

Abstract
Objective
This study aims to investigate the clinical characteristics and identify risk factors associated with progressive pulmonary fibrosis (PPF) in individuals diagnosed with primary Sjögren’s syndrome (pSS).
Methods
A retrospective case-control study was conducted from individuals with pSS-associated interstitial lung disease (pSS-ILD) registered in the Chinese Rheumatism Data Center between June 2010 and October 2023. Participants were categorized into two groups: those with PPF (pSS-PPF) and those without PPF (pSS-non-PPF). Comparative analyses were performed on clinical manifestations, laboratory parameters, pulmonary function, and treatment history between the two groups.
Results
Sixty-six individuals with pSS-ILD were included, of whom 29 met the criteria for PPF. Compared to pSS-non-PPF group, the pSS-PPF group demonstrated a higher rate of expectoration (48.3% vs 16.2%, p = 0.005) and crackles on auscultation (41.4% vs 13.5%, p = 0.01), but lower rates of parotid gland enlargement (3.4% vs 32.4%, p = 0.003), and arthritis (6.9% vs 27%, p = 0.035). Additionally, the incidence rate of the subjects suffering xerophthalmia and xerostomia in the PPF group was lower (24.1% vs 2.7%, p = 0.023). Pulmonary function testing showed significantly reduced forced vital capacity percentage predicted (83.6±15.6 vs 91.3±14.6, p = 0.042) and diffusing capacity of the lung for carbon monoxide percentage predicted (DLCO%, 54.2±21.5 vs 68.5±13.6, p = 0.003) in the PPF group. Multivariate logistic regression identified a baseline DLCO% < 60% as an independent risk factor for PPF. Parotid gland enlargement and arthritis were potentially protective. The predictive model demonstrated good performance, with an area under the curve of 0.821 (95% CI: 0.716~0.925, p < 0.001). The sensitivity was 58.6% and the specificity was 91.7%.
Conclusion
A baseline DLCO% < 60% is an independent predictor of PPF in individuals with pSS. The developed predictive model shows strong discriminatory ability, while further validation in larger cohorts is warranted.
Keywords: interstitial lung disease, predictive model, primary Sjögren’s syndrome, pSS, progressive pulmonary fibrosis, PPF, risk factors
Introduction
Primary Sjogren’s syndrome (pSS) is among the most common multisystem autoimmune diseases, with a global estimated prevalence of 60.82 cases per 100,000 individuals.1 In China, prevalence rates range from 0.29 to 0.77%, depending on the classification criteria applied.2 Over 80% of individuals with pSS experience dry mouth, dry eyes, fatigue, and pain, while approximately 30–40% exhibit extra-glandular involvement affecting organs such as the skin, lungs, heart, kidneys, and hematologic system. Among the extra-glandular manifestations, interstitial lung disease (ILD) is one of the most common, with a reported prevalence of 13%–21%. Non-specific interstitial pneumonia (NSIP) is the most frequently observed subtype in this context.3–5 Studies in China have confirmed comparable ILD prevalence rates (19.34%) and clinical manifestations in pSS patients, with NSIP similarly predominating as the most common subtype.6 Previous studies in China have identified various risk factors for the development of pSS-associated ILD (pSS-ILD), including older age, longer disease duration, higher disease activity, elevated serum KL-6 levels, and increased inflammation markers.6,7 The clinical course of pSS-ILD displays considerable variability. While some individuals maintain stable pulmonary function or respond favorably to treatment, others experience progression to progressive pulmonary fibrosis (PPF). This condition is characterized by worsening respiratory symptoms, a decline in pulmonary function, evidenced of fibrosis, and in severe cases, respiratory failure.8 Notably, the five-year survival rate for individuals with pSS-ILD is approximately 84%, representing a 2.5-fold increase in mortality risk compared to those without ILD. Key predictors of mortality include advanced age, prior malignancy, extensive lung involvement, and marked declines in forced vital capacity (FVC).9,10 Reported three-year survival rates for pSS-ILD and progressive fibrotic ILD are 91.2% and 82.4%, respectively, indicating a poorer prognosis in individuals with PPF.11
Early identification of individuals at high risk for developing PPF is essential for enabling individualized management strategies, mitigating fibrotic progression, and optimizing quality of life. However, data on the clinical predictors of progression in pSS-ILD remain limited. The absence of a standardized definition for disease progression has historically challenged pulmonary fibrosis research. Adopting the 2022 ATS/ERS/JRS/ALAT guidelines, this multicenter case-control study utilizes the term progressive pulmonary fibrosis (PPF) to ensure diagnostic consistency. We investigate clinical characteristics and risk factors for PPF in pSS-ILD, aiming to inform clinical practice and guide future research.
Methods
Study Design and Participants
A retrospective analysis was conducted using clinical data from individuals diagnosed with pSS and concomitant pulmonary interstitial fibrosis, registered in the Chinese Rheumatism Data Center (CRDC) between June 2010 and October 2023. Eligibility criteria included fulfilment of either the 2002 American-European Consensus Group (AECG) classification or the 2016 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria for pSS.12,13 In addition, participants were required to have a confirmed diagnosis of pulmonary interstitial fibrosis based on baseline high-resolution computed tomography (HRCT) of the chest and pulmonary function test (PFT), with follow-up assessments conducted at 12-months (±3 months) using repeat HRCT and PFT. Individuals with pulmonary interstitial fibrosis attributed to alternative etiologies—such as infections, tumors, medications, or occupational exposures—were excluded from the study.
