

Abstract
Extensive research has demonstrated endurance exercise to be neuroprotective. Whether these neuroprotective benefits are mediated, in part, by hepatic ketone production remains unclear. To investigate the role of hepatic ketone production on brain health during exercise, healthy 6-month-old female rats underwent viral knockdown of the rate-limiting enzyme in the liver that catalyses the first reaction in ketogenesis: 3-hydroxymethylglutaryl-CoA synthase 2 (HMGCS2). Rats were then subjected to either a bout of acute exercise or 4 weeks of chronic treadmill running (5 days/week) and cognitive behavioural testing. Acute exercise elevated ketone plasma concentration 1 h following exercise. Hepatic HMGCS2 knockdown, verified by protein expression, reduced ketone plasma concentration 1 h after acute exercise and 48 h after chronic exercise. Proteomic analysis and enrichment of the frontal cortex revealed hepatic HMGCS2 knockdown reduced markers of mitochondrial function 1 h after acute exercise. HMGCS2 knockdown significantly reduced state 3 complex I + II respiration in isolated mitochondria from the frontal cortex after chronic exercise. Spatial memory and protein markers of synaptic plasticity were significantly reduced by HMGCS2 knockdown. These deficiencies were prevented by chronic endurance exercise training. In summary, these are the first data to propose that hepatic ketogenesis is required to maintain cognition and mitochondrial function, irrespective of training status, and that endurance exercise can overcome neuropathology caused by insufficient hepatic ketogenesis. These results establish a mechanistic link between liver and brain health that enhance our understanding of how peripheral tissue metabolism influences brain health.
Keywords: cerebral cortex, cognitio, exercise, HMGCS2, ketogenesis, liver, mitochondria, proteomics
Abstract figure
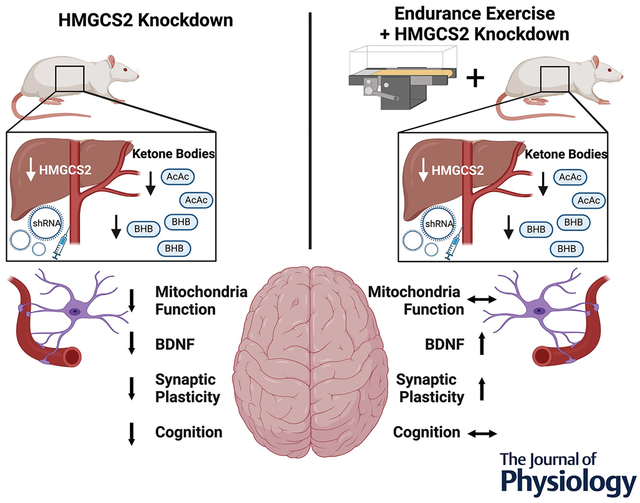
Knockdown of hepatic HMGCS2 by AAV-mediated shRNA resulted in decreased mitochondrial function, BDNF, synaptic plasticity and cognition in sedentary rats. Conversely, chronic exercise training mitigated the losses in synaptic plasticity and BDNF caused by deficient hepatic HMGCS2. HMGCS2, 3-hydroxymethylglutaryl-CoA synthase 2; AAV, adeno-associated virus; BDNF, brain derived neurotrophic factor.
Introduction
Decades of literature indicate endurance exercise improves cognition and provide neuroprotection against ageing and disease (Kirk-Sanchez & McGough, 2014; Mahalakshmi et al., 2020). Aerobic exercise improves cognition in younger adults (Stern et al., 2019), older adults (Guadagni et al., 2020) and patients with neurodegenerative disease (David et al., 2015; Zhang et al., 2022). Moreover, endurance exercise stimulates production of neurotrophic factors (Cassilhas et al., 2012; Choi et al., 2018), increases long-term potentiation (Dao et al., 2013; van Praag et al., 1999) and enhances adult hippocampal neurogenesis (Choi et al., 2018; van Praag et al., 1999) in both healthy mouse models and models of neurodegeneration. Endurance exercise also reduces oxidative stress, improves mitochondrial function and increases expression of markers for mitochondrial biogenesis in the brain of both healthy and neurodegenerative rodent models (Lau et al., 2011; Marques-Aleixo et al., 2015).
Ketone bodies (ketones), energy substrates synthesized in the liver during exercise (Evans et al., 2017; Johnson et al., 1969), can cross the blood–brain barrier (BBB) (Hasselbalch et al., 1995) and elicit similar neuroprotective effects observed from endurance exercise (Kovács et al., 2021). β-Hydroxybutyrate (BHB) is the most abundant ketone body in mammals (Newman & Verdin, 2017) and is increasingly recognized as a signalling metabolite. Administration of BHB increased mitochondrial ATP production (Tieu et al., 2003), reduced oxidative stress through enhanced complex I-driven mitochondrial respiration (Maalouf et al., 2009), increased adenosine signalling (Kovács et al., 2021) and elevated anti-oxidative genes through inhibition of class I histone deacetylases (HDACs) (Shimazu et al., 2013). Furthermore, BHB is an endogenous ligand for hydroxy-carboxylic acid receptor 2 (HCAR2)- (Taggart et al., 2005) activated microglial protective responses and attenuate amyloid-induced pathology (Moutinho et al., 2022). Further, voluntary wheel running in mice promoted the expression of brain-derived neurotrophic factor (BDNF) and increased glutamatergic transmission at CA3–CA1 through the action of BHB (Sleiman et al., 2016).
The overall goal of the Molecular Transducers of Physical Activity Consortium (MoTrPAC) is to map the multi-omic response to exercise and training across tissues (Sanford et al., 2020). MoTrPAC recently characterized the multi-omic changes across tissues in 6-month-old female and male rats that were endurance exercise trained for 1, 2, 4 and 8 weeks (MoTrPAC, 2024). Furthermore the temporal training response as it relates to mitochondrial analytes (Amar et al., 2024), transcriptomic and epigenetic signatures (Nair et al., 2024), complex trait genetics (Vetr et al., 2024) and subcutaneous white adipose tissue (Many et al., 2024) were characterized all from the same rats. Considering exercise induces ketosis, combined with the overlapping neuroprotective effects of exercise and ketones, the goal of this MoTrPAC study was to determine the role of exercise-induced ketone production on brain health. We hypothesized that loss of hepatic ketone production would attenuate cognitive improvements and neuroprotective adaptations induced by endurance exercise training. Here, we knocked down (KD) the rate-limiting enzyme that catalyses the first reaction in ketogenesis (Cotter et al., 2014; d’Avignon et al., 2018): 3-hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) in the liver of healthy 6-month-old female rats using an adeno-associated virus (AAV) short hairpin RNA (shRNA). Rats either underwent an acute bout of exercise (EX) or 4 weeks of chronic treadmill running (5 days/week) followed by cognitive behavioural testing, high-resolution respirometry and global proteomics to determine the effects of hepatic HMGCS2 knockdown on exercise-mediated neuroprotection.
Methods
Animal husbandry
Inbred female Fischer 344 rats (6 months of age) obtained from the National Institute of Aging (NIA) rodent colony were pair housed on a reverse dark–light cycle, kept at a temperature of 22–26°C, and fed a normal chow diet (Lab Diet 5L79). For chronic exercise studies, females were divided into four groups (n = 9–10): (1) rats that received scramble virus and were sedentary controls (SCR-SED), (2) rats that received HMGCS2 shRNA virus and were sedentary controls (HMGCS2 KD-SED), (3) rats that received scramble virus and chronic exercise (SCR-EX) and (4) rats that received HMGCS2 shRNA virus and chronic exercise (HMGCS2 KD-EX). We also performed a separate acute study in females only, where they were separated into two groups (n = 6): (1) rats that received scramble virus and acute exercise (SCR-EX) and (2) rats that received HMGCS2 shRNA virus and acute exercise (HMGCS2 KD-EX). Tissue collection occurred 1 h after a single exercise bout for acute studies and 48 h after the last bout of training for chronic studies. Prior to being killed, each rat was placed under 2–5% isoflurane [2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-ethane] delivered in oxygen with complete anaesthesia being ensured by assessing toe pinch reaction before bilateral pneumothorax. All animal procedures took place during the dark cycle and were approved by the Institutional Animal Care and Use Committee at the University of Missouri (protocol number – 43056).
Portal vein surgery and recovery
Each rat was placed under 2–2.5% isoflurane delivered in oxygen. Complete anaesthesia was ensured by assessing toe pinch reaction. A single 1- to 1.5-inch incision was made into the skin between the median and sagittal planes on the left side of the rat starting below the ribs and ending above the plane of the fourth inguinal mammary gland teat followed by a second similar 1- to 1.5-inch incision into the peritoneum. The portal vein was located and a 32 gauge needle containing AAV was delivered. The peritoneal lining was sutured and skin was closed using wound clips. A 0.8 ml dose of bupivacaine (2.5 mg/ml) was injected along the incision site for local pain management. One millilitre of sterile saline was injected subcutaneously for hydration support. Rats were given 2.5 mg/kg s.c. banamine for pain management and were monitored daily during post-surgical recovery and began training 7–10 days following surgery.
AAV knockdown of hepatocellular HMGCS2
AAV- shRNA and AAV-scramble virus was purchased from Vector Builder (Chicago, IL, USA). The AAV-shRNA was designed with the U6 promoter, a type III RNA polymerase III promoter commonly used for transcription of shRNA expression (Nie et al., 2010). A vector with an AAV8 serotype has been shown to effectively and stably transfect hepatocytes (Sands, 2011). To centralize virus uptake into the liver, anaesthetized rats received 100 μl of AAV8[shRNA]-EGFP-U6>rHMGCS2 (or AAV8[shRNA]-U6-SCR as a negative control) at a viral dose of 1 × 1012 genomic copies (GC)/ml into the portal vein at an approximate rate of 500 μl/min followed by a 10 s waiting period for the virus to be carried into the liver.
Acute and chronic exercise training protocols
Rats underwent familiarization on a Panlab 5-lane rat treadmill (Model LE8710RTS, Harvard Instruments, Holliston, MA, USA) for 12 days for both acute and chronic exercise protocols that occurred prior to surgery. Acute exercise training consisted of a single 30 min bout of treadmill exercise at 5 degrees (8.7%) grade 30 cm/s. The acute exercise session was equivalent to 70% of of the rats and equates to a session of moderate-intensity exercise in humans. Rats began chronic exercise training at 6 months of age that lasted 4 weeks. Rats were exercised 5 days per week using a progressive protocol designed to maintain an intensity of 70% of [increasing grade and speed, details described previously by the MoTrPAC study group (MoTrPAC, 2024)]. Sedentary rats were exposed to the treadmill but did not receive any training.
