

Abstract
Amyloid is the result of an anomalous protein and peptide aggregation, leading to the formation of insoluble fibril deposits. At present, 18 human diseases have been associated with amyloid deposits—e.g., Alzheimer's disease and Prion-transmissible Spongiform Encephalopathies. The molecular structure of amyloid is to a large extent unknown, because of lack of high-resolution structural information within the amyloid state. However, from other experimental data it has been established that amyloid fibrils predominantly consist of β-strands arranged perpendicular to the fibril axis. Identification of residues involved in these secondary structural elements is therefore of vital importance to rationally designing appropriate inhibitors. We have designed a hydrogen/deuterium exchange NMR experiment that can be applied on mature amyloid to enable identification of the residues located inside the fibril core. Using a highly amyloidogenic peptide, corresponding to residues 25–35 within the Alzheimer Aβ(1–43) peptide, we could establish that residues 28–35 constitute the amyloid core, with residues 31 and 32 being the most protected. In addition, quantitative values for the solvent accessibility for each involved residue could be obtained. Based on our data, two models of peptide assembly are proposed. The method provides a general way to identify the core of amyloid structures and thereby pinpoint areas suitable for design of inhibitors.
Amyloid diseases have in common an abnormal folding of normally soluble proteins resulting in the formation of extracellular amyloid deposits (1). Examples are Alzheimer's Disease, Prion-transmissible Spongiform Encephalopathies, and Familial Amyloidotic Polyneuropathy (2). Through self-assembly these proteins produce regular fibrillar structures possessing a predominant β-sheet conformation (3). Structural information on the core of fibrils has mainly been elucidated from fiber-diffraction studies (4), mass-spectrometry (5), and solid-state NMR spectroscopy (6), resulting in a general picture consistent with parallel or antiparallel β-strands, placed perpendicularly to the fibril axis. However, a detailed structure of a fibril at the atomic level is still lacking. In this respect, more structural information—e.g., which specific amino acids make up the amyloid-forming core—becomes crucially important.
In this article, we describe a NMR approach to determining the sequence-specific structural elements in the fibril stage of amyloid forming proteins. The method relies on the partial solvent protection of hydrogen-bonded amide protons connecting the β-strands throughout the length of the fibril. In aqueous solutions it is expected that amide protons located on the exterior of the fibril are more accessible to solvent and therefore experience a higher hydrogen exchange rate than amide protons buried within the fibril interior. Studies using mass spectrometry, in combination with deuterium exchange, have already pointed to this possibility (7). However, mass spectrometry does not allow specific assignment of the particular amino acids involved in solvent protection.
To obtain more sequence-specific information, the technique of a pulsed hydrogen/deuterium (H/D) exchange NMR experiment (8–10) was modified. Amyloid fibrils were incubated in excess of D2O during a given time. The H/D pattern, as governed by the amyloid, could then be indirectly measured using high-resolution proton NMR through rapid conversion of the fibrils into a monomeric and detectable state. By plotting the integrated peak intensity of amide protons for each residue over time, the post-trap exchange pattern could be fitted to a curve, and extrapolation to time 0 of solubilization yielded the peak intensity of amide protons present in the amyloid fibril. Internal calibration against nonexchangeable methyl groups was made and gives the relative amide-hydrogen solvent-protection value.
The proposed method provides a general and quantitative tool for structural mapping of amyloid fibrils. We have used this approach to study amyloid fibrils of the highly amyloidogenic Alzheimer β-peptide (Aβ25–35; ref. 11), comprising amino acids 25–35 of Aβ. The full-length Alzheimer Aβ (1–43) is responsible for Alzheimer's disease and associated dementia. Because of its similar amyloid-forming properties, the Aβ25–35 peptide is frequently used as a convenient model system for studies of Alzheimer amyloid formation and its effects on cellular systems (12). The structural consequences of the findings and its generality are discussed.
Materials and Methods
Sample Preparation Amyloid Fibrils from Aβ25–35.
Thirty microliters of a concentrated (8 mg/ml) solution of the Aβ25–35 peptide (Sigma–Aldrich) was incubated for 5 days at 21°C in 10 mM sodium-acetate, pH 5.4. After 5 days the initially clear solution had changed to an opaque, highly viscous gel, a characteristic feature for the formation of amyloid fibrils.
