

Abstract
Genes regulating responses in mammalian cells are often difficult to identify by functional cloning strategies limited to a single round of selection. Here we describe a strategy, cyclical packaging rescue (CPR), which allows rapid recovery and retransmission of retroviral cDNA libraries. CPR can be used not only with immortalized cell lines such as fibroblasts and Jurkat T cells, but also with primary B lymphocytes, which can be maintained only in short-term cultures. CPR allows for multiple rounds of selection and enrichment to identify cDNAs regulating responses in mammalian cells. Using CPR, five cDNAs were functionally cloned, which conferred protection against tumor necrosis factor α (TNFα)-induced apoptosis in RelA−/− fibroblasts. Three of the genes, RelA, cellular FLICE-like inhibitory protein (c-FLIP), and a dominant-negative mutant of TNF receptor 1 arising through CPR afforded strong protection against apoptosis. Two of the genes identified, Dbs and Fas-associated death domain protein (FADD), previously identified as a proapoptotic molecule, afforded partial protection against TNFα-induced apoptosis. These results suggest that CPR is a versatile method that permits functional identification of both wild-type and dominant-negative gene products that regulate cellular responses.
Retroviral cDNA libraries have been used in a number of functional screens to identify unique mammalian genes that regulate cellular responses (1–6). These screens typically have used only a single round of selection to identify positive cells expressing cDNAs that fulfill a selection criterion. To ensure that positive cDNA clones are successfully isolated from complex libraries in a single round of selection, functional screens using mammalian cells have been restricted to cellular assays in which the background of false-positive cells meeting a selection criterion is low, typically 1 in 105 false-positive cells. Unfortunately, most functional responses of mammalian cells do not meet this stringent criterion.
Mammalian expression cloning methods that permit multiple rounds of selection and enrichment have been extremely successful in identifying cDNA clones from plasmid libraries that correspond to unknown receptors or ligands (7). Unfortunately, the requirement for large T antigen-driven replication of plasmid libraries in mammalian cells has severely limited adaptation of expression cloning methods to functional screens with only highly transfectable cell lines. In a recent adaptation of expression cloning methods termed the MaRX system, Hannon et al. (8) redesigned retroviral cDNA vectors to incorporate loxP sites and a bacterial origin of replication. These retroviral vectors allow integrated proviruses to be excised as circular plasmids by Cre-mediated recombination permitting recovery of circular DNA that can be transformed into competent bacteria. Plasmids isolated from bacteria are then transfected into a retroviral packaging line to regenerate retroviral stocks.
Here we describe an alternative method called cyclical packaging rescue (CPR) that uses direct repackaging of retroviral RNAs into new infectious virions to identify genes regulating functional responses in mammalian cells (Fig. 1A). In CPR, stably integrated helper-free retroviral libraries are recovered rapidly from mammalian cells as infectious helper-free retroviral supernatants 24 h after infection with adenoviruses expressing retroviral gag-pol and env genes. Recovered retroviral supernatants are then used to reinfect fresh target cells. When performed in concert with selection using functional assays, cDNAs regulating functional responses can be identified by enrichment through multiple rounds of retroviral library recovery and retransmission. Using CPR, we identified five cDNAs that conferred resistance to tumor necrosis factor α (TNFα)-induced apoptosis in RelA−/− fibroblasts, where ≈20% of cells survive when treated with TNFα (9–12). The genes identified in the screen include both modifiers that only partially protected RelA−/− cells against TNFα and a dominant-negative mutant of TNF receptor 1 (TNFR1) that was generated during the process of CPR. These results suggest that CPR is a rapid and versatile approach that should facilitate functional analysis of gene function in mammalian cells.
Figure 1.

Rapid recovery and controlled retransmission of retroviruses from multiple cell lineages by using CPR. (A) Scheme for packaging rescue of stably integrated helper-free retroviruses from cells. Retrovirally transduced cells are infected with Adgag-pol and Adenv and washed. After 24 h, retroviral supernatants are recovered and used to retransduce fresh cells. (B) Retroviruses can be rapidly recovered from fibroblasts, primary B cells, and Jurkat T cells and retransmitted efficiently to fresh cells. Retroviral supernatants recovered 24 h after infection with Adgag-pol and Adenv were used to transduce a fresh lot fibroblasts, which can be enriched for retroviral marker expression and used to retransduce primary B cells or Jurkat T cells expressing the mCAT1 receptor. Gray-shaded plots represent controls in which one or both of the adenoviruses were omitted. The efficiency of packaging rescue is represented as the starting number of transduced cells required to retrovirally infect 105 target cells. (C) Rare marker retroviruses can be maintained through one round of CPR in fibroblasts and primary B cells. For fibroblasts, MSCV-GFP-RelA-IRES-puro-transduced cells were mixed with MSCV-IRES-puro-infected cells at ratios of ≈1:100 or 1:1,000, retroviruses were rescued by using CPR, and transferred to a fresh lot of RelA−/− fibroblasts. GFP-expressing cells were quantified by flow cytometry after puromycin selection. For primary B cells, MSCV-Thy1.1-IRES-PLAP-transduced cells were mixed with MSCV-IRES-Thy1.1-infected cells at a ratio of 1:10,000 and were subjected to one round of CPR by using fibroblast intermediates followed by flow cytometric analysis of Thy1.1 and enzymatic detection of PLAP.