Cognitive behavioural testing
Y-maze forced alternation testing was used to measure reference memory similar to others (Batalha et al., 2016). Testing was performed in the final (4th week) of endurance training during the rat’s dark cycle. The Y-shaped maze (Stolting Co., Wood Dale, IL, USA) contains three arms (each of 35 cm length × 10 cm width × 20 cm height) angled at 120°. The Y-maze forced alternation task was performed in two phases. During the first phase (learning phase), the animal explored the maze for 10 min with only two arms opened (start and other arm). The learning phase was repeated three times over 3 days. The following day (24 h later) the animal was re-exposed to the maze for 10 min (test trial) with the novel arm available, and the preference (i.e. number of seconds spent) for the novel arm is considered a measure of reference memory. The number of transitions and distance travelled were used to evaluate motor performance. The maze was cleaned with a 70% ethanol solution between each animal. Rat tracings during the learning task were continuously monitored by an automated tracking system (ANY-maze) and the time spent exploring each arm was quantified. Rats were not trained on the days of the learning phase, but training was resumed on the day of their testing phase following completion of the cognitive testing. Approximately 25% of rats failed to explore the arms in the Y-maze during the testing period and were excluded from analyses.
Gastrocnemius and frontal cortex mitochondrial isolation and respiration
Frontal cortex mitochondria were isolated as previously reported (Kelty et al., 2023; Kerr et al., 2023; Rector et al., 2010). The whole gastrocnemius muscle or frontal cortex sections of the left hemisphere (4 mm thick) were placed in mitochondria isolation buffer, homogenized with a Polytron Ultra-Turrax rotor/stator-type (Duluth, GA, USA) homogenizer (gastrocnemius only) and Teflon pestle, and isolated using a percoll gradient (for frontal cortex only) and a series of centrifugations (7000–96,000 g). Mitochondrial respiration was evaluated using high-resolution respirometry (Oroboros Oxygraph-2k; Oroboros Instruments, Innsbruck, Austria). Basal respiration was measured by loading ~50 μg of gastrocnemius and 150 μg of frontal cortex mitochondria without the addition of substrates. State 2 respiration was stimulated by the addition of malate (2 mM) and glutamate (5 mM), State 3 Complex I by titrated ADP (250—1000 μM ADP), State 3 Complex I and II by succinate (10 mM), and maximally uncoupled with the addition of carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (0.25 μM). Mitochondrial respiration was normalized to protein concentration obtained from a BCA assay kit (Thermo Fisher Scientific #23227, Waltham, MA, USA) as per the manufacturer’s instructions.
Fatty acid oxidation in isolated gastrocnemius mitochondria
Fatty acid oxidation was performed with radio-labelled [1-14C]palmitate (American Radiochemicals, St Louis, MO, USA) in the freshly isolated mixed gastrocnemius mitochondria preparation using previously described methods (Rector et al., 2010). Both 14CO2, representing complete fatty acid oxidation, and 14C-labelled acid-soluble metabolites (ASMs), representing incomplete fatty acid oxidation, were collected in a previously described trapping device, and then counted on a liquid scintillation counter.
Western blotting
As previously described (Rushing et al., 2023), 20 μg of protein lysate from whole liver homogenates or precision plus protein standard (Bio-Rad #1610374, Hercules, CA, USA) were loaded in a criterion gel, separated, transferred, blocked and washed. Blots were imaged using SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific #34096). Protein loading was normalized by amido black staining (Sigma-Aldrich #100563, St Louis, MO, USA). Amido black staining band densitometry was quantified using a ChemiDoc XRS+ System (Bio-Rad). Primary antibodies were incubated overnight at a 1:1000 dilution and included: HGMCS2 (Cell Signaling #40364, Danvers, MA, USA), 3-hydroxy-3-methylglutaryl-CoA lyase (HMGCL) (Cell Signaling #39913), 3-hydroxybutyrate dehydrogenase 1 (BDH1) (Thermo Fischer Scientific #PA556598), 3-oxoacid CoA-transferase 1 (OXCT1) (Thermo Fisher Scientific #PA552899) and acetyl-CoA acetyltransferase 1 (ACAT1) (Cell Signaling #44276).
Protein digest for proteomic and phospho-proteomic analyses
Homogenized frontal cortex sections of the right hemisphere were sent to Gehrke Proteomics Center at the University of Missouri for proteomic processing and analyses. Rat brain tissue was ground with 2.8 mm ceramic beads (OMNI – PerkinElmer, Waltham, MA, USA) in a denatured buffer (comprising 5% SDS, 20 mM Hepes at pH 7.5, and 10 mM DTT). Samples were then heated at 65°C for 20 min and spun at 16,000 g for 20 min to eliminate tissue debris. The samples were alkylated with 30 mM iodoacetamide (IAA) at room temperature for 30 min in the dark. The proteins were subsequently precipitated with cold acetone at −20°C overnight.
The protein pellets were retrieved by centrifugation at 16,100 g for 20 min at 4°C and washed twice with 80% acetone. The final protein pellets were suspended in 150 μl of 6 M urea, 2 M thiourea and 100 mM ammonium bicarbonate. The protein concentration was determined using the Pierce 660 nm Protein Assay (Thermo Fisher Scientific) following the manufacturer’s protocol. Proteins (500 μg) were digested with Trypsin (Thermo Fischer Scientific) at a ratio of 1:75 (enzyme:substrate) overnight and then digested again with Trypsine (Thermo) at a ratio of 1:200 for another 3 h. The digestion was stopped with 0.5% TFA at a final concentration. Digested peptides (500 ng) were used for total protein quantification. Digested peptides (500 ng) were used for total protein quantification, and the rest of the digested peptides were used for phospho-proteomics. The digested peptides were purified using Sep-Pak C18 1 ml Vac Cartridge according to the manufacturer’s protocol (Waters, Milford, MA, USA). The desalted peptides were then enriched using the Fe-NTA Phosphopeptide Enrichment Kit (Thermo) in accordance with the manufacturer’s instructions. The dried phosphopeptides were then dissolved in 35 μl of 0.1% formic acid in water and 8 μl of the sample was injected into an LCMS device.
Proteomic and phospho-proteomics MS
An EvoSep One liquid chromatography system was used for all proteomic analyses. The samples were processed with a 44 min gradient for peptide elution (or Evosep One 30 SPD program) and went through a 15 cm × 150 μm ID column with 1.5 μm C18 beads (Bruker PepSep) and a 20 μm ID zero dead volume electrospray emitter (Bruker Daltonik, Bremen, Germany). Mobile phases A and B consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile (ACN), respectively. The Evosep One was coupled online to a modified trapped ion mobility spectrometry quadrupole time-of-flight mass spectrometer (timsTOF Pro 2, Bruker Daltonik) via a nanoelectrospray ion source (Captive spray, Bruker Daltonik).
To conduct data-independent acquisition (DIA-PASEF), the timsTOF Pro2 was operated in long gradient DIA-PASEF mode. Sixteen scans with narrow 25 m/z isolation windows (resulting in 32 windows) covered an m/z range of 400–1200 and an ion mobility range of 0.6–1.43 Vs/cm2. The duty cycle was maintained at 100%. This setup results in a total cycle time of 1.8 s (1 × 100 ms MS1 survey scan, 16 × 100 ms dia-PASEF scans).
Proteomic and phospho-proteomic data analysis
The DIA-PASEF raw data were analysed using the directDIA workflow of Spectronaut version 18.5 with default settings: trypsin digestion set to 2 allowed missed cleavages, cysteine carbamidomethylation as a fixed modification, methionine oxidation, and acetylation at the protein N-terminus as variable modifications, and phosphorylation at STY for phospho-proteomics data. The phospho-proteomics data were exported from Spectronaut for further analysis using Perseus version 1.6.15.0. All searches were conducted against the rat database (Rattus norvegicus Uniprot #UP000002494). For statistical analysis, the proteome was analysed using Spectronaut with post-analysis based on both MS levels. Proteome candidates were filtered based on a Q-value of 0.05. Protein clustering and enrichment was performed with ingenuity pathway analysis (IPA) software (Qiagen, Germantown, MD, USA).
Quantification of ketone bodies in serum
Plasma ketone concentration of acetoacetate (AcAc) and BHB were determined in each experiment (n = 6 per group). Diluted serum samples (5 μl) were extracted by the addition of 20 μl of cold ACN:methanol (1:1, v/v) containing known concentrations of internal standards Sodium DL-3-hydroxybutyrate [3,4,4,4-D4]. β-Hydroxybutyric acid (βOHB) and [U-13C4]AcAc (concentration of each internal standards is 50 μM). Serum samples were vortexed, and centrifuged at 4°C, 15,000 g for 10 min. Supernatant was analysed using the UPLC-MS/MS quantification analysis setup described below. Analysis was performed using a Vanquish LC system using a Cortecs UPLC T3 column (100 × 2.1 mm 1.6 μm). For all analyses, the following mobile phases were used: (A) 98% water/2% methanol with 0.0125% acetic acid (AA) and (B) 60% water/40% methanol with 0.0125% AA. Samples were separated using the following binary gradient: 0–20% B for 3 min, 20–90% B for 0.5 min, 90% B for 1.5 min, 90–0% B for 0.5 min and 0% B for 2 min at 0.3 ml/min. The column was maintained at 30°C and the injection volume was 0.5 μl. The LC system was linked to a Thermo Q Exactive Plus MS equipped with heated electrospray ionization (ESI) source. The MS system was operated in PRM modes using negative ionization mode with appropriate inclusion list (see Table 1). Resolution and automatic gain control (AGC) targets were set to 70,000 and 1e5, respectively, while the isolation window was set to m/z 1.0 and collision energy 30 (arbitrary units). Common ESI parameters were the same in both approaches: auxiliary gas: 10, sheath gas flow 35, sweep gas 1, spray voltage 3 kV, capillary temperature 275°C, S-lens 50 and auxiliary gas temperature 150°C.
Table 1.
Optimized UPLC-MS/MS parameters for analytes
| Negative mode | ||||
|---|---|---|---|---|
| Analyte | RT (min) | (m/z) | (N)CE | PRM transition with formula |
| β-Hydroxybutyrate (BHB) | 2.6 | 103.0401 | 30 | 59.0133(C2H3O2) |
| Acetoacetate (AcAc) | 1.8 | 101.0244 | 30 | 57.0340(C3H5O) |
| [U-13C4]AcAc (I.S.) | 1.8 | 105.0378 | 30 | 60.0441(13C3H5O) |
| [3,4,4,4-D4]BHB (I.S.) | 2.6 | 107.0646 | 30 | 59.0133(C2H3O2) |
RT, retention time (min) on UPLC Cortecs column using optimal chromatographic separation; N(CE), normalized collision energy; PRM, parallel reaction monitoring; I.S., internal standard.
PC12 cell culture and differentiation
PC12 (pheochromocytoma) cells were purchase from American Type Culture Collection and cultured and differentiated according to the manufacturer’s recommendations, and further optimization guided by our previously established methodology (Grigsby et al., 2019). Prior to differentiation, PC12 cells were seeded on collagen-coated plates [0.01% collagen stock solution (Gibco #A1048301) that contained 0.1 M acetic acid solution] in RPMI-1640 (ATCC, #1187 093), supplemented with 10% inactivated horse serum (Sigma, #16050130) and 5% fetal bovine serum (Sigma #F2442) at 37°C in a humidified atmosphere (5% CO2, 95% air). Medium was changed every 2–3 days and cells were plated at ~25% confluency. Differentiation of PC12 cells was initiated 1 day after cells reached 40% confluency with Opti-MEM supplemented with 50 ng/ml of nerve growth factor (Gibco #13257019) and 0.5% fetal bovine serum until transfection, which occurred 8 days later.