Atomic Force Microscopy (AFM).
The aggregated peptide solution of Aβ25–35 was diluted to 0.05 mg/ml with distilled water and applied onto freshly cleaved ruby red mica (Goodfellow, Cambridge, U.K.). Samples were allowed to adsorb for 30 s, washed three times with distilled water, and air-dried. The bound material was imaged with a Nanoscope IIIa multimode AFM (Digital Instruments, Santa Barbara, CA), using Tapping Mode in air. A silicon probe was oscillated at 300–325 kHz, and images were collected at an optimized scan rate corresponding to 0.7–1 Hz. All images were flattened and presented in height mode by using nanoscope software (Digital Instruments).
NMR Spectroscopy.
Proton assignment and structural characterization of Aβ25–35.
To assign the eleven amide protons of the peptide Aβ25–35 nuclear Overhauser effect spectroscopy (NOESY) spectra were recorded in both deuterated trifluoroethanol (TFE-d3)/60% H2O (vol/vol) (pH 3) and deuterated DMSO (DMSO-d6) by using a Bruker DRX-600 MHz spectrometer. Typically 48–80 scans over 420–512 increments were collected, and in the case of TFE-d3/H2O the water resonance was selectively suppressed by applying a watergate pulse sequence (13) placed at the position of the last detection pulse. The NOESY mixing times used were 500 ms and 600 ms, respectively, for the TFE-d3/H2O and DMSO-d6 sample. D2O (99.9%), DMSO-d6 (99.96%), and TFE-d3 (>99%) were obtained from Cambridge Isotope Labs (Andover, MA). Fig. 1 shows part of the 600 MHz 1H-1H NOESY, recorded at 17°C in 40% TFE-d3/60% H2O, containing the connections between amide NH-amide NH and between amide NH-nonexchangeable side chain protons. Sequential assignment through NH(n)–NH(n+1) and NH(n+1)–Hα(n) NOEs was straightforward also in DMSO-d6, and proton chemical shifts of the peptide in the two solvents used are collected in Table 1.
Figure 1.

Part of the two-dimensional 1H-1H NOESY spectrum of Aβ25–35 in 40% TFE-d3/60% H2O, containing the NH–NH and NH–Hα cross peaks. The arrows indicate the sequential backbone assignment via intramolecular NH–Hα NOE connections. The horizontal line corresponds to the position of the combined residual solvent of H2O and the hydroxyl proton signal of TFE. Cross peaks located on this line indicate a high solvent exchange rate for the connected amide proton.
Table 1.
Nonstereospecific 1H chemical shift assignments (ppm) for Aβ25–35 in the two different solvents used
| Residue | HN | Hα | Hβ | Hψ | Hδ | Hɛ | NH2 |
|---|---|---|---|---|---|---|---|
| Assignment of Aβ25–35 in 40% TFE-d3/60% H2O (600 MHz, T = 289.9 K) | |||||||
| G1 | 3.886 | ||||||
| S2 | 8.642 | 4.526 | 3.901; 3.852 | ||||
| N3 | 8.556 | 4.757 | 2.843 | 7.559; 6.832 | |||
| K4 | 8.366 | 4.251 | 1.882; 1.773 | 1.481; 1.432 | 1.685 | 2.983 | 7.594 |
| G5 | 8.318 | 3.898 | |||||
| A6 | 7.873 | 4.299 | 1.396 | ||||
| I7 | 7.801 | 4.11 | 1.892 | 1.508; 1.188; 0.893 | 0.865 | ||
| I8 | 7.768 | 4.107 | 1.879 | 1.502; 1.194; 0.911 | 0.867 | ||
| G9 | 8.09 | 3.968; 3.876 | |||||
| L10 | 7.752 | 4.411 | 1.627 | 1.349 | 0.912; 0.869 | ||
| M11 | 7.822 | 4.416 | 2.155; 2.018 | 2.580; 2.515 | |||
| Assignment of Aβ25–35 in DMSO-d6 (600 MHz, T = 304.2 K) | |||||||
| G1 | 7.965 | 3.608 | |||||
| S2 | 8.572 | 4.483 | 3.57 | 5.19 | |||
| N3 | 8.419 | 4.576 | 2.588; 2.479 | 7.463; 7.031 | |||
| K4 | 8.017 | 4.176 | 1.704 | 1.316 | 1.516 | 2.762 | 7.632 |
| G5 | 8.177 | 3.703 | |||||
| A6 | 7.928 | 4.345 | 1.189 | ||||
| I7 | 7.96 | 4.162 | 1.733 | 1.433; 1.055; 0.82 | 0.8 | ||
| I8 | 7.759 | 4.174 | 1.715 | 1.438; 1.094; 0.82 | 0.81 | ||
| G9 | 8.147 | 3.737 | |||||
| L10 | 7.9 | 4.354 | 1.473; 1.408 | 1.606 | 0.888; 0.846 | ||
| M11 | 8.217 | 4.264 | 1.960; 1.865 | 2.507; 2.447 | |||
Chemical shifts referenced to external DSS, 0 ppm. Chemical shifts referenced to DMSO-d6 2.500 ppm.