Materials and Methods
Plasmid Construction.
An env-internal ribosomal entry site-green fluorescent protein (IRES-GFP) cassette was generated in pBSSKII (Stratagene) by sequential subcloning of the IRES-GFP insert from murine stem cell virus (MSCV)-IRES-GFP (13) into the EcoRI site followed by the Moloney murine leukemia virus (MMLV) env gene into the SmaI site. This env-IRES-GFP cassette was excised with EcoRV–NotI, Klenow-blunted, and cloned into the PmeI site of pQBIAdCMV5 (Qbiogene, Carlsbad, CA), producing Adenv. The MMLV gag-pol genes were cloned into the PmeI site of PQBIAdCMV5 to yield Adgag-pol. To create the GFP-RelA fusion protein, RelA was cloned into the NotI–SalI sites of MSCV-IRES-GFP. The IRES-GFP cassette was then removed with EcoRI, the enhanced GFP gene was excised from pEGFP-N1 (CLONTECH) as a XhoI–NotI fragment, and ligated upstream of MSCV-RelA to create an in-frame fusion protein. MSCV-IRES-Thy1.1 was provided by T. Mitchell (University of Louisville, Louisville, KY); MSCV-IRES-hCD4 was a gift from K. Murphy (Washington University, St. Louis); MSCV-IRES-placental alkaline phosphatase (PLAP) and MSCV-IRES-puro were provided by W. Pear (University of Pennsylvania, Philadelphia). The wild-type Fas-associated death domain protein (FADD) cDNA was provided by A. Winoto (University of California, Berkeley).
Cell Culture.
The 293A (Qbiogene) and Bosc23 cells were grown in DMEM with 10% FBS (JRH Biosciences, Lenexa, KS). RelA−/− fibroblasts (9) were grown in DMEM plus 10% donor bovine serum (JRH Biosciences). Jurkat T cells were cultured in RPMI medium 1640 with 10% FBS (HyClone) containing 50 μM β-mercaptoethanol. Primary splenic B cells were cultured in RPMI medium 1640 with 10% FBS (HyClone) containing 50 μM β-mercaptoethanol and 30 μg/ml bacterial lipopolysaccharide (Sigma).
Adenovirus Production and Infection.
The 293A cells were cotransfected with FspI-linearized Adenv and 5 μg of Qbiogene-adenoviral DNA by using calcium phosphate coprecipitation. After overnight incubation, cells were overlaid with DMEM plus 5% FBS (JRH Biosciences) plus 1.25% Seaplaque agarose. Approximately 2 wk after transfection, GFP+ plaques were picked and eluted overnight in 0.5 ml of DMEM plus 5% FBS. Adgag-pol was produced with ClaI-linearized Adgag-pol by Qbiogene custom service. Plaque eluates were screened for their ability to complement retroviral production in MSCV-IRES-GFP-transfected 293A cells, and positive clones were amplified. Retrovirally infected RelA−/− fibroblasts were plated at 2 × 105 cells/well of a 6-well plate and infected with 109 Adgag-pol and 5 × 108 Adenv particles as determined by optical A at 260 nm (14) in 2 ml of growth media by spinning at 1,000 × g for 1 h. Cells were washed with PBS, 2 ml of growth media was replaced, and retrovirus was harvested 24 h after adenoviral infection for retransduction of 1 × 105 RelA−/− fibroblasts. For transfer of retrovirus from fibroblasts to B cells, retrovirus recovered from 1.2 × 106 RelA−/− fibroblasts/well of a 6-well plate was used to transduce 5 × 105 B cells. For transfer of retrovirus from B cells to fibroblasts, 106 retrovirally infected human coxsackie-adenovirus receptor-transgenic B cells were resuspended in 100 μl of DMEM plus 10% FBS and incubated with 109 viral particles of Adenv and 2.5 × 109 viral particles of Adgag-pol and incubated at 37°C with occasional agitation for 30 min. Cells were washed, resuspended in 1 ml of media containing 30 μg/ml bacterial lipopolysaccharide (Sigma), and incubated for 24 h before harvesting retroviral supernatants. Supernatants from 106 B cells were used to transduce 2 × 105 RelA−/− fibroblasts. For transfer of retrovirus from Jurkat T cells to fibroblasts, 5 × 106 cells were incubated in 100 μl of DMEM plus 10% FBS containing 109 viral particles of Adenv and 2.5 × 109 particles of Adgag-pol for 30 min. After extensive washing, Jurkat cells were treated with 3 ng/ml phorbol myristic acid (Sigma) and 12.5 nM ionomycin (Sigma) for 24 h, and supernatants were transferred to 2 × 105 RelA−/− fibroblasts. For transfer from fibroblasts to Jurkat T cells, retrovirus recovered from 1.2 × 106 RelA−/− fibroblasts in one well of a 6-well plate was used to transduce 105 Jurkat T cells expressing the murine cationic amino acid transporter 1 (mCAT1) ecotropic receptor (15).