Small interfering RNA transfection of PC12 cells
Non-differentiated PC12 cells were transfected at ~40% confluency with Silencer Select small interfering (si)RNAs (Thermo Fisher Scientific) targeting OXCT1 (#4390771) or scramble negative control (#430843) according to the manufacturer’s instructions. At a ratio of 0.875:1, lipofectamine RNAi Max (#13778, Thermo Fischer Scientific) was complexed to siRNAs (200 nM) in the appropriate volume of Opti-MEM (Thermo Fischer Scientific, #31985070) for 30 min. PC12 cells were resuspended in growth media without antibiotics with the appropriate siRNA–lipofectamine complex in Opti-mem. Following a 24 h incubation to allow cell transfection, medium was replaced with differentiation media. All siRNA experiments were conducted 5 days later, after the PC12 cells had differentiated.
RNA isolation, cDNA synthesis and qRT-PCR
As described previously (Kelty et al., 2023), RNA was extracted from PC12 cells using an Rneasy kit as per the manufacturer’s instructions (Qiagen #74104) and a cDNA library was synthesized (Promega #A3802. Madison, WI, USA). A Nanodrop spectrometer was used to ensure RNA and cDNA purity. Quantitative real-time PCR (qRT-PRC) was performed in triplicate using SYBR Green reagents (Bio-Rad Laboratories #1725121) and constructed primer pairs. Data were normalized to peptidyl-prolyl cis-trans isomerase (PPIB) using the 2−ΔΔCT method. Forward and reverse primers are listed in Table 2.
Table 2.
PCR primer sequences
| Primer | Forward sequence | Reverse sequence |
|---|---|---|
| PPIB | 5′-CTTAGCTACAGGAGAGAAAGG | 5′-TTCAGCTTGAAGTTCTCATC |
| OXCT1 | 5′-AACACTCCGCAAAGGGGAAT | 5′-TCCACGTCAAACACACCCTT |
Measurements of respiration
Cellular oxygen consumption in differentiated PC12 cells was assessed using a Seahorse XF96 analyser (Agilent, Santa Clara, CA, USA) as previously described (Cunningham et al., 2021). Briefly, cells were plated in seeding medium for 8 days in collagen-coated 96-well XF96 cell culture microplates. Cells were washed and incubated for 1 h at 37°C without CO2 in serum-free, phenol red-free DMEM (Agilent #103575) containing glucose (10 mM), pyruvate (1 mM) and glutamine (2 mM) at a pH of 7.4. Oligomycin (1.0 μM), FCCP (1.5 μM) and rotenone with antimycin A (0.5 μM) were loaded into ports A–C, respectively, and delivered to the cells in that order. The basal oxygen consumption rate (OCR) was measured three times in 3 min intervals before the initial port injection and after each of the port injections. The medium was mixed before each measurement. Each sample was measured in duplicate. Basal mitochondrial respiration was determined by taking OCR before oligomycin injection minus OCR after rotenone with antimycin A. Maximal mitochondrial respiration was determined by taking OCR after FCCP injection minus OCR after rotenone with antimycin A. Spare capacity was determine by taking OCR after FCCP injection minus OCR before oligomycin injection. Non-mitochondrial respiration was determined by the OCR after rotenone with antimycin A.
Statistical analysis
Statistical analyses were performed with GraphPad Prism version 10.1.2 (Prism, La Jolla, CA, USA) with an α level of P < 0.05. Two-way analysis of variance (ANOVA) was used for all chronic datasets. A one-way ANOVA was used to determine differences in ketone bodies in acute datasets. A Brown–Forsythe ANOVA test was used to determine if a significant difference among standard deviation exists between groups. A Welch’s ANOVA test was used to determine significance between means if a significant difference among standard deviations existed between groups. An unpaired two-tailed Student’s t test was used to determine HMGCS2 knockdown in acute datasets and for in vitro studies. A Holm–Šidák test (one-way ANOVA) or Fisher least significant difference test (two-way ANOVA) was applied post hoc for pairwise multiple comparison procedures. Where possible, individual data are presented, and mean data are expressed as means ± SD). Samples 2SD from the mean were excluded based on our a priori exclusion criteria. Proteomic statistical analyses are described above.
Results
Hepatic HMGCS2 knockdown reduced circulating ketones
Hepatic HMGCS2 protein expression was significantly reduced (unpaired t test, P < 0.0001; Fig. 1A) 1 h after an acute bout of exercise (5 weeks after AAV injection) in the female HMGCS2 KD-EX (71.00 ± 148.6) compared to SCR-EX (2034 ± 586.9). There was a main effect for plasma AcAc (one-way ANOVA, P < 0.0001; Fig. 1B), BHB (Welch ANOVA, P < 0.0001; Fig. 1B) and TKB (one-way ANOVA, P < 0.0001; Fig. 1B), where exercise significantly elevated plasma ketones 1 h after an acute bout of exercise (AcAc, P < 0.0001; BHB, P = 0.0002; and TKB P < 0.0001) in SCR-EX (AcAc, 170.2 ± 40.65; BHB, 930.9 ± 196.2; and TKB, 1101 ± 231.5) compared with SCR-SED (AcAc, 39.68 ± 9.840; BHB, 174.1 ± 25.33; and TKB, 211.3 ± 30.00). Knockdown of HMGCS2 significantly attenuated plasma ketone concentration 1 h after an acute bout of exercise (AcAc, P < 0.0311; BHB, P = 0.1556; and TKB, P < 0.0712) in HMGCS2 KD-EX (AcAc, 120.0 ± 47.55; BHB, 699.8 ± 305.8; and TKB, 819.9 ± 346.4) compared with SCR-EX (AcAc, 170.2 ± 40.65; BHB, 930.9 ± 196.2; and TKB, 1101 ± 231.5).
Figure 1. Hepatic HMGCS2 knockdown reduced circulating ketones after acute and chronic exercise.
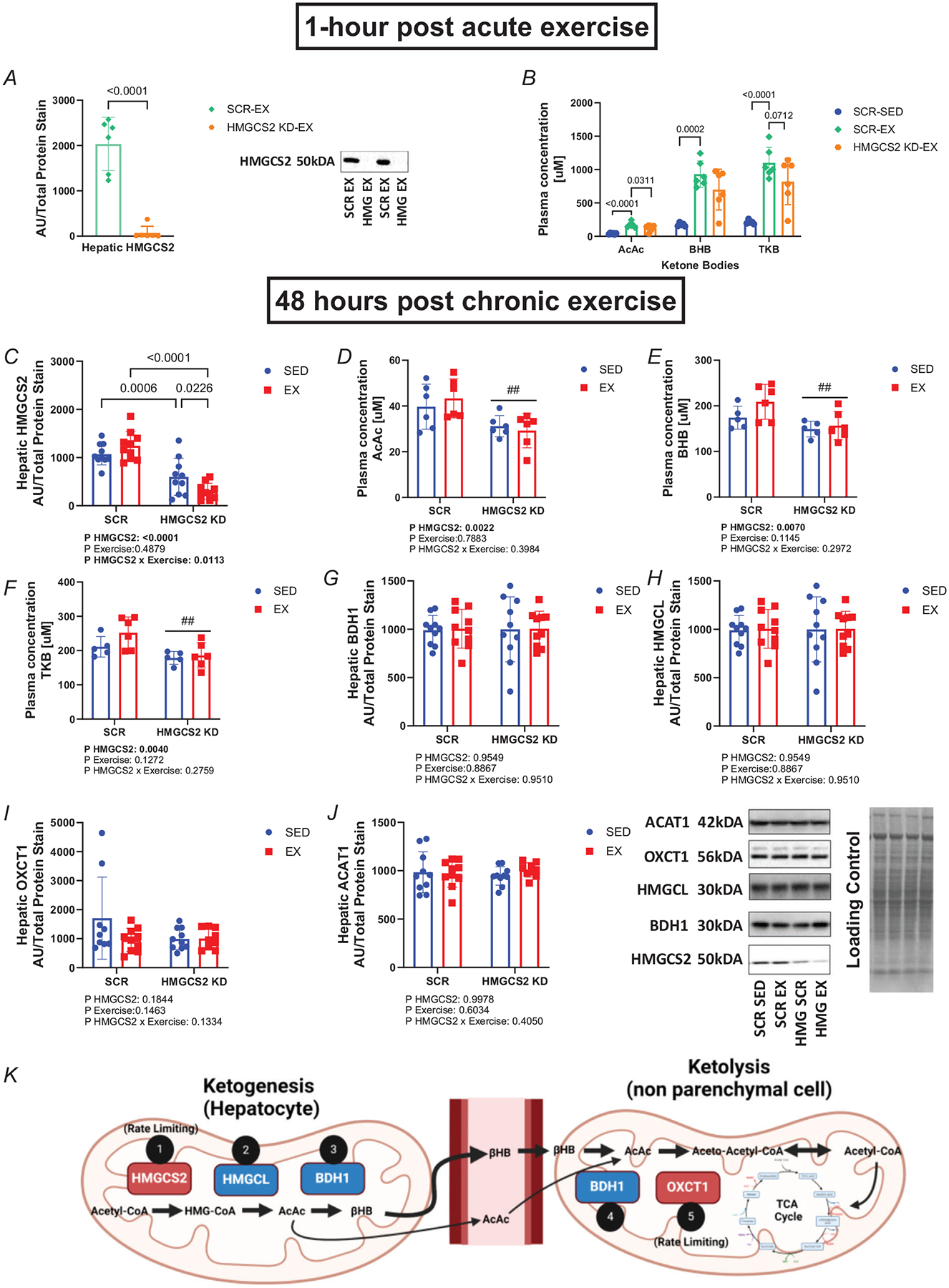
A, hepatic HMGCS2 protein expression normalized to total protein stain. B, AcAc, BHB and TKB plasma concentration (μM) 1 h after an acute bout of exercise (n = 6). Exercise groups were compared to SCR-SED values from chronic training group measurements. C, hepatic HMGCS2 protein expression normalized to total protein stain (n = 10). D–F, AcAc (D), BHB (E) and TKB (F) plasma concentration (μM) 48 h after the last bout of exercise following chronic exercise training (5 weeks after AAV injection) (n = 5–6). G–J, 3-hydroxybutyrate dehydrogenase 1 (BDH1) (G), 3-hydroxymethyl-3-methylglutaryl-CoA, lyase (HMGCL) (H), succinyl-CoA:3-ketoacid CoA transferase (SCOT) (I) and acetyl-CoA acetyltransferase 1 (ACAT1) (J) protein expression in the liver 48 h after the last bout of exercise during chronic exercise training (5 weeks after AAV injection) (n = 9–10). Representative image of western blots and their respective weight (kDA) are shown. K, schematic of ketogenesis and ketolysis pathway and key enzymes involved. Image generated in BioRender. Main effect of HMGCS2 KD represented by ##P ≤ 0.01. Values are presented as mean ± SE. EX, exercise. KD, knockdown. Values presented as mean ± SD. Arbitrary units, AU; acetoacetate (AcAc); β-hydroxybutyrate (BHB); total ketone bodies (TKB).