Deuterium exchange measurements in TFE-d3/D2O.
After rigorous vortexing, the aggregated peptide solution was allowed to exchange with D2O for 31 min at 21°C, pH 5.4. The solution was subsequently quenched in a mixture of 100 μl D2O and 200 μl TFE-d3. To minimize post-trap solvent exchange the final pH was preset to 3 by using DCl (14). The first proton spectrum was accumulated 2.5 min after the initial addition of the low pH quench-solution. One-dimensional 1H spectra (160 FID's of each 256 scans) were continuously collected over a period of 20 h and stored on the computer as a semi2D serial file. The same procedure was repeated using a longer 135 min interval for deuterium exchange. Exchange measurements in TFE-d3/D2O were performed at 17°C. Deuterium abundances of amide protons were calculated on a molar ratio.
Deuterium exchange experiment in DMSO-d6.
A similar exchange experiment was performed where Aβ25–35 amyloid was preincubated in a final mixture of 20% (vol/vol) H2O/80% D2O, using an 18 h exchange interval at 21°C. Here the sample was instead quenched in liquid N2, freeze-dried for >24 h at 10−5 bar, and suspended in dry DMSO-d6. The DMSO-d6 requires no solvent suppression and the first sequential one-dimensional spectrum was recorded after 2.5 min following dissolution, yielding a usable integrated signal after 7 min in the data collection. Integration of the water resonance in the proton spectra showed a content of 80 mM residual water in the final peptide/DMSO-d6 sample. This leads to a gradual substitution of amide deuterons in the Aβ25–35 peptides to protons during the post-trap exchange time. The temperature was set to 31°C.
Data Analysis and Fitting.
Exchange data were fitted using CURVEEXPERT 1.37 software (D. Hyams, Starkville, MS). Appropriate equations were incorporated into the program and curves were fitted using weighted Y values (weighing function 1/Y).
Results
The applicability of the H/D exchange NMR method to determine the specific amino acid residues comprising the core of mature amyloid fibrils has been investigated by two approaches. The first relies on rapid solubilization of incubated fibrils through addition of a TFE-d3/D2O mixture. This mixed solvent contained free deuterons and the post-trap exchange was therefore minimized by measuring at a pH value around 3 (8). In the second approach, the bulk part of protons and deuterons were removed through lyophilization after the initial exchange period. The freeze-dried fibrils were then dissolved in DMSO-d6, an aprotic solvent capable of dissociating amyloid fibrils.
AFM Studies.
The Aβ25–35 peptide rapidly forms a highly viscous gel, containing massive amounts of amyloid fibrils, when kept in aqueous solution at pH 5.4. Presence of fibrils was verified using AFM, as shown in Fig. 2, and the image demonstrates fibrils, which are straight and display indefinite length. Fibril heights were measured to be around 3 nm and the lateral arrangement of protofilament substructures can be discerned. From the obtained picture we estimate the amount of fibrillar structures to be around 90%.
Figure 2.

AFM image showing Aβ25–35 amyloid fibrils. The peptide was incubated at pH 5.4 for 5 days. The average fibril length was measured to >1 μm and the height corresponded to ≈3 nm.
Proton Assignment in TFE-d3/H2O and DMSO-d6.