Baseline characteristics were collected, including sex, age at baseline, age at diagnosis of pSS, age at diagnosis of ILD, and ILD duration. Clinical manifestations documented comprised ILD-related symptoms and signs, as well as glandular and extra-glandular features associated with pSS. Laboratory parameters included immunoglobulin levels, serum complement concentrations, and autoantibody profiles. Baseline PFT results were recorded, along with disease activity and damage assessments using the EULAR Sjögren’s Syndrome Disease Activity Index (ESSDAI), Sjögren’s Syndrome Disease Damage Index (SSDDI), and the European League Against Rheumatism Sjögren’s Syndrome Patient Reported Index (ESSPRI). Treatment regimens at baseline were also documented.
Participants were stratified into two groups based on the criteria for PPF, which require fulfillment of the following within a 12-month period. (1) Worsening of respiratory symptoms; (2) Decline in pulmonary function, defined as either an absolute reduction in FVC% of ≥ 5% or an absolute reduction in hemoglobin-adjusted diffusing capacity of the lung for carbon monoxide percentage predicted (DLCO%) ≥ 10%; and (3) Radiological evidence of HRCT progression, including one or more of the following: increased reticulation and bronchiectasis, new traction bronchiectasis accompanied by ground-glass opacities, new reticular opacities, increased severity or coarseness of reticular abnormalities, new or worsening honeycombing, or volume loss in the lungs.14 A diagnosis of PPF was established when all criteria were met over the one-year period, excluding cases in which progression could be attributed to alternative causes.
The collected data from both groups were compared and analyzed to identify potential risk factors associated with the development of PPF in individuals with pSS and to construct a clinical prediction model for PPF occurrence.
Statistical Analysis
Statistical analyses were conducted using IBM SPSS Statistics version 27. The distribution of continuous variables was assessed using histograms in conjunction with the Shapiro–Wilk test. Continuous variables following a normal distribution were presented as mean ± standard deviation (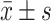 ), whereas non-normally distributed data were reported as median with interquartile range [M (P25, P75)]. Categorical variables were expressed as frequencies and percentages.
), whereas non-normally distributed data were reported as median with interquartile range [M (P25, P75)]. Categorical variables were expressed as frequencies and percentages.
Group comparisons were conducted using independent-samples t-tests for normally distributed continuous variables with equal variances or t’-tests for those with unequal variances. The Mann–Whitney U-test was employed for non-normally distributed continuous variables. Categorical variables were analyzed using the chi-squared test, the continuity corrected chi-squared test, or Fisher’s exact test. Variables with a p-value < 0.05 in univariate analysis were considered potential predictors of pSS-PPF and were subsequently included in a multivariate analysis using binary logistic regression with forward likelihood ratio (LR) stepwise selection to identify independent risk factors. Statistical significance level was defined as p < 0.05. The goodness-of-fit of the final model was evaluated using the Hosmer–Lemeshow test, and model performance was assessed by constructing receiver operating characteristic (ROC) curves.
Due to limited sample size, cases with missing data were excluded from the analysis. Given the retrospective design of the study, the sample size was based on available data, and a no a priori power calculation was performed.
Results
Participant Characteristics
A total of 1,392 individuals with a diagnosis of pSS and ILD were identified in the Chinese Rheumatism Data Center. Of these, 66 individuals from 12 clinical centers met the eligibility criteria and were included in the analysis. Twenty-nine participants were classified into the pSS-PPF group and 37 into the pSS-non-PPF group.
The proportion of female participants in the pSS-PPF group and pSS-non-PPF group was 27 cases (93.1%) and 36 cases (97.3%), respectively. The median follow-up duration was 26.5 months [interquartile range (IQR): 14.8, 51.6] in the pSS-PPF group and 30.8 months [IQR: 22.5, 60.6] in the pSS-non-PPF group. The mean baseline age was 52.4±12.7 years in the pSS-PPF group and 55.4±10.1 years in the pSS-non-PPF group. The median time to PPF development during follow-up in the PPF group was 14.9 months [IQR: 9.9, 35.2].
Compared with the pSS-non-PPF group, individuals in the pSS-PPF group were younger at the time of ILD diagnosis (48.9 ± 13.0 years vs 53.7 ± 10.5 years, p = 0.102) and had a longer duration at baseline (15.5 months [IQR: 2.1, 51.8] vs 5.5 months [IQR: 0, 20.4], p = 0.227). A higher proportion of participants in the pSS-PPF group presented with ILD as the initial clinical manifestation (31% vs 13.5%, p = 0.084). However, none of these differences reached statistical significance. No significant differences were observed between the two groups in terms of body mass index (BMI), smoking history, environmental exposure history, or comorbid conditions. Detailed comparisons are presented in Table 1 and Table 2.
Table 1.
The General Characteristics of pSS-PPF and pSS-Non-PPF
| pSS-PPF(n=29) | pSS-non-PPF (n=37) | t/t’/χ²/χ²c /U | P value | |
|---|---|---|---|---|
| Gender Male [cases (%)] |
2 (6.9) | 1 (2.7) | 0.047a | 0.829 |
| Baseline age (years) | 52.4±12.7 | 55.4±10.1 | −1.056b | 0.295 |
| Age at diagnosis of pSS (years) | 49.1±13.0 | 51.7±10.4 | −0.888b | 0.378 |
| Age at diagnosis of ILD (years) | 48.9±13.0 | 53.7±10.5 | −1.661b | 0.102 |
| Follow-up time (months) | 26.5 [14.8,51.6] | 30.8 [22.5,60.6] | 630 c | 0.227 |
| ILD duration at baseline (months) | 15.5 [2.1, 51.8] | 5.5 [0,20.4] | 416 c | 0.116 |
| ILD as initial presentation [cases (%)] | 9 (31) | 5 (13.5) | 2.986 d | 0.084 |
| BMI | 22.1±3.4 | 23.1±2.5 | 60.50 e | 0.191 |
| Smoking history [cases (%)] | 2 (6.9) | 2 (5.4) | 0 a | 1 |
| Environmental exposure history [cases (%)] | 2 (6.9) | 3 (8.1) | 0 a | 1 |
Note: aχ²c value (Continuous Correction Chi-Square Test), bt value (t-test), cU value (Mann–Whitney U-Test), dχ² value (Chi-Square Test), et’ value (used when variance is unequal).