Analysis of hepatic HMGCS2 protein expression 48 h after the last bout of exercise during chronic exercise training (5 weeks after AAV injection) revealed an interaction between HMGCS2 knockdown and exercise (two-way ANOVA, P = 0.0113; Fig. 1C) and a main effect of HMGCS2 knockdown (two-way ANOVA, P < 0.0001; Fig. 1C). Post hoc analysis revealed HMGCS2 knockdown significantly reduced HMGCS2 protein (P = 0.0006) in HMGCS2 KD-SED (599.7 ± 385.2) compared with SCR-SED (1072 ± 222.5) and significantly reduced HMGCS2 protein (P < 0.0001) in HMGCS2 KD-EX (301.3 ± 165.9) compared with SCR-EX (1246 ± 297.4). Analysis of plasma ketone concentration 48 h after the last bout of exercise during chronic exercise (5 weeks after AAV injection) revealed a main effect of HMGCS2 knockdown for AcAc (two-way ANOVA, P = 0.022; Fig. 1D), BHB (two-way ANOVA, P = 0.0070; Fig. 1E) and TKB (two-way ANOVA, P = 0.0040, Fig. 1F). Neither HMGCS2 knockdown nor exercise altered other enzymes in the ketogenesis and ketolysis pathways (two-way ANOVA, P > 0.05; Fig. 1G–J). Figure 1K displays the steps in these ketogenesis and ketolysis pathways.
Hepatic HMGCS2 knockdown increased markers of mitochondrial dysfunction 1 h after an acute bout of exercise
Proteomic analysis of frontal cortex between HMGCS2 KD-EX and SCR-EX identified 7303 unique proteins that were further filtered (Q-value < 0.05) yielding 2645 differentially expressed proteins (DEPs) displayed in a volcano plot (Fig. 2A). Among other conical pathways, IPA enrichment of these DEPs revealed a significant upregulation (z-score > 2) in mitochondrial dysfunction (z-score = 4.134; Fig. 2B) and autophagy (z-score = 2.832; Fig. 2B) in HMGCS2 KD-EX compared with SCR-EX 1 h after an acute bout of exercise in female rats. Furthermore, IPA enrichment revealed a significant downregulation (z-score < −2) in electron transport, ATP synthesis and heat production by uncoupling proteins (z-score = −2.967; Fig. 2B), oxidative phosphorylation (z-score = −2.785; Fig. 2B) and the citric acid (TCA) cycle and respiratory electron transport (z-score = −3.13; Fig. 2B). The −log(P-values) and z-scores for all the conical pathways displayed in Fig. 2B can also be found in Table 3. Predictive analysis revealed a significant downregulation for the top-stream regulator PPARGC1A (z-score < −2; Fig. 2C) based on inputted DEP clustering. Specific DEPs related to oxidative phosphorylation are displayed in Fig. 2D. The log2 fold change expression and Q-value for Fig. 2C and D can be found in Tables 4 and 5, respectively.
Figure 2. Hepatic HMGCS2 knockdown increases markers of mitochondrial dysfunction 1 h after an acute bout of exercise.

A, volcano plot for comparison between HMGCS2-EX and SCR-EX (n = 6). Red dots represent DEPs (Q-value < 0.05). B, top upregulated and downregulated canonical pathways from DEPs (n = 6). C, predicted upstream regulators from protein clustering analysis. Log2-fold change of DEPs used for predictive calculation are shown below each protein. D, image of proteins from each mitochondrial complex used for calculation of z-score in the oxidative phosphorylation canonical pathway. Orange indicates a positive z-score in the numerator and blue a negative z-score (absolute z-score = 2 for significance). IPA graphical figure key is shown to the right of each panel. Differentially expressed protein, DEP.
Table 3.
Canonical pathways altered in the frontal cortex of HMGCS2 KD-EX compared to SCR-EX 1 h after an acute bout of exercise.
| Ingenuity canonical pathways | −log(P-value) | z-score |
|---|---|---|
| Mitochondrial dysfunction | 18.5 | 4.134 |
| EIF2 signalling | 13.8 | 2.137 |
| Endocannabinoid neuronal synapse pathway | 13.5 | 2.03 |
| Ephrin B signalling | 12.8 | 2.132 |
| Electron transport, ATP synthesis and heat production by uncoupling proteins | 11.6 | −2.967 |
| Amyloid processing | 10.2 | 2.121 |
| MHC class II antigen presentation | 9.31 | 2.466 |
| Eukaryotic translation initiation | 7.97 | 3.43 |
| GABA receptor activation | 7.74 | 2.558 |
| Oxidative phosphorylation | 7.25 | −2.785 |
| The citric acid (TCA) cycle and respiratory electron transport | 7.02 | −3.13 |
| Autophagy | 6.91 | 2.832 |
Table 4.
DEPs identified for predicted upstream regulator: PPARGC1A in the frontal cortex of HMGCS2 KD-EX compared with SCR-EX 1 h after an acute bout of exercise.
| Symbol | Protein name/description | Log2(fold change) | False discovery rate (q-value) |
|---|---|---|---|
| ATP5F1B | ATP synthase F1 subunit beta | −0.0904 | 0.0001 |
| CACNA1H | Calcium voltage-gated channel subunit alpha1 H | 0.6431 | 0.0367 |
| CACNA2D2 | Calcium voltage-gated channel auxiliary subunit alpha2delta 2 | 0.4035 | 0.0179 |
| COX7B | Cytochrome c oxidase subunit 7B | 0.4005 | 0.0287 |
| DLAT | Dihydrolipoamide S-acetyltransferase | −0.0171 | 0.0055 |
| MAPT | Microtubule-associated protein tau | 0.4641 | 0.0179 |
| MFN2 | Mitofusin 2 | −0.0300 | 0.0010 |
| NDUFV1 | NADH:ubiquinone oxidoreductase core subunit V1 | −0.0093 | 0.0184 |
| NDUFV2 | NADH:ubiquinone oxidoreductase core subunit V2 | 0.0611 | 0.0370 |
| PRKAA2 | Protein kinase AMP-activated catalytic subunit alpha 2 | 0.0750 | 0.0075 |
| SOD2 | Superoxide dismutase 2 | −0.0374 | 0.0219 |
Table 5.
DEPs identified for oxidative phosphorylation canonical pathway in the frontal cortex of HMGCS2 KD-EX compared with SCR-EX 1 h after an acute bout of exercise
| Symbol | Protein name/cescription | Log2(fold change) | False discovery rate (q-value) |
|---|---|---|---|
| ATP5A1 | ATP synthase F1 subunit alpha | −0.0444 | 0.0421 |
| ATP5B | ATP synthase F1 subunit beta | −0.0904 | 0.0001 |
| ATP5H | ATP synthase peripheral stalk subunit d | −0.0346 | 0.0303 |
| ATP5J | ATP synthase peripheral stalk subunit F6 | 0.2185 | 0.0170 |
| COX4I1 | Cytochrome c oxidase subunit 4I1 | −0.0470 | 0.0148 |
| COX7B | Cytochrome c oxidase subunit 7B | 0.4005 | 0.0287 |
| NDUFA1 | NADH:ubiquinone oxidoreductase subunit A1 | 0.1737 | 0.0087 |
| NDUFA10 | NADH:ubiquinone oxidoreductase subunit A10 | −0.0903 | 0.0129 |
| NDUFA11 | NADH:ubiquinone oxidoreductase subunit A11 | −0.1815 | 0.0001 |
| NDUFA12 | NADH:ubiquinone oxidoreductase subunit A12 | −0.0930 | 0.0022 |
| NDUFA13 | NADH:ubiquinone oxidoreductase subunit A13 | −0.1300 | 0.0037 |
| NDUFA5 | NADH:ubiquinone oxidoreductase subunit A5 | −0.0477 | 0.0060 |
| NDUFA6 | NADH:ubiquinone oxidoreductase subunit A6 | −0.0101 | 0.0149 |
| NDUFAB1 | NADH:ubiquinone oxidoreductase subunit AB1 | −0.2031 | 0.0000 |
| NDUFB10 | NADH:ubiquinone oxidoreductase subunit B10 | −0.0502 | 0.0027 |
| NDUFB4 | NADH:ubiquinone oxidoreductase subunit B4 | −0.4149 | 0.0003 |
| NDUFB6 | NADH:ubiquinone oxidoreductase subunit B6 | −0.1170 | 0.0037 |
| NDUFB7 | NADH:ubiquinone oxidoreductase subunit B7 | −0.0240 | 0.0321 |
| NDUFS2 | NADH:ubiquinone oxidoreductase core subunit S2 | −0.0208 | 0.0137 |
| NDUFS7 | NADH:ubiquinone oxidoreductase core subunit S7 | −0.0643 | 0.0020 |
| NDUFS8 | NADH:ubiquinone oxidoreductase core subunit S8 | 0.1214 | 0.0207 |
| NDUFV1 | NADH:ubiquinone oxidoreductase core subunit V1 | −0.0093 | 0.0184 |
| NDUFV2 | NADH:ubiquinone oxidoreductase core subunit V2 | 0.0611 | 0.0370 |
| NDUFV3 | NADH:ubiquinone oxidoreductase subunit V3 | −0.0845 | 0.0009 |
| SDHB | Succinate dehydrogenase complex iron sulphur subunit B | −0.0321 | 0.0345 |
Four weeks of treadmill running conferred skeletal muscle endurance exercise adaptations
Analysis of mitochondrial state respiration 48 h after the last bout of exercise during chronic exercise training (5 weeks after AAV injection) revealed a main effect of exercise for basal (two-way ANOVA, P = 0.0316; Fig. 3A), state 2 (two-way ANOVA, P = 0.0076; Fig. 3B), state 3 complex I (two-way ANOVA, P = 0.0016; Fig. 3C), state 3 complex I and II (two-way ANOVA, P = 0.0012; Fig. 3D) and uncoupled respiration (two-way ANOVA, P = 0.0089; Fig. 3E) in skeletal muscle of female rats.
Figure 3. Four weeks of treadmill running confers mitochondrial endurance exercise adaptations in the gastrocnemius.

A–E, basal (A), state 2 (B), state 3 Complex I (C), state 3 Complex I + II (D) and uncoupled respiration (E) in mitochondrial isolates from the gastrocnemius muscle 48 h after the last bout of exercise during chronic exercise training (5 weeks after AAV injection). O2 flux normalized to total protein (n = 8–9). F–H, complete (F), incomplete (G) and total (H) [1-14C] palmitate oxidation in mitochondrial isolates from the gastrocnemius muscle 48 h after the last bout of exercise during chronic exercise training (5 weeks after AAV injection). Palmitate oxidation normalized to total protein (n = 8–9). Main effect of exercise represented by *P ≤ 0.05 or **P ≤ 0.01. Values presented as mean ± SD. Oxygen, (O2).
Analysis of mitochondrial palmitate oxidation 48 h after the last bout of exercise during chronic exercise training (5 weeks after AAV injection) revealed a main effect of exercise for complete palmitate oxidation (two-way ANOVA, P < 0.0001; Fig. 3F), incomplete palmitate oxidation (two-way ANOVA, P = 0.0434; Fig. 3G) and total palmitate oxidation (two-way ANOVA, P = 0.0035; Fig. 3H) in female rats. Post hoc analysis revealed exercise significantly increased complete palmitate oxidation (P < 0.0001) in HMGCS2 KD-EX (1326 ± 242.9) compared with HMGCS2 KD-SED (738.6 ± 218.5). No significant main effects for HMGCS2 KD or interactions were noted for other outcome measures in the gastrocnemius muscle.