Assignment of most Aβ25–35 protons in both TFE-d3 and DMSO-d6 solutions could be made by NOESY spectra, in combination with standard total correlation spectroscopy (TOCSY) spectra. Proton chemical shifts of the peptide are displayed in Table 1. Although TFE is known to induce the formation of α-helices in peptides, we did not see clear evidence for that in 40% TFE-d3. Although the presence of sequential HN to HN NOEs would suggest some helix formation—e.g., presence of helical turns—the lack of long-range backbone NOE patterns indicates overall a rather unstructured, random coil-like state. A similar observation was made for Aβ25–35 in DMSO-d6 solution, with no apparent detectable secondary structure based on NOESY spectra. In addition, the chemical shift in DMSO-d6 versus temperature profiles displayed almost uniform straight slopes (−4 parts per million per degree) over the temperature range of 20–96°C degrees, indicating that there are no internal hydrogen bonds present to stabilize the structure in DMSO-d6 (data not shown).
H/D Exchange NMR Analysis in the Fibril Stage by Using TFE-d3/D2O.
In aqueous solution we found that 40% (vol/vol) TFE-d3/60% water mixtures were the best choice to rapidly dissolve the amyloid fibrils and at the same time obtain good resolved NMR spectra of the soluble monomeric form. Growth of monomeric population in solution as a function of post-trap exchange time (tsolv) is monitored by the integral intensity of methyl resonances (Imethyl) and showed a monotonic increase with time (Fig. 3). The time curve of Imethyl can minimally be described by a bi-exponential function with two rate constants, 0.0172 and 0.1244 min−1 (Fig. 3). However, the conversion from large aggregates to monomers must be kinetically more complex. The integral intensity of amide protons (INH) is affected by the increase in monomer population and decreases by amide proton exchange with D2O. To internally correct for the unknown time-dependence of slow solubilization, the INH values at each time point were therefore normalized against the Imethyl at that time point (the average integral intensity of Ile-7, Ile-8, and Leu-9 was used) giving INHnorm (= INH/Imethyl). As shown Fig. 4, the INHnorm curves for Asn-3 to Met-11 now show the expected monotonic decrease with time (except at very small tsolv). The amino proton of Gly-1 and amide proton of Ser-2 were exchanged to deuterium within the dead time of the experiment (2.5–7 min).
Figure 3.

Measured integrals corresponding to the sum of methyl groups of Leu-10, Ile-7, and Ile-8, together with integrals of NH Ile-8, and NH2 Asn-3, plotted as a function of time, after dissolving amyloid aggregates formed by Aβ25–35 into a solution of 40% TFE-d3/60% D2O. The integrals are influenced by the combination of recovery of aggregates into NMR detectable monomers (leading to an increased signal) and H/D exchange of exchangeable protons (resulting in a decreased signal).
Figure 4.

Graphical overview of the H/D exchange (TFE-d3/H2O) experiments performed on the Aβ25–35 fibrils sample, after incubation for 31 min in D2O. Points indicate the experimentally observed proton integral of amide protons in the monomeric state, directly after solubilization of the amyloid in 40% TFE-d3/60% D2O. Integrals are internally corrected for the slow conversion of aggregates into monomers. The black fitted lines indicate the single exponential fit of the proton integrals according to Eq. 1, giving the amide proton solvent exchange rates of the monomer, displayed in Table 2. In the case that time points (between the 0–30-min intervals) corresponded an integral value lower than the following point in time, data points were omitted from the curve fitting. The broken line represents the best fit according to a fifth polynomial function, to better account for the nonlinear interpolation of Io at short exchange times.