Abbreviations: pSS, Primary Sjögren’s Syndrome; ILD, Interstitial Lung Disease; BMI, Body Mass Index; PPF, progressive pulmonary fibrosis.
Table 2.
Comparison of Complications Between pSS-PPF and pSS-Non-PPF [Number of Positive Cases/Total Sample Size (%)]
| pSS-PPF | pSS-non-PPF | χ²/χ²c | P value | |
|---|---|---|---|---|
| Hypertension | 6/26 (23.1) | 6/34 (17.6) | 0.271 a | 0.602 |
| Diabetes | 2/26 (7.7) | 1/34 (2.9) | 0.057 b | 0.811 |
| Hyperlipidemia | 5/26 (19.2) | 6/33 (18.2) | 0.011 a | 0.918 |
| Coronary heart disease | 1/29 (3.4) | 3/37 (8.1) | 0.072 a | 0.789 |
| Stroke | 0/29 (0) | 3/37 (8.1) | 0.25 c | |
| Fragility fracture | 0/29 (0) | 2/37 (5.4) | 0.5 c | |
| Tumor | 2/29 (6.9) | 0/37 (0) | 0.189c | |
| Thyroiditis | 4/29 (13.8) | 5/37 (13.5) | 0 b | 1 |
Note: aχ² value (Chi-square test), bχ²c value (Continuity corrected Chi-square test), cFisher’s exact probability method.
Abbreviations: pSS, Primary Sjogren’s syndrome; PPF, progressive pulmonary fibrosis.
Clinical Characteristics
Pulmonary Symptoms and Signs
As shown in Table 3, among the 29 participants in the pSS-PPF group, 26 (89.7%) presented with ILD-related symptoms at the time of diagnosis. Dry cough was reported in 10 cases (34.5%), sputum production in 14 cases (48.3%), and shortness of breath in 20 cases (69%). ILD-related physical signs were observed in 13 participants (44.8%), including cyanosis in 2 cases (6.9%), digital clubbing in 1 case (3.4%), and crackles on auscultation in 12 cases (41.4%).
Table 3.
Clinical Characteristics of pSS-PPF and pSS-Non-PPF [Cases (%)]
| pSS-PPF (n=29) | pSS-non-PPF (n=37) | χ²/χ²c | P value | |
|---|---|---|---|---|
| ILD-related symptoms | 26 (89.7) | 33 (89.2) | 0 a | 1 |
| Dry cough | 10 (34.5) | 16 (43.2) | 0.523 b | 0.47 |
| Sputum | 14 (48.3) | 6 (16.2) | 7.912 b | 0.005 |
| Dyspnea | 20 (69) | 25 (67.6) | 0.015 b | 0.904 |
| ILD-related signs | 13 (44.8) | 7 (18.9) | 5.167 b | 0.023 |
| Cyanosis | 2 (6.9) | 0 (0) | 0.189 c | |
| Digital clubbing | 1 (3.4) | 2 (5.4) | 0 a | 1 |
| Crackles | 12 (41.4) | 5 (13.5) | 6.601 b | 0.01 |
| System involvement | ||||
| No sicca | 7 (24.1) | 1 (2.7) | 5.145 a | 0.023 |
| Xerostomia | 21 (72.4) | 32 (86.5) | 2.036 b | 0.154 |
| Xerophthalmia | 21 (72.4) | 24 (64.9) | 0.427 b | 0.513 |
| Purpura rash | 1 (3.4) | 1 (2.7) | 0 a | 1 |
| Parotid enlargement | 1 (3.4) | 12 (32.4) | 8.635 b | 0.003 |
| Arthritis | 2 (6.9) | 10 (27) | 4.429 b | 0.035 |
| Myositis | 0 (0) | 1 (2.7) | 1 c | |
| Renal injury | 1 (3.4) | 0 (0) | 0.439 c | |
| PAH | 4 (13.8) | 1 (2.7) | 1.492 a | 0.222 |
| Pulmonary bullae | 5 (17.2) | 6 (16.2) | 0.012 b | 0.912 |
| PBC | 0 (0) | 1 (2.7) | 1 c | |
| PN | 0 (0) | 3 (8.1) | 0.25 c | |
| Leukocytopenia | 2 (6.9) | 4 (10.8) | 0.014 a | 0.906 |
| Thrombocytopenia | 0 (0) | 1 (2.7) | 1 c |
Note: aχ²c value (Continuity-corrected Chi-Square test), bχ² value (Chi-Square test), cFisher’s exact probability method.
Abbreviations: ILD, Interstitial Lung Disease; PAH, Primary Pulmonary Hypertension; PBC, Primary Biliary Cirrhosis; PN, peripheral neuropathy; pSS, Primary Sjogren’s syndrome, PPF, progressive pulmonary fibrosis.
When compared to the pSS-non-PPF group, individuals in the pSS-PPF group exhibited a significantly higher frequency of sputum production (48.3% vs 16.2%, p = 0.005). Signs indicative of pulmonary interstitial abnormalities—including cyanosis, digital clubbing, and crackles—were prevalent in the pSS-PPF group, (44.8% vs 18.9%, p = 0.023), with crackles alone showing a statistically significant difference (41.4% vs 13.5%, p = 0.01).
Systemic Manifestations
In terms of systemic involvement (Table 3), the pSS-PPF group showed significantly lower frequencies of parotid gland enlargement (3.4% vs 32.4%, p = 0.003) and arthritis (6.9% vs 27%, p = 0.035) compared to the PSS-non-PPF group. Additionally, the proportion of participants without symptoms (xerostomia and xerophthalmia) was significantly higher in the pSS- PPF group (24.1% vs 2.7%, p = 0.023).