Hepatic HMGCS2 knockdown (in vivo) or neuronal OXCT1 (in vitro) impairs mitochondrial function
Analysis of frontal cortex mitochondrial state respiration revealed significant reductions in state 3 complex I + II respiration with HMGCS3 KD (main effect from two-way ANOVA, P = 0.0199; Fig. 4D), with trending decreases also seen in state 3 complex I and maximal uncoupled respiration (P = 0.10 in both cases; Fig 4C and 4E). However, no significant main effects were seen following 4 weeks of endurance exercise training in female rats 48 h after the last bout of exercise for any of these mitochondrial respiratory states.
Figure 4. Hepatic HMGCS2 knockdown (in vivo) or neuronal OXCT1 (in vitro) impairs mitochondrial function.
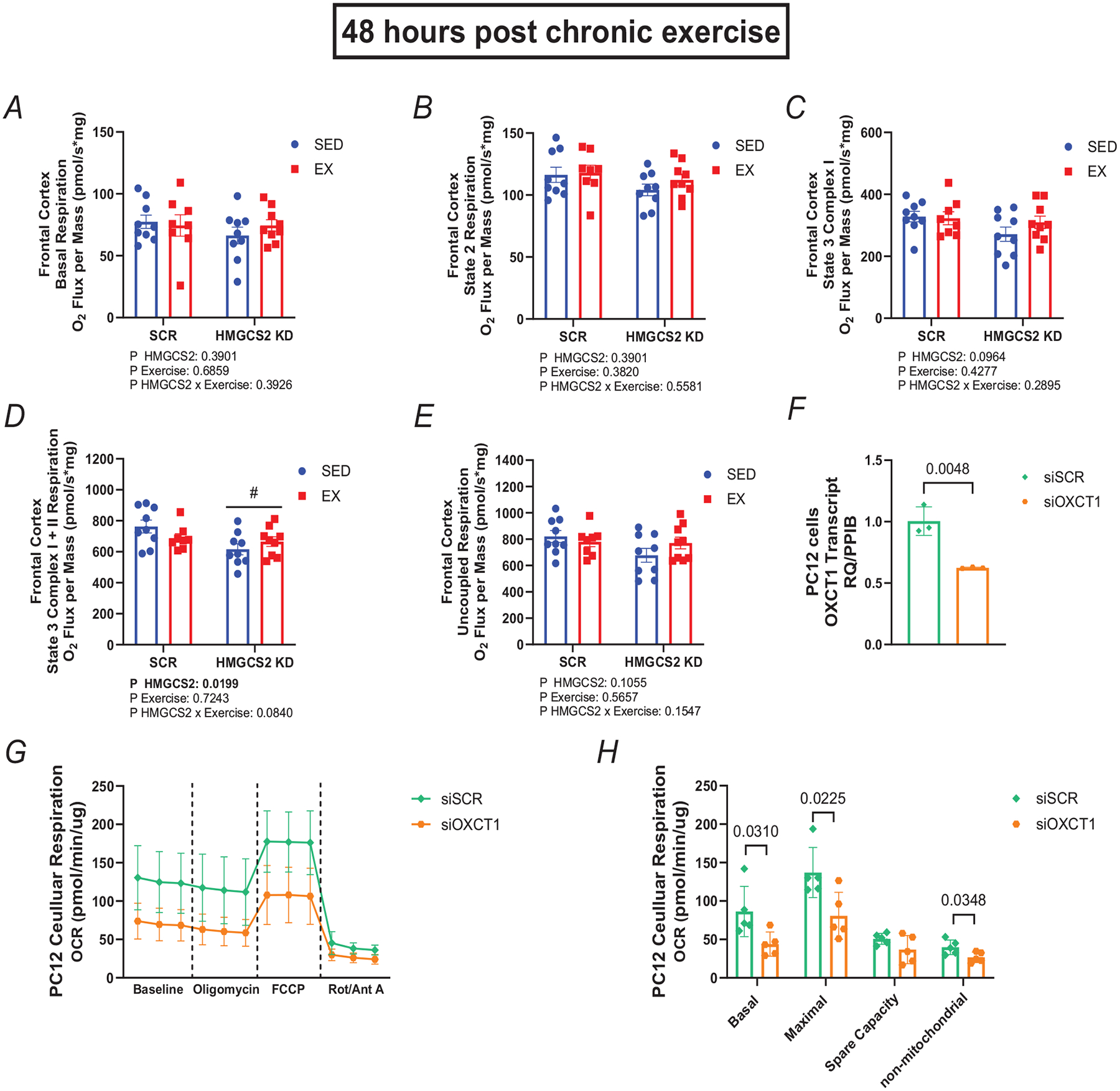
A–E, basal (A), state 2 (B), state 3 Complex I (C), state 3 Complex I + II (D) and maximal uncoupled respiration (E) in mitochondrial isolates from the frontal cortex 48 h after the last bout of exercise during chronic exercise training (5 weeks after AAV injection). O2 flux normalized to total protein (n = 8–9). F, transcript expression of OXCT1 normalized to PPIB in PC12 cells (n = 3). G, PC12 cell OCR at baseline, after oligomycin (1.0 μM), carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) (1.5 μM) and rotenone with antimycin A (0.5 μM). OCR was measured three times in 3 min intervals before the initial port injection and after each port injection and normalized to total protein (n = 5). H, average OCR across three time points in PC12 cells (n = 5). Basal mitochondrial respiration was determined by taking OCR before oligomycin injection minus OCR after rotenone with antimycin A. Maximal mitochondrial respiration was determined by taking OCR after FCCP injection minus OCR after rotenone with antimycin A. Spare capacity was determined by taking OCR after FCCP injection minus OCR before oligomycin injection. Non-mitochondrial respiration was determined by the OCR after rotenone with antimycin A. Main effect of HMGCS2 KD represented by #P ≤ 0.05. Values presented as mean ± SD. OCR, oxygen consumption rate; oxygen, O2; peptidyl-prolyl cis-trans isomerase, PPIB; RQ, relevant quotient.
Targeted OXCT1 knockdown with siRNAs in immortalized rat neuronal-like cells (PC12s) significantly reduced OXCT1 mRNA expression (unpaired t test, P = 0048; P = 0048; Fig. 4F) in siOXCT1 (0.6240 ± 0.0056) compared with siSCR control (1.004 ± 0.1164). Analysis of oxygen consumption rate in immortalized rat neuronal-like cells (PC12s) with (siOXCT1) or without OXCT1 (siSCR) revealed a significant reduction in basal respiration (unpaired t test, P = 0.0310; Fig. 1G and H) in siOXCT1 (43.89 ± 15.72) compared with siSCR control (86.29 ± 32.72), a significant reduction in maximal uncoupled respiration (unpaired t test, P = 0.0225; Fig. 4G and H) in siOXCT1 (80.71 ± 30.55) compared with siSCR control (137.1 ± 32.63) and a significant reduction in non-mitochondrial respiration (unpaired t test, P = 0.0348; Fig. 4G and H) in siOXCT1 (26.68 ± 6.685) compared with siSCR control (39.86 ± 9.499).
Endurance exercise ameliorated cognitive impairment induced by hepatic knockdown of HMGCS2
Proteomic analysis identified 7285 unique proteins between SCR-EX and SCR-SED, 7268 proteins between HMGCS2 KD-SED and SCR-SED, 7825 proteins between HMGCS2 KD-EX and HMGCS2 KD-SED, and 7286 proteins between HMGCS2 KD-EX and SCR-EX. These were further filtered (Q-value < 0.05) yielding 3676 DEPs displayed in a volcano plot between SCR-EX and SCR-SED (Fig. 5A), 2590 DEPs between HMGCS2 KD-SED and SCR-SED (Fig. 5B), 1995 DEPs between HMGCS2 KD-EX and HMGCS2 KD-SED (Fig. 5C), and 3476 DEPs between HMGCS2 KD-EX and SCR-EX (Fig. 5D).
Figure 5. Endurance exercise ameliorates cognitive impairment induced by hepatic knockdown of HMGCS2.

A–D, volcano plots for comparison between SCR-EX and SCR-SED (A), HMGCS2 KD-SED and SCR-SED (B), HMGCS2 KD-EX and HMGCS2 KD-SED (C) and HMGCS2 KD-EX and SCR-EX (D) 48 h after the last bout of exercise during chronic exercise training (5 weeks after AAV injection) (n = 6). E, time spent in the novel arm (seconds) during the testing phase (n = 7–8). Testing was performed in the final (4th) week of endurance training with any animal that did not leave the starting arm excluded from analyses. Representative tracings of each group are shown to the right of the graph. F, BDNF protein expression in the frontal cortex expressed as raw intensity (n = 6). G and H, top upregulated and downregulated proteomic (G) and phospho-proteomic (H) canonical pathways related to synaptic plasticity in HMGCS2 KD-SED compared with SCR-SED (n = 6). I and J, top upregulated and downregulated canonical pathways related to synaptic plasticity in HMGCS2 KD-EX compared to HMGCS2 KD-SED (I), and HMGCS2-KD vs. SCR- EX (J) (n = 6). Orange indicates a positive z-score in the numerator and blue a negative z-score (absolute z-score = 2 for significance). Z-score generated in IPA. Main effect of HMGCS2 KD represented by #P ≤ 0.05. Values presented as mean ± SD. Differentially expressed protein, DEP.
Analysis of cognition during the final week revealed HMGCS2 knockdown significantly reduced time spent in the novel arm indicative of reduced cognition/reference memory (main effect in two-way ANOVA, P = 0.0204; Fig. 5E), driven by the significant reduction in cognition witnessed in the HMGCS2-KD SED vs. SCR-SED controls (P = 0.0065). This impairment in cognition induced by HMGCS2 knockdown in the sedentary female rats was completely prevented with endurance exercise training (Fig. 5E).
Analysis of BDNF protein raw intensity from proteomics revealed a significant interaction between HMGCS2 knockdown and exercise (two-way ANOVA, P = 0.0010; Fig. 5F). Post hoc analysis revealed a significant decrease (P = 0.0432) in BDNF in the frontal cortex in the SCR-EX (6.655 ± 0.121) compared with SCR-SED (6.996 ± 0.313) and a significant decrease (P < 0.0001) in SCR-EX compared with HMGCS2-SED group (6.136 ± 0.289). There was also a significant increase (P = 0.0036) in BDNF in the frontal cortex in the HMGCS2 KD-EX group (6.655 ± 0.320) compared with the HMGCS2 KD-SED group.