To obtain a value of the protection level at the end of the preincubation period of the fibrils in D2O the INHnorm values were extrapolated back to tsolv = 0, by fitting as a first order approximation a single exponential function to them:
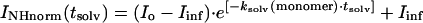 |
1 |
The fitted variables are the post-trap H/D exchange rate of amide protons in the monomeric state, ksolv(monomer), and the extrapolated proton abundance at time 0, Io, representing the percentage of protonated amide protons that survived the deuterium exchange period with D2O in the fibril state. Io will from hereon be referred to as the protection factor. By definition, Io is 100% in absence of any H/D exchange in the fibril state. It has a limiting minimum value of 15%, under conditions of complete H/D exchange in the fibril state (corresponding to 30 μl of H2O relative to 170 μl of added D2O). Iinf is the residual percentage of amide-protons in the final solution after infinite H/D post-trap exchange times. Iinf is a fixed parameter and can be calculated based on the known partial volumes of D2O/H2O and TFE-d3 added to the peptide solution. In this case, Iinf, is 9% (corresponding to 30 μl of H2O relative to deuterons originating from addition of 200 μl of TFE-d3 and 270 μl of D2O). The single exponential fits must be regarded as estimated upper limits of Io. Extrapolation using fifth-order polynomial equations provides a better approximation for the central residues, as shown in Fig. 4, and fitted values of Io with this method were used whenever exponential fits were not applicable. It is important to note that Io can now be sampled for each individual amino acid and studied as a function of the exchange time in the fibril stage.
Fig. 5A and Table 2 show the final protection factors of amide protons (Io) of Aβ25–35 amyloid fibrils in TFE-d3/D2O after 31 and 135 min of deuterium exchange, respectively. Values range between 10 and 97% and the final distribution displays a bell-shaped curve, more or less symmetrically positioned between residues Lys-4 to Met-11, and with maximum protection factors found at residues Ile-7 and Ile-8.
Figure 5.

(A) H/D exchange protection factors of individual amide protons within the Aβ25–35 amyloid fibrils, as determined by the TFE-d3/D2O resolubilization approach. Bars indicate the percentage of nonexchanged amide protons in the fibril state. Dark gray, after 31 min of incubation in D2O for 31 min; light gray, after incubation in D2O for 135 min. Quantitative H/D exchange protection factors are given in Table 2. (B) Series of one-dimensional proton spectra for the Aβ25–35 fibrils following redissolution in DMSO-d6. Fibrils where incubated for 18 h in D2O and assignments are taken from NOESY spectra. Data concerning residue 6–10 could be fitted to Eq. 2. Because of partial overlap residues 5 and 11 had to be excluded from the fit. Quantitative protection factors are given in Table 2. The asterisks indicate peaks of a minor conformational species generated by dissolving the peptide amyloid in DMSO-d6.
Table 2.
Post-trap solvent exchange rates, ksolv (in min−1), fitted for various amide protons of the monomeric form of the Aβ25–35 peptide, and shown for the two solvents used; and calculated protection factors in percent for Aβ25–35 amide protons under amyloid conditions as a function of deuterium incubation time
| Residue | 40% TFE-d3/ 60% H2O | DMSO-d6 | Residue protection factors, %
|
ksolv(amyloid) (10−3 min−1) | ||
|---|---|---|---|---|---|---|
| 31 min† | 135 min† | 18 hours‡ | ||||
| Gly-1 | >0.2 | >0.2 | n.d. | n.d. | n.d. | — |
| Ser-2 | >0.2 | >0.2 | n.d. | n.d. | n.d. | — |
| Asn-3 | 0.0285 | >0.2 | 10 | n.d. | n.d. | — |
| Lys-4 | 0.0185 | >0.2 | 46 | 23 | n.d. | — |
| Gly-5 | 0.0122 | * | 67 | 35 | § | — |
| Ala-6 | 0.0118 | 0.0158 | 80 | 59 | 58 | 0.5–7.2 |
| Ile-7 | 0.0061 | 0.0458 | 93 | 81 | 56 | 0.55–2.2 |
| Ile-8 | 0.0058 | 0.0204 | 97 | 79 | 47 | 0.7–1.7 |
| Gly-9 | 0.0091 | 0.0370 | 82 | 62 | 47 | 0.7–6.4 |
| Leu-10 | 0.0109 | 0.0067 | 74 | 49 | 49 | 0.65–10.0 |
| Met-11 | 0.0144 | * | 54 | 44 | § | — |
Values are corrected for isotope dilution effects. The last column gives the upper and lower limits of the solvent exchange rates for amide protons in the amyloid state ksolv(amyloid), based on the data in the three previous columns. n.d., Not determined because post-trap solvent exchange reached detection limit before first measured time point.
Partial overlap for Gly-5 and Met-11.
Data obtained from TFE-d3/D2O H/D exchange experiment.