Serological Characteristics
As presented in Table 4, no statistically significant differences were observed between the pSS-PPF group and PSS-non-PPF group with respect to serum immunoglobulin levels or the positivity rates of autoantibodies.
Table 4.
Serological Characteristics of pSS-PPF and pSS-Non-PPF
| pSS-PPF | pSS-non-PPF | t/χ² /χ²c | P value | |
|---|---|---|---|---|
| (Number of Positive Cases/Actual Sample Size (%) or Mean ± Standard Deviation) | ||||
| ANA | 22/29 (75.9) | 33/37 (89.2) | 1.23 a | 0.267 |
| Anti-SSA Ab | 25/29 (86.2) | 91.9 (34/37) | 0.117 a | 0.733 |
| Anti-SSB Ab | 8/29 (27.6) | 35.1 (13/37) | 0.427 b | 0.513 |
| Anti-RO-52 Ab | 22/27 (81.5) | 70.3 (26/37) | 1.046 b | 0.306 |
| ACA | 1/28 (3.6) | 0 (0/34) | 0.452 c | |
| AMA-M2 | 0/27 (0) | 9.4 (3/32) | 0.243 c | |
| Anti-nRNP Ab | 2/29 (6.9) | 13.5 (5/37) | 0.215 a | 0.643 |
| Anti-Sm Ab | 0/29 (0) | 2.7 (1/37) | 1 c | |
| RF | 8/24 (33.3) | 53.3 (16/30) | 2.16 b | 0.142 |
| IgG | 19.68±9.38 | 18.37±7.34 | 0.640 d | 0.524 |
| IgA | 2.97±1.37 | 2.97±1.46 | 0.019 d | 0.985 |
| IgM | 1.16±0.78 | 1.22±0.82 | 0.292 d | 0.771 |
Note: aχ²c value (continuity corrected chi-square test), bχ² value (chi-square test), cFisher exact probability method, dt value (t-test).
Abbreviations: ANA, Antinuclear Antibody; Ab, Antibody; AMA-M2, Anti-Mitochondrial Antibody subtype M2; ACA, Anti-Centromere Antibody; RF, Rheumatoid Factor; IgG/A/M, Immunoglobulin G,A,M.
Pulmonary Function and Disease Activity Indices
Pulmonary function test results (Table 5), demonstrated that individuals in the pSS-PPF group had significantly lower baseline FVC% (83.6±15.6 vs 91.3±14.6, p = 0.042) and DLCO% (54.2±21.5 vs 68.5±13.6, p = 0.003) compared to those in the pSS-non-PPF group. No statistically significant differences were observed between the two groups in ESSDAI, SSDDI, and ESSPRI.
Table 5.
Baseline Pulmonary Function and Related Assessment of pSS-PPF and pSS-Non-PPF
| pSS-PPF (n=29) |
pSS-non-PPF (n=37) |
t/t’ /U | P value | |
|---|---|---|---|---|
| FVC% | 83.6±15.6 | 91.3±14.6 | −2.076 a | 0.042 |
| DLCO% | 54.2±21.5 | 68.5±13.6 | −3.117 b | 0.003 |
| ESSDAI | 9 (0.5,15) | 6 (0.5,10.5) | 463 c | 0.338 |
| SSDDI | 4 (3,4.5) | 4 (2.5,4) | 474.5 c | 0.406 |
| ESSPRI-sicca | 38.6±22.2 | 45.4±24.9 | −1.16 a | 0.25 |
| ESSPRI-fatigue | 29.5±20.0 | 28.4±23.0 | 0.216 a | 0.83 |
| ESSPRI-pain | 8 (0,30) | 8 (0,30.5) | 544.5 c | 0.913 |
Note: at value (t-test), bt’ value (t’ test, used when variance is not equal), cU value (Mann–Whitney U-test).
Abbreviations: FVC%, Forced Vital Capacity percentage predicted; DLCO%, Diffusing Capacity of the Lung for Carbon Monoxide percentage predicted; ESSDAI, EULAR Sjögren’s Syndrome Disease Activity Index; SSDDI, Sjögren’s Syndrome Disease Damage Index; pSS, Primary Sjogren’s syndrome, PPF, progressive pulmonary fibrosis; ESSPRI, European League Against Rheumatism Sjögren’s Syndrome Patient Reported Index.
Therapeutic Parameters
As shown in Table 6, no statistically significant differences in therapeutic regimens were observed between the pSS-PPF and pSS-non-PPF groups during the follow-up period. Glucocorticoid therapy was administered to 23 participants (79.3%) in the pSS-PPF group and 26 participants (70.3%) in the pSS-non-PPF group, respectively (χ² = 0.695, p = 0.405). Immunosuppressive therapy was administered to 20 (69%) and 24 (64.9%) participants, respectively (χ² = 0.123, p = 0.726). A small proportion of participants in both groups received more than two immunosuppressive agents concurrently: 3 participants (10.3%) in the pSS-PPF group and 7 participants (18.9%) in the PSS-non-PPF group (χ² = 0.382, p = 0.536). Tripterygium glycosides was administered to 10 participants (34.5%) in the pSS-PPF group and 14 participants (37.8%) in the PSS-non-PPF group (χ² = 0.079, p = 0.779). The overall use of long-term antifibrotic therapy (Pirfenidone) was low across both groups: 4 cases (13.8%) in the pSS-PPF group and 5 cases (13.5%) in the pSS-non-PPF group (χ²c =0, p = l). Four participants in the pSS-non-PPF group received Janus kinase (JAK) inhibitors during follow-up, whereas none of the participants in the pSS-PPF group were treated with JAK inhibitors prior to the onset of fibrosis progression.
Table 6.