DEPs from the frontal cortex of HMGCS2 KD-SED and SCR-SED rats were inputted into IPA for enrichment and revealed a significant downregulation (z-score < −2) in synaptic long-term potentiation (z-score = −2.921; Fig. 5G), synaptic adhesion-like molecules (z-score = −3.000; Fig. 5G) and synaptic long-term depression (z-score = −2.910; Fig. 5G) in HMGCS2 KD-SED compared with SCR-SED. Differentially expressed phosphorylation sites obtained were inputted into IPA for enrichment and revealed a significant downregulation (z-score < −2) in development of neurons (z = 2.01; Fig. 5H), memory (z = 2.10; Fig. 5H) and neuritogenesis (z = −2.15; Fig. 5H) in HMGCS2 KD-SED compared with SCR-SED in female rats. Additionally, DEPs from the frontal cortex of HMGCS2 KD-EX and HMGCS2 KD-SED were inputted into IPA for enrichment and revealed a significant upregulation (z-score > 2) in synaptogenesis signalling pathway (z-score = 5.115; Fig. 5I), synaptic long-term depression (z-score = 4.352; Fig. 5I), synaptic long-term potentiation (z-score = 4.271; Fig. 5I), activation of NMDA receptors and postsynaptic events (z-score = 3.300; Fig. 5I), glutamate binding, activation of AMPA receptors and synaptic plasticity (z-score = 2.309; Fig. 5I) in HMGCS2 KD-EX compared with HMGCS2 KD-SED. No significant phosphorylation sites were identified by phospho-proteomics in HMGCS2 KD-EX compared with HMGCS2 KD-SED.
DEPs from the frontal cortex of HMGCS2 KD-EX and SCR-EX 48 h were also inputted into IPA for enrichment and revealed a significant upregulation (z-score > 2) in synaptogenesis signalling pathway (z-score = 9.192; Fig. 5J), synaptic long-term depression (z-score = 6.197; Fig. 5J), synaptic long-term potentiation (z-score = 5.477; Fig. 5J), activation of NMDA receptors and postsynaptic events (z-score = 5.477; Fig. 5J), glutamate binding, activation of AMPA receptors and synaptic plasticity (z-score = 4.796; Fig. 5J) in HMGCS2 KD-EX compared with SCR-EX.
To examine potential mechanisms that account for the reduced frontal cortex mitochondrial respiration by hepatic HMGCS2 knockdown, we explored potential canonical signalling pathways from the proteomic and phospho-proteomic datasets that compared HMGCS2 KD-SED with SCR-SED. These analyses revealed reduced cytosolic calcium signalling with increased apoptotic signalling (Fig. 6A and B), suggesting altered calcium regulation by the mitochondria. ADP/ATP translocase 2 (ANT2) is hypothesized as a component of the mitochondrial permeability transition pore that causes calcium efflux (Karch et al., 2019) and was upregulated with hepatic knockdown of HMGCS2 (Fig. 6C). We further examined the mitochondrial Ca2+ uniporter (MCU) (Fig. 6D), which is regulated by EMRE (essential MCU regulator) encoded by the single-pass membrane protein with aspartate-rich tail (SMDT1) gene (Bulthuis et al., 2023), which was upregulated with hepatic knockdown of HMGCS2 and inversely correlated (P = 0.05) with state 3 mitochondrial respiration (Fig. 6E).
Figure 6. AAV-mediated knockdown of hepatic HMGCS2 causes mitochondrial calcium overload inversely correlated with state 3 mitochondrial respiration.
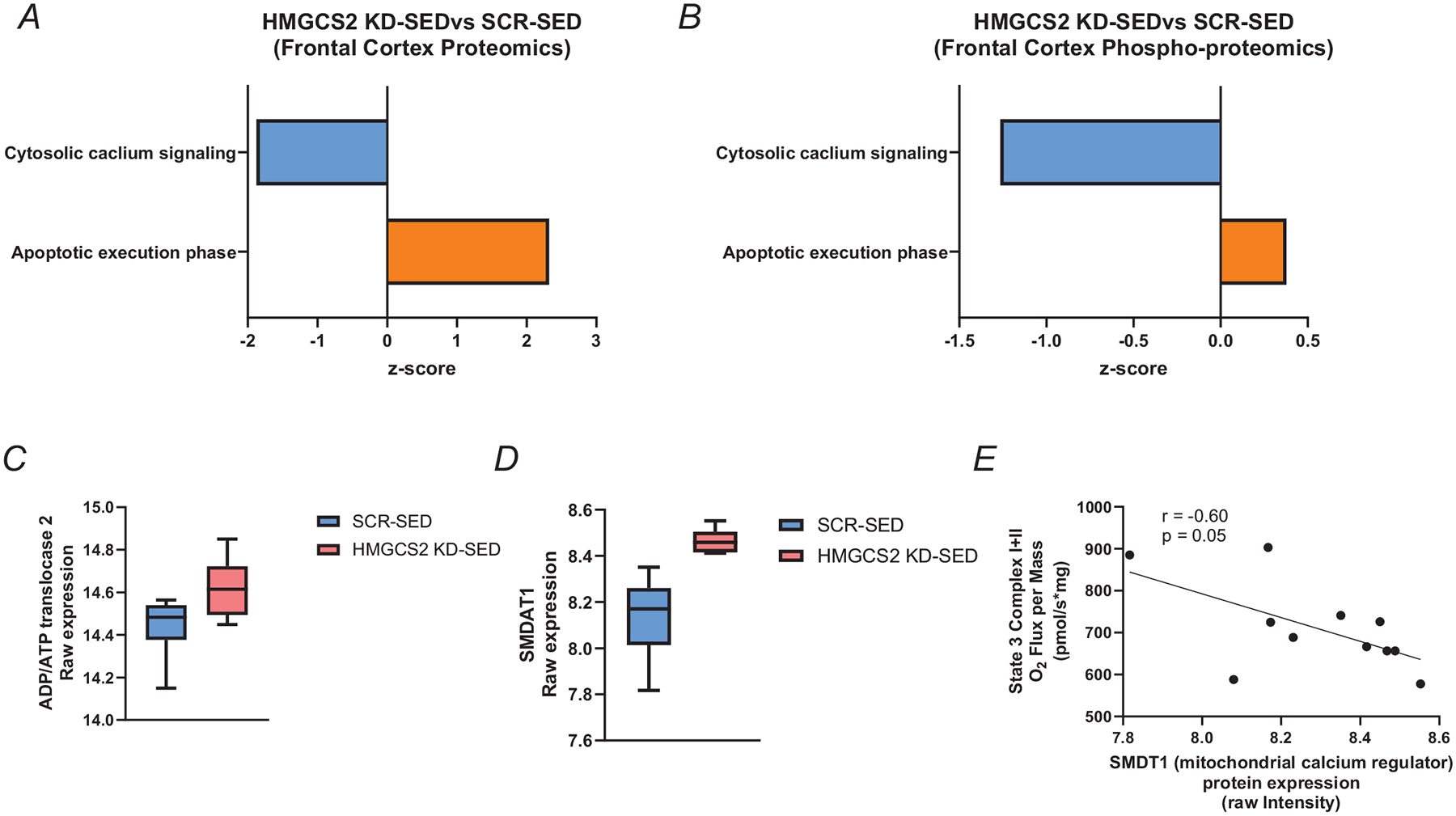
A and B, canonical signalling pathways revealed from proteomic (A) and phosphor-proteomic (B) IPA analyses. Blue indicates a decrease and orange an increase in HMGCS2-KD compared with SCR-SED rats. C, ADP/ATP translocase 2 (ANT2) protein expression in the frontal cortex expressed as raw intensity (n = 6). D, single-pass membrane protein with aspartate-rich tail (SMDT1) protein expression in the frontal cortex expressed as raw intensity (n = 6). E, correlation between State 3 Complex I + II mitochondrial respiration and SMDAT1 raw intensity in HMGCS2 KD-SED and SCR-SED rats. Z-score generated in IPA. Values presented as mean ± SD.
Discussion
Endurance exercise improves cognition (Stern et al., 2019), offers neuroprotection against ageing (Kirk-Sanchez & McGough, 2014) and is a top strategy to protect against neurodegenerative disease risk (Hill & Gammie, 2022). To our knowledge, this is the first study to demonstrate the necessity of hepatic ketone production in the maintenance of brain health, which we also show is overcome by chronic aerobic exercise training. Specifically, our novel findings show viral knockdown of hepatic HMGCS2, the rate-limiting enzyme in ketogenesis, increased markers of mitochondrial dysfunction and reduced markers of oxidative phosphorylation and electron transport 1 h after an acute bout of endurance exercise in the frontal cortex of female rats. Furthermore, our study demonstrated knockdown of hepatic HMGCS2 reduced mitochondrial respiration, impaired cognition and decreased protein markers of frontal cortex synaptic plasticity. Concurrently, we show that 4 weeks of endurance exercise upregulated protein markers of synaptic plasticity and ameliorated the cognitive impairment caused by viral knockdown of HMGCS2.
Based on our findings from three independent experiments, we propose maintenance of mitochondrial function in neurons is mediated by hepatic ketone production and subsequent oxidation. First, proteomic analyses of the frontal cortex revealed a protein signature of increased mitochondrial dysfunction, decreased oxidative phosphorylation and decreased respiratory electron transport with loss of hepatic HMGCS2 1 h after an acute bout of exercise. Second, state 3 frontal cortex mitochondrial respiration was significantly reduced with loss of hepatic HMGCS2. Third, in vitro knockdown of OXCT1, the rate-limiting step in ketone oxidation and subsequent ketone utilization, significantly reduced basal and maximal respiration in differentiated PC12 cells (immortalized rat neuronal-like cells), suggesting a loss of ketone oxidizing capacity reduces respiration in vitro. Combined, these results from our study strongly suggest hepatic HMGCS2 is crucial to maintain mitochondrial function in the brain.
Our a priori hypothesis was that loss of hepatic HMGCS2 and loss of subsequent hepatic ketone production would attenuate cognitive improvements and neuroprotective adaptations induced by endurance exercise. Instead, endurance exercise reversed protein markers of cognitive impairment in rats rendered HMGCS2 deficient in the liver. Surprisingly, endurance exercise training also prevented the cognitive impairment caused by hepatic HMGCS2 knockdown, but there was no significant interaction such that exercising animals were significantly different from sedentary animals (Fig. 5E) although there was a 3.5-fold increase in cognition compared to the HMGCS2 KD that remained sedentary. Furthermore, proteomic analyses of the frontal cortex of the brain revealed loss of hepatic HMGCS2 was associated with decreased markers of synaptic plasticity, which was reversed with exercise.
Consistent with others (Adams & Koeslag, 1988; Cassilhas et al., 2012; Radahmadi et al., 2016), endurance exercise increased ketones in the circulation, BDNF signalling, markers of synaptic plasticity and long-term potentiation in the HMGCS2 KD group. BDNF was also reduced in the HMGCS2 KD group compared to the SCR in sedentary conditions. Curiously, we also observed a modest decrease in BDNF in the SCR-EX compared to SCR-SED group, possibly caused by a feedback mechanism that takes place in healthy trained rats. Others report ketones increase BDNF expression in the brain by inhibiting histone deacetylase activity (Sleiman et al., 2016). In addition, neurotrophic factors, such as BDNF, have been shown to increase long-term potentiation (Ying et al., 2002), a primary mechanism for memory encoding (Bliss & Collingridge, 1993). We also observed an increase in markers of synaptic plasticity within the HMGCS2 KD-EX group compared with the SCR-EX, which could be an exaggerated exercise response to compensate for the loss of HMGCS2 in the liver. Consequently, we postulate loss of hepatic HMGCS2 caused lower levels of ketones circulating in the blood that resulted in reduced synaptic plasticity in the frontal cortex, contributing to the cognitive impairment observed in the HMGCS2-SED cohort.