Retroviral Production and Infection.
Bosc23 cells were transfected with retroviral constructs or a mouse embryo cDNA library (CLONTECH) by using calcium phosphate coprecipitation, and retroviral supernatants were obtained after 48 h. Primary B cells were stimulated for 24 h with 30 μg/ml bacterial lipopolysaccharide and spin-infected at 106 cells/ml of retroviral supernatant for 1.5 h at 1,000 × g. Cells were then cultured for 2 d in the presence of 30 μg/ml bacterial lipopolysaccharide to allow retroviral expression.
TNFα Treatment.
In library screening experiments, RelA−/− fibroblasts were plated at ≈30% confluency and treated for 2 d with 10 ng/ml TNFα (R&D Systems).
Abs and Detection Reagents.
Anti-Thy1.1 Ab (PharMingen, clone OX-7), anti-PLAP Ab (Dako), and streptavidin-coated magnetic beads (Miltenyi Biotec, Auburn, CA) were used and magnetic cell sorting was performed according to manufacturer's instructions. Flow cytometry was performed on an Epics Coulter XL. PLAP+ fibroblasts also were detected with SigmaFast NitroBlue tablets after fixation with 0.05% glutaraldehyde.
PCR, Sequencing, and Southern Blotting.
Genomic DNA isolated from RelA−/− fibroblasts was used as a template for PCR with CLONTECH retroviral-specific primers 5′-LIB (5′-AGCCCTCACTCCTTCTCTAG-3′) and 3′-LIB (5′-ACCTACAGGTGGGGTCTTTCATTCCC-3′). PCR was performed on 2 μg of genomic DNA by using Expand high fidelity PCR system (Roche Molecular Biochemicals) with the following cycling conditions: 94°C-2 min, (94°C-15 sec, 55°C-30 sec, 68°C-8 min) × 10 cycles (94°C-15 sec, 55°C-30 sec, 68°C-8 min plus 5 sec per cycle) × 20 cycles; 72°C-7 min. Sequencing reactions were performed by using the same primers as above on an Applied Biosystems Prism 310 genetic analyzer. For TNFR1-specific PCR, an initial PCR was run on library or genomic DNA with the retroviral primers as described above. A fraction of this sample (1/25) was then subjected to PCR with the TNFR1-specific primers below and the same temperature program as above: 5′-TTGTGCCTACCTCCTCCGCTT-3′ and 5′-TCACCCACAGGGAGTAGGGCA-3′. These reactions were digested with XmnI and subjected to Southern blotting by using the TNFR1-specific 32P-labeled probe 5′-GCCTGGCGGCGCCGCACGCCG-3′.
Results
We examined whether mammalian cells containing stably integrated helper-free retroviruses could be transiently induced to produce infectious helper-free retroviruses by controlled adenoviral expression of MMLV gag-pol and env (Fig. 1A). Two separate recombinant adenoviruses, Adgag-pol and Adenv, were used to complement retroviral packaging function to prevent emergence of replication-competent retroviruses through recombination. The Adenv adenovirus contained an IRES-GFP marker to allow adenoviral infection to be monitored.