Data obtained from the DMSO-d6 H/D exchange experiment.
Partial overlap for amide resonances of Gly-5 and Met-11.
Deuterium Exchange Studies Using Lyophilized Fibrils and DMSO-d6.
The second approach used was to first remove the bulk part of the D2O/H2O mixture through lyophilization and dissolve the freeze-dried material in the aprotic solvent DMSO-d6. The freeze-dried Aβ25–35 amyloid deposits readily dissolved in DMSO-d6 with a measured rate of 0.04–0.05 min−1 at 31°C, sufficiently fast to allow the analysis of H/D exchange patterns by using one-dimensional 1H NMR spectroscopy (Fig. 5B). Because the final DMSO-d6 sample cannot be made completely free from water, exchange from residual H2O to partially deuterated amide protons will occur slowly over time during data collection. The analytical approach is from here on quite similar to the method using TFE-d3/D2O, but now an inverse exponential relationship occurs whose function can be fitted using Eq. 2.
 |
2 |
Io represents the extrapolated amide proton integral at time 0 of solubilization of the fibrils in DMSO-d6 (containing H2O), and Iinf the amide proton integral after infinite post-trap exchange times. The method using DMSO-d6 indicates a relatively constant solvent protection level for residues Ala-6 to Leu-10, with Io varying between 58% for Ala-6 to 47% for both Ile-8 and Gly-9, after 18 h of H/D exchange (Table 2). Partial overlap between Gly-5 and Met-11 prevents accurate fits for these two residues. Note that the detection limit to discriminate between pre- and post-trap exchange is located at a ratio H/D of 20% in the DMSO-d6 experiment. Amide protons from residues Gly-1 to Lys-4 are exchanged within the dead time (2.5–7 min) of the post-trap exchange period and therefore no protection values can be given for these amino acids.
Discussion
New methods are required for studies of amyloid structures, because a detailed knowledge of the fibril structure is of interest for development of amyloid inhibitors. We have designed a H/D exchange experiment measured by NMR, where mature fibrils are exposed to D2O solvent. Surface-exposed amide protons in the fibrils are rapidly exchanged for deuterons, while residues within the fibril core remain partially protected as a result of the intensive hydrogen-bonding networks throughout β-sheets. By rapid dissociation of the fibril assembly into its building blocks, the exchanged proton pattern of the original fibril is trapped within the soluble monomer. The trapped H/D ratio can then subsequently be analyzed, either by one- or two-dimensional high-resolution solution 1H NMR.
To exploit the method, we used a model peptide derived from the full-length Alzheimer β-peptide (1–43), represented by amino acids 25–35. The results clearly reveals a protected core in the amyloid fibril comprising amino acids Lys-4 to Met-11 [Lys-28 to Met-35 in the Aβ(1–43) numbering]. Apart from pinpointing exactly the residues involved in secondary structure within the amyloid, quantitative values of solvent protection at a single amino acid level were also obtained.
The success of this assay relies both on the ability to rapidly dissociate the mature fibrils to soluble monomers and at the same time minimize post-trap exchange within the monomeric state. When the post-trap amide-proton exchange is sufficiently slower than the solubilization rate of the amyloid, the method enables extrapolation of experimental integrals to exactly time 0. In other words, the H/D distribution within the amyloid aggregates just before solubilization can be extracted, even though at that time all peptides are aggregated and undetectable by using NMR.
In general, the amide exchange depends on pH, temperature, and the protection factor of secondary structure. In aqueous solutions the pH parameter shows a logarithmic dependency to the exchange rate and exhibits a minimum between 3 and 4 (14). An increased rate additionally occurs on an increase in temperature. An optimum temperature is therefore low enough to reduce intrinsic solvent exchange rates, but still high enough to record high-resolution-type NMR spectra. Post-trap solvent exchange can also be significantly slowed down by induction of secondary structure within the monomer. In our case, stabilization of secondary structure by α-helices of Aβ25–35 seems moderate, as indicated by the absence of long-range NOEs.