Comparison of Treatments for pSS-PPF and pSS-Non-PPF [Cases (%)]
| pSS-PPF (n=29) | pSS-non-PPF (n=37) | χ²/χ²c | P value | |
|---|---|---|---|---|
| Glucocorticoid | 23 (79.3) | 26 (70.3) | 0.695 a | 0.405 |
| HCQ | 10 (34.5) | 19 (51.4) | 1.878 a | 0.171 |
| Immunosuppressant | 20 (69) | 24 (64.9) | 0.123 a | 0.726 |
| CYC | 4 (13.8) | 7 (18.9) | 0.049 b | 0.824 |
| MMF | 9 (31) | 15 (40.5) | 0.635 a | 0.426 |
| CNI | 4 (13.8) | 4 (10.8) | 0 b | 1 |
| AZA | 2 (6.9) | 0 | 0.189 c | |
| Others IS | 4 (13.8) | 5 (13.5) | 0 a | 1 |
| Pirfenidone | 4 (13.8) | 5 (13.5) | 0b | 1 |
| JAKi | 0 | 4 (10.8) | 0.125 c | |
| TG | 10 (34.5) | 14 (37.8) | 0.079 a | 0.779 |
Note: aχ² value (chi-square test), bχ²c value (continuity corrected chi-square test), cFisher exact probability method.
Abbreviations: HCQ, Hydroxychloroquine Sulfate; CYC, Cyclophosphamide; MMF, Mycophenolate Mofetil; Calcineurin Inhibitors; AZA, Azathioprine; others IS, Methotrexate, Leflunomide, Iguratimod; JAKi, Tofacitinib, Baricitinib; TG, Tripterygium glycosides; pSS, Primary Sjogren’s syndrome, PPF, progressive pulmonary fibrosis.
Risk Factors for PPF in pSS
Binary logistic regression analysis was conducted with the presence of PPF as the dependent variable to identify independent risk factors in participants with pSS-associated ILD. Variables included in the model were selected from the univariate analysis based on statistical significance and clinical relevance. These variables comprised baseline FVC%, baseline DLCO%, presence of crackles at the lung bases, cough, parotid gland enlargement, arthritis, and absence of xerostomia and xerophthalmia. During multivariate modeling, FVC%, crackles at lung bases, cough, and absence of xerostomia and xerophthalmia were excluded from the final model. To facilitate the interpretation of results and their clinical application, an ROC curve was plotted for baseline DLCO%. Using Youden’s index, an optimal cutoff value of 58.25% was determined. However, to improve clinical utility, a threshold of DLCO% < 60% was adopted for binary classification.
Subsequently, baseline DLCO% < 60% was identified as an independent risk factor for PPF, conferring a 4.362-fold increased risk of disease progression. In contrast, the presence of parotid gland enlargement and arthritis were identified as protective factors for pSS-PPF with risk reductions of 0.062 and 0.096 times, respectively (Table 7). The model achieved a negative predictive value (NPV) of 77.8%, a positive predictive value (PPV) of 72.4%, and an overall accuracy of 76.9%. Hosmer–Lemeshow goodness-of-fit testing indicated acceptable calibration (χ² = 6.469, p= 0.486). The area under the curve (AUC) was 0.821 (95% CI: 0.716–0.925; p < 0.001), exceeding the prespecified threshold of 0.7. At the optimal cut-off of 0.627 determined by maximizing Youden’s index (J=0.503), sensitivity was 58.6% and specificity 91.7% (Figure 1 and Table 8).
Table 7.
Binary Logistic Regression Analysis of Risk Factors for pSS-PPF
| Variable | β Value | Standard Error | P value | OR Value | 95% CI Value |
|---|---|---|---|---|---|
| Parotid enlargement | −2.779 | 1.126 | 0.014 | 0.062 | 0.007–0.565 |
| Arthritis | −2.345 | 0.904 | 0.010 | 0.096 | 0.016–0.564 |
| Baseline DLCO% < 60% | 1.473 | 0.627 | 0.019 | 4.362 | 1.277–14.904 |
Note: DLCO%: Diffusing Capacity of the Lung for Carbon Monoxide percentage predicted.
Abbreviations: pSS, Primary Sjogren’s syndrome, PPF, progressive pulmonary fibrosis.
Figure 1.

Area Under the Curve (AUC) of the risk model.
Table 8.
Predictive Model Performance Metrics
| AUC/95% CI | Cut-off | PPV | NPV | Sensitivity | Specificity |
|---|---|---|---|---|---|
| 0.821 (0.716–0.925) | 0.627 | 72.40% | 77.80% | 58.60% | 91.70% |
Abbreviations: AUC, Area Under Curve; 95% CI, 95% confidence interval; PPV, Positive Predictive Value; NPV, Negative Predictive Value.
Discussion
The clinical course of ILD in individuals with pSS displays significant variability. Reported rates of progression to PPF among pSS-ILD populations range from 24% to 48.7%.15–18 Current evidence consistently demonstrates that the usual interstitial pneumonia (UIP) pattern on HRCT is associated with a higher likelihood of fibrotic progression in connective tissue disease-associated ILD, including pSS.15–17,19 However, recent findings from a Chinese cohort study indicate that PSS-UIP may represent a more indolent phenotype compared to other connective tissue disease-related UIP subtypes. Specifically, the rate of FVC decline in pSS-UIP was approximately 25.9 mL/year—significantly slower than that observed in rheumatoid arthritis-UIP (88.1 mL/year) and ANCA-associated vasculitis-UIP (72.9 mL/year).20 The radiologic manifestation of pSS-ILD is variable and often demonstrates overlapping features,21 which, coupled with interobserver variability in ILD subtype classification, even in experienced ILD centers, poses a challenge for predicting PPF based on CT pattern alone. Developing imaging-independent predictive tools offers greater practical value for broader applicability.