Packaging rescue with Adgag-pol and Adenv infection was a rapid and highly efficient means to recover and retransduce stably integrated retroviruses from fibroblasts (Fig. 1B). Supernatants recovered from fibroblasts with a stably integrated MSCV-IRES-PLAP retrovirus contained sufficient infectious retrovirus to retransduce an equivalent number of fresh fibroblasts only 24 h after infection with Adgag-pol and Adenv. In multiple experiments with fibroblasts, this efficiency of retroviral recovery and retransmission was between 50% and 100% (data not shown).
Packaging rescue also could be used with primary B cells under conditions consistent with adaptation to functional screens by using in vitro assays of B cell differentiation (Fig. 1B). Because primary B cells have very low endogenous levels of the coxsackie-adenovirus receptor, B cells from transgenic mice expressing the human coxsackie-adenovirus receptor in lymphocytes were used (16–18). Splenic B cells were polyclonally activated with bacterial lipopolysaccharide and transduced with an MSCV-IRES-Thy1.1 retrovirus. After 3 or 4 days in culture, primary B cells were infected with Adgag-pol and Adenv and supernatants containing repackaged retrovirus were recovered after 24 h. These retroviral supernatants were used to transduce fibroblasts. In multiple experiments, the efficiency of recovery and retransmission of retroviruses from equivalent numbers of transduced B cells to fibroblasts was between 2% and 5%. Transduced fibroblasts were enriched by magnetic bead selection with the Thy1.1 marker and a second round of retroviral recovery by adenoviral infection was used to retransduce fresh polyclonally activated primary B cells. Use of an immortalized RelA−/− fibroblast intermediate, in contrast to short-lived primary B cells, allowed us to establish reproducible conditions for highly efficient transfer of retroviruses from fibroblasts back to primary B cells. Thus, integrated retroviruses recovered from transduced B cells at the endpoint of in vitro assays for B cell differentiation can be retransmitted to fresh undifferentiated primary B cells by using two rounds of Adgag-pol and Adenv infection with a fibroblast intermediate.
Packaging rescue also could be adapted to the human Jurkat T cell line, used extensively in the study of T cell signaling (19, 20). After infection with Adgag-pol and Adenv, ≈3% of the integrated retroviruses could be transferred from Jurkat T cells to fibroblasts (Fig. 1B). Analogously to primary B lymphocytes, retroviruses could then be efficiently transferred back from fibroblasts to Jurkat T cells expressing the murine ecotropic receptor, mCAT1 (15). Integrated retroviruses could also be transferred directly from Jurkat T cells to fresh Jurkat T cells, although the efficiency was somewhat lower (0.8%).
For all three cell types described above, the efficiency of retroviral repackaging and transfer appeared sufficient to allow for adaptation of packaging rescue to functional screens designed to identify rare cDNAs from cells transduced with complex retroviral cDNA libraries. To determine whether rare cDNAs could be maintained by packaging rescue, we subjected both fibroblasts and primary B cells to one round of packaging rescue by using cells transduced with different ratios of a rare marker gene-expressing retrovirus to a control retrovirus. At a ratio of 1:102 or 1:103 with a control retrovirus, a GFP retrovirus could be readily detected at equivalent ratios after packaging rescue from fibroblasts to fibroblasts (Fig. 1C). At a ratio of 1:104 with a control retrovirus, a PLAP retrovirus also could be maintained at equivalent ratios after packaging rescue from primary B cells to primary B cells by using a fibroblast intermediate. Thus, rare marker genes expressed by retroviruses could be maintained in both fibroblasts and primary B cells after transfer by packaging rescue.
These results suggested that multiple rounds of packaging rescue by Adgag-pol- and Adenv-mediated retroviral recovery could be performed in concert with functional assays of cellular differentiation to functionally enrich and identify cDNAs from retroviral libraries. We term this strategy of selecting retroviral libraries through multiple rounds of functional assays CPR. To test this CPR strategy, we selected a functional assay in which the background of cells surviving a selection criterion was so high that repeated rounds of cellular selection were necessary.
Approximately 20% of RelA−/− fibroblasts survive treatment with the inflammatory cytokine TNFα (9–12), necessitating multiple rounds of functional selection to enrich for and identify rare cDNAs that confer resistance to TNFα from complex retroviral cDNA libraries. We first examined whether CPR was effective in enriching through multiple rounds of selection for a retrovirus expressing a GFP-RelA fusion protein that conferred resistance to TNFα-induced apoptosis of RelA−/− fibroblasts. Under conditions in which cells were treated twice each round with TNFα to reduce background cell survival to 2–5% per round, a GFP-RelA retrovirus was enriched 50,000-fold in four rounds of CPR selection from an initial ratio of 1:1.5 × 105 to a final ratio of 1:3 (Fig. 2). At each round, enrichment for GFP+ cells detected by flow cytometry depended on TNFα selection; no enrichment after packaging rescue was observed in the absence of TNFα selection (data not shown). A RelA immunoblot demonstrated that RelA was successively enriched in cell pools confirming that the increase in GFP+ cells detected by flow cytometry was caused by enrichment for the GFP-RelA retrovirus and not by GFP marker expression from Adenv (data not shown). These results indicated that genes conferring resistance to TNFα could be identified after four rounds of selection with CPR.