Another approach to reducing the rate of post-trap exchange is to remove the bulk part of H2O/D2O and dissolve the fibrils in an aprotic solvent. In this work, cold lyophilization followed by dissolution in DMSO-d6 was used (10). A complete removal of residual water, both from freeze-dried samples and from DMSO-d6 solutions, was difficult to achieve. The exchange rate was however sufficiently slow and the residual water additionally enabled monitoring of a post-trap exchange and extrapolation of proton-integrals to time 0.
Analysis of the data as described above gives the state of the fibril structure over a given incubation time. Although a single preincubation time alone is highly informative, one step further is to take more time points, thereby monitoring the H/D exchange within the fibril over increasingly longer periods. This allows for a more detailed analysis of solvent exchange rates permitting the possibility to further map structural stability and solvent accessibility in fibrils. In the course of investigation we noticed nonexponential decay in the protection factors (Io, Eqs. 1 and 2) when the time of preincubation in D2O was increased (Table 2). Depending on the residue, the protection factor translates into solvent exchange rates [ksolv(amyloid)] for amide protons in the amyloid between 1.7–10 × 10−3 min−1 at 31 min incubation time and up to 0.5–0.7 × 10−3 min−1 at 18 h incubation time (Table 2). The solvent exchange rates were reproducible within 10%, as measured at 31 and 135 min incubation time. The initial exchange rates are surprisingly high. However, the exchange rates observed after 18 h preincubation in D2O are of the same order of magnitude as derived for other amyloids through mass spectroscopy (15).
The nonexponential decay could either be attributed to the existence in the incubation solution of a mixture of amorphous aggregates and amyloid fibril or to the heterogeneity of amyloids themselves at a molecular level. In AFM microscopy only less than 10% amorphous aggregates were observed (see Results). We therefore conclude that the amyloid fibrils are heterogeneous. This finding is in accordance with other investigations, which show that the affinities of free peptides onto already formed fibrils change as a function of incubation time (16) and by the fact that fibrils need to mature. Often observed kinks and bending of the fibrils (17) can also explain the finding. The fact that the sequence-specific pattern of the protection factors does not change significantly with incubation time shows that the structural features between more solvent-accessible parts compared with less solvent-accessible parts are rather similar.
The protection factors (Fig. 5) show that residues 1–3 are highly solvent exposed. Because residues 1 and 2 becomes exchanged within the dead time of the experiments, the assumption of a solvent-exposed position within the fibril is indirect and derived from the bell-shaped distribution of protection factors. Interestingly, in the complete Alzheimer β peptide (1–43) the corresponding residues (25–27) are suggested to form part of a bend (18). The bell-shaped distribution of protection factors centered around residues Ile-7 and Ile-8 suggests an out-of-register antiparallel β-sheet core where Lys-4 is arranged opposite to Met-11 (Fig. 6A). This leads to maximum protection at the core residues Ile-7 and Ile-8, and a lower averaged solvent protection for residues positioned left and right of Ile-7 and Ile-8. Such an arrangement generates favorable hydrophobic interactions, as well as charge interaction between Lys-4 and the negatively charged C terminus. However, alternatively an in-register parallel assembly, as illustrated in Fig. 6B, cannot be excluded and has previously been reported with regard to Aβ10–35 (19) and Aβ1–40 (20).
Figure 6.

Schematic models of putative hydrogen-bond assemblies within the fibril. The arrangements correspond to the observed protection factors and optimal hydrophobic and charged interactions. Residues with hydrophobic side chains are depicted with filled rings, whereas hydrophilic residues are depicted with open rings. Charged residues are indicated. (A) Out-of-register antiparallel arrangement of Aβ25–35 peptide. Note that Lys-4 and the C terminus form a favorable electrostatic interaction. (B) In-register parallel arrangement of the Aβ25–35 peptide. Note the improved alignment of strongly hydrophobic residues (e.g., Ala-6, Ile-7, and Ile 8) when compared with the arrangement in A. On the contrary, unfavorable electrostatic interactions are seen between Lys-4 residues and between C terminus residues.
The method presented within this work provides a general and powerful approach that enables identification and quantification of core residues involved in amyloid structures. The ability to quantitatively measure solvent protection of each participating residue provides an advantage compared with mass spectroscopy analysis, which only provides an averaged sum of the exchanged protons. The information gained may lead to a more rational design of amyloid inhibitors.