A recent cohort study conducted in China also identified extensive lung involvement (> 30% of lung area) as an independent risk factor for both disease progression (OR = 4.143) and mortality (HR = 3.450) in pSS-ILD.18 Consistent with this radiological-prognostic relationship, we observed significantly higher prevalence of inspiratory crackles in the pSS-PPF group versus non-PPF controls (p<0.05). Mechanistically, crackles reflect underlying extensive fibrosis and UIP patterns,22 providing a clinical correlate to severe parenchymal involvement. Although direct HRCT quantification was unavailable, this physical sign concordance indirectly validates disease extent as a key prognostic determinant. Notably, sputum production—atypical in classic ILD—was also elevated in PPF patients (p<0.05). This may indicate active inflammation or secondary infection, both of which may exacerbate ILD. Both findings reinforce that thorough physical examination (crackles/sputum assessment) and symptom documentation are critical for identifying high-risk phenotypes, particularly where advanced imaging access is limited.
Previous studies have suggested that older age is associated with an increased risk of pulmonary function decline and fibrotic progression in ILD.15,17 In the present cohort, the mean age at ILD diagnosis was slightly lower in the pSS-PPF group (48.9±13.0 years), compared to the pSS-non-PPF group (53.7±10.5 years); however, this difference did not reach statistical significance. A prior study comparing HRCT findings and pulmonary function changes between individuals with onset of pSS-ILD at age 65 or older and those with adult-onset disease reported that, in the older-onset group, pulmonary function declined more slowly, primarily presenting as restrictive ventilatory impairment.23 Imaging findings demonstrated more pronounced fibrotic changes in the older-onset group, suggesting that ILD may develop earlier in this population but remain undetected until fibrosis has advanced.
Additionally, retrospective evidence has indicated that ILD progression is not solely related to the presence of a UIP pattern, but also correlates with follow-up duration, indicating that the likelihood of progression increases over time.16 Therefore, in assessing the association between age and progression to PPF, the potential confounding influence of ILD duration must be considered. In the current study, while the duration of ILD was numerically longer in the pSS-PPF group than in the pSS-non-PPF group, the difference was not statistically significant.
Previous studies indicate that approximately 10% to 51% of patients develop ILD several years prior to the clinical diagnosis of Sjögren’s syndrome.24 The prevalence of xerostomia and xerophthalmia is notably lower in individuals with pSS-associated fibrotic ILD compared to those without fibrotic involvement.25 Moreover, the absence of sicca symptoms in pSS-ILD has been associated with earlier disease progression and greater declines in total lung capacity and FVC%.26 Prior studies have identified pSS onset without sicca symptoms, elevated lactate dehydrogenase levels, and low baseline FVC% as independent risk factors for ILD progression.27
The findings of the current study align with these observations. A higher proportion of participants in the pSS-PPF group presented with ILD as the initial manifestation compared to the pSS-non-PPF group (31% vs 13.5%), with a trend towards statistical difference (p = 0.084). Additionally, the pSS-PPF group had a significantly higher rate of patients without sicca symptoms (24.1% vs 2.7%, *p* = 0.023) and a markedly lower frequency of parotid enlargement (3.4% vs 32.4%, *p* = 0.003). This phenotype corresponds to the lung-onset pSS reported in Chinese cohorts, characterized by the absence of oral/ocular dryness but more severe lung impairment than sicca-onset pSS.28 Potential mechanisms underlying this lung-predominant phenotype may involve: (1) lung-specific antigen exposure triggering autoimmunity; (2) the lungs being the initial affected organ with delayed salivary gland involvement; or (3) tissue-specific immune microenvironments driving localized pulmonary inflammation and fibrosis progression.29 Furthermore, the absence of classic sicca symptoms may delay the diagnosis of the primary disease, thereby postponing the initiation of immunotherapy. Further studies, particularly in human subjects, are warranted to validate these mechanisms.
The management of pSS-ILD generally involves the administration of glucocorticoids, either as monotherapy or in combination with immunosuppressive agents such as cyclophosphamide, mycophenolate mofetil, or azathioprine. However, current evidence remains insufficient to establish an optimal, standardized treatment protocol for pSS-ILD.30 For patients with pSS-PPF who show limited responsiveness to immunosuppressive therapies, antifibrotic agents such as pirfenidone and nintedanib may offer potential clinical benefit. Nonetheless, further validation through large-scale clinical studies are necessary to confirm their efficacy.31,32
In the present study, there were no statistically significant differences between the pSS-PPF and pSS-non-PPF groups regarding the use of glucocorticoids, immunosuppressive agents, or antifibrotic therapy. All participants who received antifibrotic treatment were prescribed pirfenidone, which may be attributed to factors such as cost and availability.
JAK inhibitors represent a potential therapeutic strategy for pSS-ILD, given their ability to modulate the JAK-STAT pathway, which is involved in both inflammatory and fibrotic processes. Preliminary evidence from other connective tissue disease-associated ILDs suggests that JAK inhibitors may influence immune regulation through multiple pathways and exert a therapeutic effect in pSS-ILD.29 In the present cohort, four participants in the pSS-non-PPF group received treatment with JAK inhibitors, whereas no participants in the pSS-PPF group were exposed to this therapy. This observation warrants further investigation.
HRCT and pulmonary function parameters, particularly FVC% and DLCO%, are critical indicators for assessing fibrotic progression in ILD. Decreased FVC% and DLCO% are strong predictors of mortality across various fibrotic ILD subtypes. However, a prior study indicated that a decline in FVC% alone may not reliably predict subsequent deterioration in pulmonary function.33 In the present study, univariate analysis indicated that baseline FVC% and DLCO% values were significantly lower in the pSS-PPF group compared to the pSS-non-PPF group. Subsequent multivariate logistic regression analysis identified baseline DLCO% < 60% as an independent risk factor for the development of PPF is pSS-ILD, increasing the risk by 4.362 times. The exceptional specificity (91.7%) positions this model as a reliable rule-in tool for PPF confirmation, effectively reducing unnecessary interventions in predicted negative cases and enhancing precision in treatment initiation while conserving medical resources particularly for patients with DLCO%<60%. The suboptimal sensitivity (58.6%) poses a substantial missed-diagnosis risk, necessitating complementary high-sensitivity tests when applied in screening scenarios.