Figure 2.
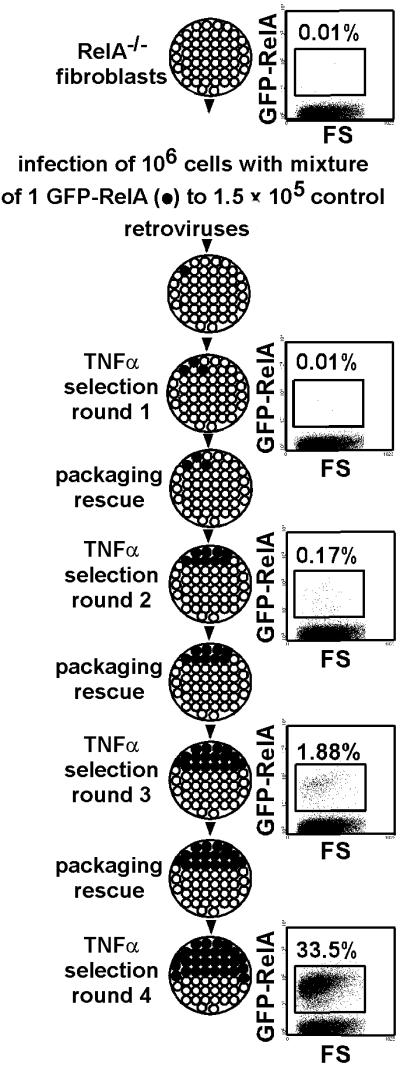
Multiple rounds of a functional assay can be used by using CPR to amplify and identify rare regulating responses of cDNAs. Multiple rounds of selection for cDNAs conferring resistance to TNFα-induced apoptosis can be performed with CPR. RelA−/− fibroblasts with an integrated retrovirus expressing a GFP-RelA fusion protein were initially seeded at a frequency of 1:156,000 with RelA−/− fibroblasts with a control retrovirus. CPR was performed and after four rounds of selection, cells transduced with the GFP-RelA retrovirus were enriched 50,000-fold to a frequency of 1:3.
We next used a retroviral mouse embryo cDNA library to screen by CPR for genes that conferred resistance to TNFα-induced apoptosis of RelA−/− fibroblasts. After four rounds of selection, 11 of 17 individual pools of library-transduced fibroblasts demonstrated increased resistance to TNFα. Genomic DNA isolated from these pools was subjected to PCR for retrovirally expressed cDNAs from representative pools (Fig. 3A). Specific PCR bands from TNFα-resistant pools were subcloned into a retroviral vector upstream of an IRES-Thy1.1 cassette. Retroviral stocks for each cDNA clone were used to infect RelA−/− fibroblasts at a frequency of 10–15% to assay for ability of cDNAs to confer resistance to TNFα. Transduced cells were split, then left untreated or treated with TNFα for 24 h, and analyzed by flow cytometry (Fig. 4). In this assay, the frequency of cells expressing neutral genes without any antiapoptotic properties decreased slightly relative to uninfected cells upon treatment with TNFα. Five cDNAs (Table 1) were identified that were capable of protecting RelA−/− fibroblasts to varying degrees from TNFα-induced apoptosis.
Figure 3.

Genes functionally identified by CPR include dominant-negative mutants generated during the process of CPR. (A) Genomic DNA from TNFα-sensitive and TNFα-resistant pools of cells was subjected to PCR using retroviral-specific primers. “Initial” represents unselected retroviral library-transduced cells before TNFα treatment. Pool 12 remained sensitive to TNFα treatment after four rounds of CPR. Pools 13, 15, and 17 are representative of pools that became resistant to TNFα treatment. (B) The enriched cDNA for TNFR1 is a dominant-negative mutant that was generated during CPR in round 1. The region mutated in the TNFR1* clone identified in pool 13 was amplified by PCR and digested with XmnI. The mutant TNFR1* cDNA is missing an XmnI restriction site found in the wild-type TNFR1 cDNA.
Figure 4.