Conversely, exercise was able to override the loss of ketones from hepatic HGMCS2 deficiency, possibly through another independent mechanism (i.e. increased IGF-1, FGF21 or irisin in the circulation to increase synaptic plasticity) (Cefis et al., 2023). Another possibility is that the cognitive impairment caused by HMGCS2 deficiency resulted from reduced mitochondrial function. There is a strong association between mitochondrial dysfunction, long-term potentiation and cognition (Todorova & Blokland, 2017). Therefore, the inability to rely on ketones as an additional source of fuel that can be shuttled into the TCA cycle for neuronal ATP production could reduce the neurons’ ability to produce synaptic events, resulting in decreased long-term potentiation. Ketones may increase energy availability ameliorating mitochondrial dysfunction in neurons, increasing synaptic plasticity and long-term potentiation, and ultimately restoring cognition. A precedent for hepatic afferent nerves affecting the brain also exists (Soty et al., 2017). Interestingly, these afferent neurons express G protein-coupled receptor 41 (GPR41 also known as HCAR2) (Cook et al., 2021), a known receptor of BHB (Kimura et al., 2011), and therefore afferent signalling may also contribute to the cognitive and brain mitochondrial impairments observed with hepatic HMGCS2 deficiency.
The inclusion of comparisons after both acute and chronic training periods provided us with a more comprehensive examination of the contribution of HMGCS2 in exercise responses. The addition of the acute exercise experiments allowed us to confirm our exercise protocol acutely increased ketones in the circulation and provided insight into the extent of ketogenesis during exercise with HMGCS2 knockdown. A portion of ketones were still present in the circulation after HMGCS2 knockdown, which could be an artefact from the remaining HMGCS2 to produce ketones in the liver or another tissue that had a compensation of ketone synthesis during exercise. Our mildly decreased plasma ketone concentration from HMGCS2 KD in sedentary conditions is consistent with other reports that show a similar decrease in ketones in mice treated with Hmgcs2-targeted antisense oligonucleotides (Cotter et al., 2014), However, we should also point out that ketone levels can be the same between sedentary and exercise conditions despite high extrahepatic flux of ketones as measured by tracers (Féry & Balasse, 1983). Therefore, although ketone levels were not dramatically different due to HMGCS2 knockdown, circulating measures of ketones do not measure turnover, and we suspect that the significant hepatic knockdown of HMGCS2 reduced ketone turnover and utilization rates far more than absolute circulating levels indicate, as evidenced by the strong effect on changing protein content in the brain. More specifically, despite a portion of ketones remaining in the circulation, there was still significant mitochondrial dysfunction in the frontal cortex based on the non-biased proteomic analyses, suggesting neuronal mitochondria rely strongly on plasma ketone concentration and that the knockdown of HMGCS2 had significant biological effects in the brain. Considering ketones are also reduced in sedentary conditions (Fig. 1D–F), our data would suggest chronic and continued reduction of plasma ketones may explain the loss of mitochondrial function and other HMGCS2-induced deficits.
Comparison of HMGCS2 KD-SED to SCR-SED proteomic and phospho-proteomic datasets revealed a decrease in cytosolic calcium signalling with increased apoptotic signalling, suggesting altered calcium regulation by the mitochondria. Calcium influx into the mitochondria stimulates multiple mitochondrial enzymes of the TCA cycle, increasing production of NADH and ultimately leading to increased mitochondrial respiration and ATP synthesis (Lee et al., 2023). The inverse correlation between SMDT1, which regulates calcium influx (Bulthuis et al., 2023), and state 3 mitochondrial respiration suggests increased calcium is no longer enhancing mitochondrial respiration, but impairing respiration instead. Considering that we also see an upregulation of ANT2 that is hypothesized to be a component of the mitochondrial permeability transition pore for calcium efflux (Karch et al., 2019), we hypothesize that the loss of ATP production could be due to mitochondrial calcium overload.
Even though we did not observe an improvement in cognition from the exercise training protocol in the SCR-EX cohort, our exercise paradigm recapitulated well-known skeletal muscle exercise adaptions including increased mitochondrial respiration and palmitate oxidation in the gastrocnemius (Fletcher et al., 2014; Holloszy, 1967), demonstrating a robust exercise protocol. Therefore, an exercise-induced increase in cognition in the SCR-EX cohort was probably not due to the lack of exercise intensity, but possibly due to limitations in the Y-maze testing. Although great for teasing out cognitive deficits, as observed with the HMGCS2 knockdown, the Y-maze is a single-point-two-choice maze making the probability of success innately and high causing the Y-maze to be less reliable in detecting cognitive improvements (as would be expected with exercise). Notably, this study concentrated on females, who are known to exhibit greater cognitive deterioration compared to males at the same stage of neurodegenerative disease (Cerri et al., 2019; Williamson et al., 2022), enhancing the translatability to populations disproportionately affected by these conditions. In summary, these data indicate hepatic ketogenesis is required to maintain cognition and mitochondrial function in endurance trained and untrained rats, establishing a mechanistic link between liver and brain health. Furthermore, we show that endurance exercise can overcome neuropathology caused by insufficient hepatic ketogenesis. Collectively, these findings highlight the potential for novel targets in maximizing ketone delivery and utilization to improve neuronal mitochondrial health and cognition.
Key points.
Decades of literature demonstrate endurance exercise to be neuroprotective.
Whether neuroprotective benefits are mediated, in part, by hepatic ketone production remains unclear.
This study provides the first set of data that suggest hepatic ketogenesis is required to maintain cognition, synaptic plasticity and mitochondrial function.
These data indicate endurance exercise can protect against cognitive decline caused by compromised hepatic ketogenesis.
These results establish a mechanistic link between liver and brain function, prompting further investigation of how hepatic metabolism influences brain health.
Acknowledgements
This work is part of the Molecular Transducers of Physical Activity Consortium (MoTrPAC) Preclinical Animal Study Sites (PASS) 2. Proteomics data analysis and MS services were provided by the Proteomics Core at the University of Missouri. The authors would like to thank Dr Thao Nguyen from the Gehrke Proteomics Center for her proteomics data preparation and analysis. The authors gratefully acknowledge the support and expertise of the Core facility staff.
Funding
This MoTrPAC study is supported by NIH U01AG070928 (MoTrPAC Phase 2 Animal Studies: J.P.T., F.W.B. and R.S.R.) and in part by VA-Merit Grant I01BX003271 (Salary support for R.S.R.). This work was supported with resources and the use of facilities at the University of Missouri and Harry S. Truman Memorial Veterans Hospital in Columbia, MO, USA.
Biography

Taylor Kelty received his PhD in Biomedical Sciences from the University of Missouri. His research interests are in the implication of liver–brain crosstalk in brain health. He is currently a post-doctoral fellow in the department of Nutrition and Exercise Physiology at the University of Missouri, where he is investigating the role of hepatic ketone metabolism in neurodegenerative disease progression.
Competing interests
The authors declare no conflicts of interest.
Supporting information
Additional supporting information can be found online in the Supporting Information section at the end of the HTML view of the article. Supporting information files available:
Peer Review History
The peer review history is available in the Supporting Information section of this article (https://doi.org/10.1113/JP287573#support-information-section).
Data availability statement
Data can be shared upon request.
References
- Adams JH, & Koeslag JH (1988). Carbohydrate homeostasis and post-exercise ketosis in trained and untrained rats. The Journal of Physiology, 407(1), 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amar D, Gay NR, Jimenez-Morales D, Jean Beltran PM, Ramaker ME, Raja AN, Zhao B, Sun Y, Marwaha S, Gaul DA, Hershman SG, Ferrasse A, Xia A, Lanza I, Fernández FM, Montgomery SB, Hevener AL, Ashley EA, Walsh MJ, … Zhen J (2024). The mitochondrial multi-omic response to exercise training across rat tissues. Cell metabolism, 36(6), 1411–1429.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batalha VL, Ferreira DG, Coelho JE, Valadas JS, Gomes R, Temido-Ferreira M, Shmidt T, Baqi Y, Buée L, Müller CE, Hamdane M, Outeiro TF, Bader M, Meijsing SH, Sadri-Vakili G, Blum D, & Lopes LV (2016). The caffeine-binding adenosine A2A receptor induces age-like HPA-axis dysfunction by targeting glucocorticoid receptor function. Scientific Reports, 6(1), 31493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, & Collingridge GL (1993). A synaptic model of memory: Long-term potentiation in the hippocampus. Nature, 361(6407), 31–39. [DOI] [PubMed] [Google Scholar]
- Bulthuis EP, Adjobo-Hermans MJW, de Potter B, Hoogstraten S, Wezendonk LHT, Tutakhel OAZ, Wintjes LT, van den Heuvel B, Willems P, Kamsteeg EJ, Gozalbo MER, Sallevelt S, Koudijs SM, Nicolai J, de Bie CI, Hoogendijk JE, Koopman WJH, & Rodenburg RJ (2023). SMDT1 variants impair EMRE-mediated mitochondrial calcium uptake in patients with muscle involvement. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1869(8), 166808. [DOI] [PubMed] [Google Scholar]
- Cassilhas RC, Lee KS, Fernandes J, Oliveira MG, Tufik S, Meeusen R, & de Mello MT (2012). Spatial memory is improved by aerobic and resistance exercise through divergent molecular mechanisms. Neuroscience, 202, 309–317. [DOI] [PubMed] [Google Scholar]
- Cefis M, Chaney R, Wirtz J, Méloux A, Quirié A, Leger C, Prigent-Tessier A, & Garnier P (2023). Molecular mechanisms underlying physical exercise-induced brain BDNF overproduction. Frontiers in Molecular Neuroscience, 16, 1275924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerri S, Mus L, & Blandini F (2019). Parkinson’s Disease in Women and Men: What’s the Difference? Journal of Parkinson’s Disease, 9(3), 501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Bylykbashi E, Chatila ZK, Lee SW, Pulli B, Clemenson GD, Kim E, Rompala A, Oram MK, Asselin C, Aronson J, Zhang C, Miller SJ, Lesinski A, Chen JW, Kim DY, van Praag H, Spiegelman BM, Gage FH, & Tanzi RE (2018). Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science, 361(6406). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook TM, Gavini CK, Jesse J, Aubert G, Gornick E, Bonomo R, Gautron L, Layden BT, & Mansuy-Aubert V (2021). Vagal neuron expression of the microbiota-derived metabolite receptor, free fatty acid receptor (FFAR3), is necessary for normal feeding behavior. Molecular Metabolism, 54, 101350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter DG, Ercal B, Huang X, Leid JM, D’avignon DA, Graham MJ, Dietzen DJ, Brunt EM, Patti GJ, Crawford PA (2014). Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. Journal of Clinical Investigation, 124(12), 5175–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham RP, Moore MP, Dashek RJ, Meers GM, Takahashi T, Sheldon RD, Wheeler AA, Diaz-Arias A, Ibdah JA, Parks EJ, Thyfault JP, & Rector RS (2021). Critical Role for Hepatocyte-Specific eNOS in NAFLD and NASH. Diabetes, 70(11), 2476–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao AT, Zagaar MA, Levine AT, Salim S, Eriksen JL, & Alkadhi KA (2013). Treadmill exercise prevents learning and memory impairment in Alzheimer’s disease-like pathology. Current Alzheimer Research, 10(5), 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David FJ, Robichaud JA, Leurgans SE, Poon C, Kohrt WM, Goldman JG, Comella CL, Vaillancourt DE, & Corcos DM (2015). Exercise improves cognition in Parkinson’s disease: The PRET-PD randomized, clinical trial. Movement Disorders, 30(12), 1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Avignon DA, Puchalska P, Ercal B, Chang Y, Martin SE, Graham MJ, Patti GJ, Han X, & Crawford PA (2018). Hepatic ketogenic insufficiency reprograms hepatic glycogen metabolism and the lipidome. Journal of Clinical Investigation Insight, 3(12), e99762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M, Cogan KE, & Egan B (2017). Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. The Journal of Physiology, 595(9), 2857–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Féry F, & Balasse EO (1983). Ketone body turnover during and after exercise in overnight-fasted and starved humans. American Journal of Physiology, 245(4), E318–325. [DOI] [PubMed] [Google Scholar]