The low sensitivity of the model may be attributed to disease heterogeneity, as early symptoms are frequently mild or nonspecific, complicating timely identification. Furthermore, the retrospective design and relatively small sample size of the study may have contributed to the observed limitations in model performance.
However, this study has several limitations. First, as a retrospective case-control study, it was designed to explore associations between clinical variables and the development of PPF in individuals with pSS-ILD but could not establish causal relationships. Second, potential selection bias may have arisen due to variability in follow-up duration and incomplete data. The relatively small sample size in China may have limited the statistical power to detect certain associations. Third, although the predominance of female participants (93.1% in the pSS-PPF group) reflects the known gender distribution of pSS, the underrepresentation of male patients may limit the generalizability of the findings to the broader population.
Conclusion
In this Chinese case-control study, the risk of progression to PPF in pSS-ILD warrants attention. Our findings demonstrate that a baseline DLCO% < 60% is significantly associated with fibrotic progression. The derived predictive model serves strictly as a hypothesis-generating framework for identifying high-risk Chinese pSS-ILD phenotypes; its clinical application requires validation in independent, multi-ethnic cohorts using standardized HRCT phenotyping protocols.
Acknowledgments
We are enormously grateful to the following hospitals for providing the clinical case data that formed the backbone of this study: Beijing Chaoyang Hospital Affiliated to Capital Medical University; Shandong University QILU Hospital; The Second Affiliated Hospital of Zhejiang University School of Medicine; Guangdong Provincial People’s Hospital; The First Affiliated Hospital of China Medical University; Xinjiang Uygur Autonomous Region People’s Hospital; The affiliated hospital of Inner Mongolia medical university; The second Xiangya hospital of central south university; The Second Affiliated Hospital of Chongqing Medical University; and Mudanjiang Medical University Affiliated Hongqi Hospital.
Funding Statement
This study was supported by the Chinese National Key Technology R&D Program, Ministry of Science and Technology (2017YFC0907601, 2017YFC0907605), CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-005), National High Level Hospital Clinical Research Funding (2022-PUMCH-B-013).
Abbreviations
pSS, Primary Sjögren’s Syndrome; PPF, Progressive Pulmonary Fibrosis; ILD, Interstitial Lung Disease; NSIP, Non-Specific Interstitial Pneumonia; HRCT, High-Resolution Computed Tomography; FVC, Forced Vital Capacity; PFT, Pulmonary Function Test; FVC%, Forced Vital Capacity Percentage of Predicted; DLCO%, Diffusing Capacity of the Lung for Carbon Monoxide Percentage of Predicted; EULAR, European League Against Rheumatism; ACR, American College of Rheumatology; ESSDAI, EULAR Sjögren’s Syndrome Disease Activity Index; SSDDI, Sjögren’s Syndrome Disease Damage Index; ESSPRI, EULAR Sjögren’s Syndrome Patient Reported Index; BMI, Body Mass Index; HCQ, Hydroxychloroquine; CYC, Cyclophosphamide; MMF, Mycophenolate Mofetil; AZA, Azathioprine; JAKi, Janus Kinase Inhibitor; CNIs, Calcineurin Inhibitors; TGs, Tripterygium glycosides; UIP, Usual Interstitial Pneumonia; CTD, Connective Tissue Disease; RA, Rheumatoid Arthritis; AAV, ANCA-Associated Vasculitis; PAH, Pulmonary Arterial Hypertension; PBC, Primary Biliary Cholangitis; PN, Peripheral Neuropathy; ANA, Antinuclear Antibody; RF, Rheumatoid Factor; Ab, Antibody; IgG/A/M, Immunoglobulin G,A,M; CRDC, Chinese Rheumatism Data Center; ATS, American Thoracic Society; ERS, European Respiratory Society; JRS, Japanese Respiratory Society; ALAT, Latin American Thoracic Society; IPF, Idiopathic Pulmonary Fibrosis; ACA, Anti-Centromere Antibody; AUC, Area Under the Curve; ROC Curve, Receiver Operating Characteristic Curve.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author, Dong Xu, upon reasonable request.
Ethics Approval
This study was conducted in accordance with the declaration of Helsinki. This study was approved by the Ethics Committee of Peking Union Medical College Hospital (Approval No. JS-2038), with waived informed consent due to the retrospective use of anonymized data.
Disclosure
None of the authors have any financial disclosure or conflicts of interest for this work.