Genes functionally identified by CPR include the modifiers FADD and Dbs* that only partially protect against TNFα-induced apoptosis. Amplified PCR bands were recloned into retroviral vectors and tested for their ability to confer protection against TNFα-induced apoptosis in RelA−/− fibroblasts. The fold-survival advantage of virally transduced cells over nontransduced cells after a single round of TNFα treatment was calculated as: percentage of infected cells after TNFα treatment, percentage of uninfected cells before TNFα treatment, percentage of infected cells before TNFα treatment, and percentage of uninfected cells after TNFα treatment. SDs from a minimum of six independent experiments are shown and ranges of values obtained for fold-survival advantage are shown in parentheses.
Table 1.
Genes conferring resistance to TNFα-mediated apoptosis
| Gene | Positive pools | cDNA |
|---|---|---|
| RelA | 10/17 | Wild type |
| c-FLIP | 3/17 | Wild type |
| TNFR1 | 1/17 | Truncated, amino acids 1–371 |
| FADD | 1/17 | Wild type |
| Dbs | 1/17 | Truncated, amino acids 530–1,097 |
Of the genes functionally identified, FADD, TNFR1, and Dbs each were found only in one resistant pool, but RelA and c-FLIP were discovered in multiple pools (Table 1). RelA and c-FLIP are two well-characterized antiapoptotic genes (9–11, 21–23) that confirm the efficacy of the screen. In contrast, TNFR1, FADD, and Dbs (6) were unexpected candidates.
The TNFR1 clone contained a TGG→TGA nonsense mutation at codon 371, creating a protein similar to dominant-negative signaling mutants previously described (24). This mutation fortuitously destroyed an internal XmnI restriction site found in the wild-type TNFR1 gene, allowing us to investigate how the dominant-negative TNFR1 arose (Fig. 3B). Using TNFR1-specific primers, the region surrounding the mutation was PCR-amplified with the parent retroviral library or genomic DNA from each CPR round of pool 13. These products were digested with XmnI and subjected to Southern blot analysis. Only wild-type TNFR1 cDNAs were detected in the parental retroviral library, indicating that the mutant TNFR1 cDNA was generated by retroviral mutation during the first round of CPR.
In contrast to the other cDNAs identified, both FADD and Dbs provided only partial protection of RelA−/− fibroblasts against TNFα-induced apoptosis. Although the Dbs clone isolated was N-terminally truncated from amino acids 1–529, others have shown that amino acids 525-1097 possess full transforming and signaling capacity (25). Sequencing of the FADD clones identified in our screens demonstrated identity with the sequence of the published FADD gene. Retroviral expression of a previously characterized wild-type FADD clone also conferred partial protection of RelA−/− fibroblasts against TNFα-induced apoptosis (data not shown), indicating an unexpected role for wild-type FADD in mediating resistance to TNFα.
Discussion
In studies directed toward improving the delivery of therapeutic genes, infection of a variety of mammalian cell types with adenoviruses expressing retroviral gag-pol and env, the two genes normally deleted in most retroviral vectors, leads to efficient packaging and release of retroviral virions (26–29). The cell lines used in these studies were derived from a broad range of tissues and organs, including liver, lung, kidney, cervix, neuronal glia, and T lymphocytes. Our studies extend these findings to primary B cells and Jurkat T cells and indicate that retroviral recovery and transmission using adenoviral complementation can recover rare marker retroviruses consistent with adaptation to functional screens designed to identify gene function. For primary B cells, these screens would include in vitro assays of terminal B cell differentiation for processes such as class switch recombination and plasma cell differentiation. Jurkat T cell lines have historically proven to be extremely useful models for the study of T cell signaling (19, 20), suggesting that CPR may allow for the identification and characterization of genes involved in T cell receptor signaling.
Our results demonstrate that CPR can be used to rapidly conduct multiple rounds of selection and enrichment of cDNA retroviral libraries to identify genes regulating functional responses. Using CPR, we identified five genes from an embryonic retroviral cDNA library that conferred protection against TNFα-induced apoptosis. Two of the cDNAs, RelA and c-FLIP, were well-characterized antiapoptotic genes that were identified from multiple pools in our screen. The third cDNA that also completely protected RelA−/− fibroblasts from apoptosis was a dominant-negative mutant cDNA of TNFR1 that introduced a stop codon upstream of the death domain in the intracellular tail. Interestingly, this dominant-negative mutation arose through retroviral mutation occurring during CPR. We have recently subjected a wild-type TNFR1 clone to CPR and generated a panel of additional mutants that also confer resistance to TNFα (unpublished data). Thus, CPR has the ability to both generate and functionally select for dominant-negative mutants of wild-type genes.
