence and Genome Analysis. Plainview, NY: Cold Spring Harbor Lab. Press; 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.