Acknowledgments
This work was supported by the Swedish Medical Research Council, the patient's association FAMY, the medical faculty at Umeå University, and the County of Västerbotton (E.L.).
Abbreviations
- Aβ
Alzheimer β-peptide
- TFE-d3
deuterated trifluoroethanol
- DMSO-d6
deuterated DMSO
- H/D
hydrogen/deuterium
- NOESY
nuclear Overhauser effect spectroscopy
- AFM
atomic force microscopy
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Tan S Y, Pepys M B. Histopathology. 1994;25:403–414. doi: 10.1111/j.1365-2559.1994.tb00001.x. [DOI] [PubMed] [Google Scholar]
- 2.Rochet J C, Lansbury P T., Jr Curr Opin Struct Biol. 2000;10:60–68. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 3.Kirschner D A, Abraham C, Selkoe D J. Proc Natl Acad Sci USA. 1986;83:503–507. doi: 10.1073/pnas.83.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serpell L C, Sunde M, Fraser P E, Luther P K, Morris E P, Sangren O, Lundgren E, Blake C C. J Mol Biol. 1995;254:113–118. doi: 10.1006/jmbi.1995.0604. [DOI] [PubMed] [Google Scholar]
- 5.Kheterpal I, Zhou S, Cook K D, Wetzel R. Proc Natl Acad Sci USA. 2000;97:13597–13601. doi: 10.1073/pnas.250288897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tycko R. Methods Enzymol. 2001;339:390–413. doi: 10.1016/s0076-6879(01)39324-2. [DOI] [PubMed] [Google Scholar]
- 7.Kheterpal I, Williams A, Murphy C, Bledsoe B, Wetzel R. Biochemistry. 2001;40:11757–11767. doi: 10.1021/bi010805z. [DOI] [PubMed] [Google Scholar]
- 8.Englander S W, Mayne L. Annu Rev Biophys Biomol Struct. 1992;21:243–265. doi: 10.1146/annurev.bb.21.060192.001331. [DOI] [PubMed] [Google Scholar]
- 9.Liu K, Cho H S, Hoyt D W, Nguyen T N, Olds P, Kelly J W, Wemmer D E. J Mol Biol. 2000;303:555–565. doi: 10.1006/jmbi.2000.4164. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y Z, Paterson Y, Roder H. Protein Sci. 1995;4:804–814. doi: 10.1002/pro.5560040420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yankner B A, Duffy L K, Kirschner D A. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 12.Forloni G, Chiesa R, Smiroldo S, Verga L, Salmona M, Tagliavini F, Angeretti N. NeuroReport. 1993;4:523–526. doi: 10.1097/00001756-199305000-00015. [DOI] [PubMed] [Google Scholar]
- 13.Piotto M, Saudek V, Sklenar V. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 14.Wuthrich K. Science. 1989;243:45–50. doi: 10.1126/science.2911719. [DOI] [PubMed] [Google Scholar]
- 15.Tito P, Nettleton E J, Robinson C V. J Mol Biol. 2000;303:267–278. doi: 10.1006/jmbi.2000.4142. [DOI] [PubMed] [Google Scholar]
- 16.Esler W P, Stimson E R, Jennings J M, Vinters H V, Ghilardi J R, Lee J P, Mantyh P W, Maggio J E. Biochemistry. 2000;39:6288–6295. doi: 10.1021/bi992933h. [DOI] [PubMed] [Google Scholar]
- 17.Walsh D M, Lomakin A, Benedek G B, Condron M M, Teplow D B. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 18.Shao H, Jao S, Ma K, Zagorski M G. J Mol Biol. 1999;285:755–773. doi: 10.1006/jmbi.1998.2348. [DOI] [PubMed] [Google Scholar]
- 19.Benzinger T L, Gregory D M, Burkoth T S, Miller-Auer H, Lynn D G, Botto R E, Meredith S C. Biochemistry. 2000;39:3491–3499. doi: 10.1021/bi991527v. [DOI] [PubMed] [Google Scholar]
- 20.Antzutkin O N, Balbach J J, Leapman R D, Rizzo N W, Reed J, Tycko R. Proc Natl Acad Sci USA. 2000;97:13045–13050. doi: 10.1073/pnas.230315097. [DOI] [PMC free article] [PubMed] [Google Scholar]