Two of the cDNAs, FADD and Dbs, only conferred partial protection of RelA−/− fibroblasts against TNFα-induced apoptosis and corresponded to previously unrecognized modifiers of TNFα signaling. Although FADD has been identified as a critical intermediate in TNFα signaling, previous reports have described a proapoptotic role for overexpression of FADD. Recently, another group has reported that a mutant version of FADD retains antiapoptotic properties against TNFα-induced apoptosis when expressed in RelA−/− fibroblasts (30). In our screen, we unexpectedly identified overexpression of wild-type FADD as conferring protection of RelA−/− fibroblasts against TNFα. This ability of FADD to protect against TNFα-induced apoptosis did not result from low levels of retroviral expression, as multiple cycles of retroviral infection and overexpression of FADD resulted in a transient increase in apoptotic cells in the absence of TNFα, but still led to the protection of RelA−/− fibroblasts upon TNFα treatment (data not shown). The ability of FADD to protect against TNFα-induced apoptosis appeared specific to RelA−/− fibroblasts because FADD overexpression did not alter the TNFα sensitivity of the L929 and Wehi164 cell lines (data not shown). These data emphasize the importance of performing screens in the cell line in which the biological function of interest naturally occurs.
The second cDNA clone that conferred partial protection of RelA−/− fibroblasts against TNFα-induced apoptosis encoded for a C-terminal fragment of the Dbs protein, a putative guanine exchange factor. The C-terminal Dbs protein fragment that we identified corresponded to a truncated Dbs protein that others have shown possesses full transforming and signaling capacity (25). These results provide evidence that Dbs possesses antiapoptotic properties. The ability of CPR to repetitively enrich for and identify partial modifiers of TNFα signaling suggests that CPR is a sensitive approach that allows for the identification of subtle signaling interactions.
Prokaryotic genetic screens have for many years successfully used methods such as bacteriophage-based cloning to rapidly obtain and retransmit genetic information between multiple rounds of selection. Mammalian genetic screens, however, have lagged with respect to the ability to conduct analogous recovery and retransmission of genetic information. Here we have used commonly used adenoviral and retroviral gene delivery systems to cooperatively allow multiple rounds of selection in mammalian cells. In contrast to the MaRX system, which uses sequential bacterial and eukaryotic packaging cell intermediates to transfer retroviral cDNA libraries between host cell populations (8), our CPR method exploits the natural life cycle of retroviruses to rapidly repackage retroviral RNAs expressed within host cells directly into infectious helper-free virions. CPR can be used to identify functionally interesting gain-of-function genes, dominant-negative mutants, and partial effectors from a complex retroviral library. CPR is not limited to cells that are easily infected by both adenovirus and MMLV-pseudotyped retrovirus, as transgenic expression of exogenous adenoviral receptor on normally recalcitrant B cells or mCAT1 on human Jurkat T cells facilitates retroviral repackaging. These data suggest that CPR may be a broadly applicable method to facilitate characterization of gene function in mammalian systems.
Acknowledgments
This work was supported by National Institutes of Health Grant AI42252 and the Pew Charitable Trusts.
Abbreviations
- CPR
cyclical packaging rescue
- TNFα
tumor necrosis factor α
- TNFR1
TNF receptor 1
- FADD
Fas-associated death domain protein
- c-FLIP
cellular FLICE-like inhibitory protein
- GFP
green fluorescent protein
- MMLV
Moloney murine leukemia virus
- IRES
internal ribosomal entry site
- MSCV
murine stem cell virus
- PLAP
placental alkaline phosphatase
- mCAT1
murine cationic amino acid transporter 1
References