- Fletcher JA, Meers GM, Linden MA, Kearney ML, Morris EM, Thyfault JP, & Rector RS (2014). Impact of various exercise modalities on hepatic mitochondrial function. Medicine and Science in Sports and Exercise, 46(6), 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigsby KB, Ruegsegger GN, Childs TE, & Booth FW (2019). Overexpression of Protein Kinase Inhibitor Alpha Reverses Rat Low Voluntary Running Behavior. Molecular Neurobiology, 56(3), 1782–1797. [DOI] [PubMed] [Google Scholar]
- Guadagni V, Drogos LL, Tyndall AV, Davenport MH, Anderson TJ, Eskes GA, Longman RS, Hill MD, Hogan DB, & Poulin MJ (2020). Aerobic exercise improves cognition and cerebrovascular regulation in older adults. Neurology, 94(21), e2245–e2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselbalch SG, Knudsen GM, Jakobsen J, Hageman LP, Holm S, & Paulson OB (1995). Blood-brain barrier permeability of glucose and ketone bodies during short-term starvation in humans. American Journal of Physiology, 268(6 Pt 1), E1161–1166. [DOI] [PubMed] [Google Scholar]
- Hill MA, & Gammie SC (2022). Alzheimer’s disease large-scale gene expression portrait identifies exercise as the top theoretical treatment. Scientific Reports, 12(1), 17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloszy JO (1967). Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. Journal of Biological Chemistry, 242(9), 2278–2282. [PubMed] [Google Scholar]
- Johnson RH, Walton JL, Krebs HA, & Williamson DH (1969). Post-exercise ketosis. The Lancet, 294(7635), 1383–1385. [DOI] [PubMed] [Google Scholar]
- Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM, & Molkentin JD (2019). Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Science Advances, 5(8), eaaw4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelty TJ, Taylor CL, Wieschhaus NE, Thorne PK, Amin AR, Mueller CM, Olver TD, Tharp DL, Emter CA, Caulk AW, & Rector RS (2023). Western diet-induced obesity results in brain mitochondrial dysfunction in female Ossabaw swine. Frontiers in Molecular Neuroscience, 16, 1320879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr NR, Kelty TJ, Mao X, Childs TE, Kline DD, Rector RS, & Booth FW (2023). Selective breeding for physical inactivity produces cognitive deficits via altered hippocampal mitochondrial and synaptic function. Frontiers in Ageing Neuroscience, 15, 1147420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, Kobayashi M, Hirasawa A, & Tsujimoto G (2011). Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). The Proceedings of the National Academy of Sciences, 108(19), 8030–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk-Sanchez NJ, & McGough EL (2014). Physical exercise and cognitive performance in the elderly: Current perspectives. Central Intelligence Agency, 9, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács Z, Brunner B, & Ari C (2021). Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases. Nutrients, 13(7), 2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau YS, Patki G, Das-Panja K, Le WD & Ahmad SO (2011). Neuroprotective effects and mechanisms of exercise in a chronic mouse model of Parkinson’s disease with moderate neurodegeneration. European Journal of Neuroscience, 33(7), 1264–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Duron HE, & Chaudhuri D (2023). Beyond the TCA cycle: New insights into mitochondrial calcium regulation of oxidative phosphorylation. Biochemical Society Transactions, 51(4), 1661–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maalouf M, Rho JM, & Mattson MP (2009). The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Research Reviews, 59(2), 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalakshmi B, Maurya N, Lee SD, & Bharath Kumar V (2020). Possible Neuroprotective Mechanisms of Physical Exercise in Neurodegeneration. International Journal of Molecular Sciences, 21(16), 5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Many GM, Sanford JA, Sagendorf TJ, Hou Z, Nigro P, Whytock KL, Amar D, Caputo T, Gay NR, Gaul DA, Hirshman MF, Jimenez-Morales D, Lindholm ME, Muehlbauer MJ, Vamvini M, Bergman BC, Fernández FM, Goodyear LJ, Hevener AL, … MoTrPAC Study Group (2024). Sexual dimorphism and the multi-omic response to exercise training in rat subcutaneous white adipose tissue. Nature Metabolism, 6(5), 963–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Aleixo I, Santos-Alves E, Balça MM, Rizo-Roca D, Moreira PI, Oliveira PJ, Magalhães J, & Ascensão A (2015). Physical exercise improves brain cortex and cerebellum mitochondrial bioenergetics and alters apoptotic, dynamic and auto(mito)phagy markers. Neuroscience, 301, 480–495. [DOI] [PubMed] [Google Scholar]
- MoTrPAC (2024). Temporal dynamics of the multi-omic response to endurance exercise training. Nature, 629(8010), 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutinho M, Puntambekar SS, Tsai AP, Coronel I, Lin PB, Casali BT, Martinez P, Oblak AL, Lasagna-Reeves CA, Lamb BT, & Landreth GE (2022). The niacin receptor HCAR2 modulates microglial response and limits disease progression in a mouse model of Alzheimer’s disease. Science Translational Medicine, 14(637), eabl7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair VD, Pincas H, Smith GR, Zaslavsky E, Ge Y, Amper MAS, Vasoya M, Chikina M, Sun Y, Raja AN, Mao W, Gay NR, Esser KA, Smith KS, Zhao B, Wiel L, Singh A, Lindholm ME, Amar D, … MoTrPAC Study Group (2024). Molecular adaptations in response to exercise training are associated with tissue-specific transcriptomic and epigenomic signatures. Cell Genomics, 4(6), 100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JC, & Verdin E (2017). β-Hydroxybutyrate: A Signaling Metabolite. Annual Review of Nutrition, 37(1), 51–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie L, Thakur MD, Wang Y, Su Q, Zhao Y, & Feng Y (2010). Regulation of U6 promoter activity by transcriptional interference in viral vector-based RNAi. Genomics, Proteomics & Bioinformatics, 8(3), 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radahmadi M, Hosseini N, & Alaei H (2016). Effect of exercise, exercise withdrawal, and continued regular exercise on excitability and long-term potentiation in the dentate gyrus of hippocampus. Brain Research, 1653, 8–13. [DOI] [PubMed] [Google Scholar]
- Rector RS, Uptergrove GM, Borengasser SJ, Mikus CR, Morris EM, Naples SP, Laye MJ, Laughlin MH, Booth FW, Ibdah JA, & Thyfault JP (2010). Changes in skeletal muscle mitochondria in response to the development of type 2 diabetes or prevention by daily wheel running in hyperphagic OLETF rats. American Journal of Physiology-Endocrinology and Metabolism, 298(6), E1179–E1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushing KA, Bolyard ML, Kelty T, Wieschhaus N, Pavela G, Rector RS, & Plaisance EP (2023). Dietary ketone ester attenuates the accretion of adiposity and liver steatosis in mice fed a high-fat, high-sugar diet. Frontiers in Physiology, 14, 1165224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands MS (2011). AAV-mediated liver-directed gene therapy. Methods in Molecular Biology, 807, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JA, Nogiec CD, Lindholm ME, Adkins JN, Amar D, Dasari S, Drugan JK, Fernández FM, Radom-Aizik S, Schenk S, Snyder MP, Tracy RP, Vanderboom P, Trappe S, Walsh MJ, & Molecular Transducers of Physical Activity Consortium (2020). Molecular Transducers of Physical Activity Consortium (MoTrPAC): Mapping the Dynamic Responses to Exercise. Cell, 181(7), 1464–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr., de Cabo R, Ulrich S, Akassoglou K, & Verdin E (2013). Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science, 339(6116), 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman SF, Henry J, Al-Haddad R, El Hayek L, Abou Haidar E, Stringer T, Ulja D, Karuppagounder SS, Holson EB, Ratan RR, Ninan I, & Chao MV (2016). Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. eLife, 5, e15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soty M, Gautier-Stein A, Rajas F, & Mithieux G (2017). Gut-Brain Glucose Signaling in Energy Homeostasis. Cell metabolism, 25(6), 1231–1242. [DOI] [PubMed] [Google Scholar]
- Stern Y, MacKay-Brandt A, Lee S, McKinley P, McIntyre K, Razlighi Q, Agarunov E, Bartels M, & Sloan RP (2019). Effect of aerobic exercise on cognition in younger adults: A randomized clinical trial. Neurology, 92(9), e905–e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, Ren N, Kaplan R, Wu K, Wu TJ, Jin L, Liaw C, Chen R, Richman J, Connolly D, Offermanns S, Wright SD, & Waters MG (2005). (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. Journal of Biological Chemistry, 280(29), 26649–26652. [DOI] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson-Lewis V, Ramasamy R, & Przedborski S (2003). D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. Journal of Clinical Investigation, 112(6), 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorova V, & Blokland A (2017). Mitochondria and Synaptic Plasticity in the Mature and Aging Nervous System. Current Neuropharmacology, 15(1), 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Christie BR, Sejnowski TJ, & Gage FH (1999). Running enhances neurogenesis, learning, and long-term potentiation in mice. The Proceedings of the National Academy of Sciences, 96(23), 13427–13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetr NG, Gay NR, MoTrPAC Study Group & Montgomery SB (2024). The impact of exercise on gene regulation in association with complex trait genetics. Nature Communications, 15(1), 3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J, Yabluchanskiy A, Mukli P, Wu DH, Sonntag W, Ciro C, & Yang Y (2022). Sex differences in brain functional connectivity of hippocampus in mild cognitive impairment. Frontiers in ageing neuroscience, 14, 959394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, & Bramham CR (2002). Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: Requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. Journal of Neuroscience, 22(5), 1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhen K, Su Q, Chen Y, Lv Y, & Yu L (2022). The Effect of Aerobic Exercise on Cognitive Function in People with Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. International Journal of Environmental Research and Public Health, 19(23), 15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data can be shared upon request.