- 1.Whitehead I, Kirk H, Kay R. Mol Cell Biol. 1995;15:704–710. doi: 10.1128/mcb.15.2.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kitamura T, Onishi M, Kinoshita S, Shibuya A, Miyajima A, Nolan G P. Proc Natl Acad Sci USA. 1995;92:9146–9150. doi: 10.1073/pnas.92.20.9146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duan Y, Naruse T, Nakamura M, Yamaguchi Y, Kawashima T, Morikawa Y, Kitamura T, Suda T. Biochem Biophys Res Commun. 1998;253:401–406. doi: 10.1006/bbrc.1998.9700. [DOI] [PubMed] [Google Scholar]
- 4.He Y W, Deftos M L, Ojala E W, Bevan M J. Immunity. 1998;9:797–806. doi: 10.1016/s1074-7613(00)80645-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hitoshi Y, Lorens J, Kitada S I, Fisher J, LaBarge M, Ring H Z, Francke U, Reed J C, Kinoshita S, Nolan G P. Immunity. 1998;8:461–471. doi: 10.1016/s1074-7613(00)80551-8. [DOI] [PubMed] [Google Scholar]
- 6.Whitehead I, Kirk H, Kay R. Oncogene. 1995;10:713–721. [PubMed] [Google Scholar]
- 7.Gluzman Y. Cell. 1981;23:175–182. doi: 10.1016/0092-8674(81)90282-8. [DOI] [PubMed] [Google Scholar]
- 8.Hannon G J, Sun P, Carnero A, Xie L Y, Maestro R, Conklin D S, Beach D. Science. 1999;283:1129–1130. doi: 10.1126/science.283.5405.1129. [DOI] [PubMed] [Google Scholar]
- 9.Beg A A, Baltimore D. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 10.Wang C Y, Mayo M W, Baldwin A S., Jr Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 11.Van Antwerp D J, Martin S J, Kafri T, Green D R, Verma I M. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 12.Liu Z G, Hsu H, Goeddel D V, Karin M. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 13.Ranganath S, Ouyang W, Bhattarcharya D, Sha W C, Grupe A, Peltz G, Murphy K M. J Immunol. 1998;161:3822–3826. [PubMed] [Google Scholar]
- 14.Mittereder N, March K L, Trapnell B C. J Virol. 1996;70:7498–7509. doi: 10.1128/jvi.70.11.7498-7509.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albritton L M, Tseng L, Scadden D, Cunningham J M. Cell. 1989;57:659–666. doi: 10.1016/0092-8674(89)90134-7. [DOI] [PubMed] [Google Scholar]
- 16.Wan Y Y, Leon R P, Marks R, Cham C M, Schaack J, Gajewski T F, DeGregori J. Proc Natl Acad Sci USA. 2000;97:13784–13789. doi: 10.1073/pnas.250356297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt M R, Piekos B, Cabatingan M S, Woodland R T. J Immunol. 2000;165:4112–4119. doi: 10.4049/jimmunol.165.7.4112. [DOI] [PubMed] [Google Scholar]
- 18.Tallone T, Malin S, Samuelsson A, Wilbertz J, Miyahara M, Okamoto K, Poellinger L, Philipson L, Pettersson S. Proc Natl Acad Sci USA. 2001;98:7910–7915. doi: 10.1073/pnas.141223398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldsmith M A, Weiss A. Proc Natl Acad Sci USA. 1987;84:6879–6883. doi: 10.1073/pnas.84.19.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldsmith M A, Bockenstedt L K, Dazin P, Weiss A. Adv Exp Med Biol. 1989;254:25–33. doi: 10.1007/978-1-4757-5803-0_4. [DOI] [PubMed] [Google Scholar]
- 21.Wang C Y, Mayo M W, Korneluk R G, Goeddel D V, Baldwin A S., Jr Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 22.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer J L, Schroter M, Burns K, Mattmann C. Nature (London) 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 23.Yeh W C, Itie A, Elia A J, Ng M, Shu H B, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel D V, Mak T W. Immunity. 2000;12:633–642. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- 24.Tartaglia L A, Goeddel D V. J Biol Chem. 1992;267:4304–4307. [PubMed] [Google Scholar]
- 25.Westwick J K, Lee R J, Lambert Q T, Symons M, Pestell R G, Der C J, Whitehead I P. J Biol Chem. 1998;273:16739–16747. doi: 10.1074/jbc.273.27.16739. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida Y, Emi N, Hamada H. Biochem Biophys Res Commun. 1997;232:379–382. doi: 10.1006/bbrc.1996.5976. [DOI] [PubMed] [Google Scholar]
- 27.Ramsey W J, Caplen N J, Li Q, Higginbotham J N, Shah M, Blaese R M. Biochem Biophys Res Commun. 1998;246:912–919. doi: 10.1006/bbrc.1998.8726. [DOI] [PubMed] [Google Scholar]
- 28.Lin X. Gene Ther. 1998;5:1251–1258. doi: 10.1038/sj.gt.3300720. [DOI] [PubMed] [Google Scholar]
- 29.Duisit G, Salvetti A, Moullier P, Cosset F L. Hum Gene Ther. 1999;10:189–200. doi: 10.1089/10430349950018986. [DOI] [PubMed] [Google Scholar]
- 30.De Smaele E, Zazzeroni F, Papa S, Nguyen D U, Jin R, Jones J, Cong R, Franzoso G. Nature (London) 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]