References
- 1.Qin B, Wang J, Yang Z, et al. Epidemiology of primary Sjögren’s syndrome: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74(11):1983–1989. doi: 10.1136/annrheumdis-2014-205375 [DOI] [PubMed] [Google Scholar]
- 2.Zhang NZ, Shi CS, Yao QP, et al. Prevalence of primary Sjögren’s syndrome in China. J Rheumatol. 1995;22(4):659–661. [PubMed] [Google Scholar]
- 3.Mariette X, Criswell LA. Primary Sjögren’s Syndrome. New Eng J Med. 2018;378(10):931–939. doi: 10.1056/NEJMcp1702514 [DOI] [PubMed] [Google Scholar]
- 4.Sambataro G, Ferro F, Orlandi M, et al. Clinical, morphological features and prognostic factors associated with interstitial lung disease in primary Sjӧgren’s syndrome: a systematic review from the Italian society of rheumatology. Autoimmun Rev. 2020;19(2):102447. doi: 10.1016/j.autrev.2019.102447 [DOI] [PubMed] [Google Scholar]
- 5.Joy GM, Arbiv OA, Wong CK, et al. Prevalence, imaging patterns and risk factors of interstitial lung disease in connective tissue disease: a systematic review and meta-analysis. Eur Respir Rev. 2023;32(167):220210. doi: 10.1183/16000617.0210-2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao H, Zhang XW, He J, et al. Prevalence, risk factors, and prognosis of interstitial lung disease in a large cohort of Chinese primary Sjögren syndrome patients: a case-control study. Medicine. 2018;97(24):e11003. doi: 10.1097/MD.0000000000011003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weng L, Chen Y, Liang T, et al. Biomarkers of interstitial lung disease associated with primary Sjögren’s syndrome. Eur J Med Res. 2022;27(1):199. doi: 10.1186/s40001-022-00828-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Panagopoulos P, Goules A, Hoffmann-Vold AM, Matteson EL, Tzioufas A. Natural history and screening of interstitial lung disease in systemic autoimmune rheumatic disorders. Ther Adv Musculoskelet Dis. 2021;13:1759720x211037519. doi: 10.1177/1759720X211037519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang H, Xie W, Geng Y, Fan Y, Zhang Z. Mortality in patients with primary Sjögren’s syndrome: a systematic review and meta-analysis. Rheumatology. 2021;60(9):4029–4038. doi: 10.1093/rheumatology/keab364 [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, Qiu Y, Xie Z, et al. Risk factors and prognosis of interstitial lung disease for primary Sjögren syndrome patients: a retrospective case‒control study. Clin Rheumatol. 2023;42(11):3033–3041. doi: 10.1007/s10067-023-06596-7 [DOI] [PubMed] [Google Scholar]
- 11.Chen YH, Lee TJ, Hsieh HJ, et al. Clinical outcomes and risk factors of progressive pulmonary fibrosis in primary Sjögren’s syndrome-associated interstitial lung disease. BMC Pulm Med. 2023;23(1):268. doi: 10.1186/s12890-023-02562-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European consensus group. Ann Rheum Dis. 2002;61(6):554–558. doi: 10.1136/ard.61.6.554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiboski CH, Shiboski SC, Seror R, et al. 2016 American college of rheumatology/European league against rheumatism classification criteria for primary Sjögren’s Syndrome: a consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol. 2017;69(1):35–45. doi: 10.1002/art.39859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an Update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18–e47. doi: 10.1164/rccm.202202-0399ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang T, Yuan F, Xu L, Sun W, Liu L, Xue J. Characteristics of patients with primary Sjögren’s syndrome associated interstitial lung disease and relevant features of disease progression. Clin Rheumatol. 2020;39(5):1561–1568. doi: 10.1007/s10067-019-04906-6 [DOI] [PubMed] [Google Scholar]
- 16.Lee KA, Nam BD, Hwang JH, Kim HS. Clinical course and risk factors for development and progression of interstitial lung disease in primary Sjögren’s syndrome. Sci Rep. 2023;13(1):9189. doi: 10.1038/s41598-023-35608-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim YJ, Choe J, Kim HJ, Song JW. Long-term clinical course and outcome in patients with primary Sjögren syndrome-associated interstitial lung disease. Sci Rep. 2021;11(1):12827. doi: 10.1038/s41598-021-92024-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Y, Zhou J, Dong X, Guo X, Lu Y, Zheng Y. Risk factors for progression and prognosis of primary Sjögren’s syndrome-associated interstitial lung disease in a Chinese population. Int J Rheum Dis. 2020;23(12):1734–1740. doi: 10.1111/1756-185X.14023 [DOI] [PubMed] [Google Scholar]
- 19.Parambil JG, Myers JL, Lindell RM, Matteson EL, Ryu JH. Interstitial lung disease in primary Sjögren syndrome. Chest. 2006;130(5):1489–1495. doi: 10.1378/chest.130.5.1489 [DOI] [PubMed] [Google Scholar]
- 20.Yang S, Wang J, Sun D, Wang Y, Xue C, Ye Q. Disease progression in patients with usual interstitial pneumonia and probable UIP patterns on computed tomography with various underlying etiologies: a retrospective cohort study. Front Med. 2023;10:1246767. doi: 10.3389/fmed.2023.1246767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong X, Zhou J, Guo X, et al. A retrospective analysis of distinguishing features of chest HRCT and clinical manifestation in primary Sjögren’s syndrome-related interstitial lung disease in a Chinese population. Clin Rheumatol. 2018;37(11):2981–2988. doi: 10.1007/s10067-018-4289-6 [DOI] [PubMed] [Google Scholar]
- 22.Sgalla G, Walsh SLF, Sverzellati N, et al. “Velcro-type” crackles predict specific radiologic features of fibrotic interstitial lung disease. BMC Pulm Med. 2018;18(1):103. doi: 10.1186/s12890-018-0670-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong X, Gao Y, Li M, Wang D, Li J, Zhang Y. Characteristics of Chest HRCT and pulmonary function tests in elderly-onset primary Sjögren syndrome with interstitial lung disease. Medicine. 2023;102(8):e32952. doi: 10.1097/MD.0000000000032952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He C, Chen Z, Liu S, Chen H, Zhang F. Prevalence and risk factors of interstitial lung disease in patients with primary Sjögren’s syndrome: a systematic review and meta-analysis. Int J Rheum Dis. 2020;23(8):1009–1018. doi: 10.1111/1756-185X.13881 [DOI] [PubMed] [Google Scholar]
- 25.Luppi F, Ferrara G, Faverio P. Fibrotic or nonfibrotic interstitial lung disease in patients with primary Sjögren syndrome. Polish Arch Int med. 2023;133(2). doi: 10.20452/pamw.16428 [DOI] [PubMed] [Google Scholar]
- 26.Gao H, Zou YD, Zhang XW, et al. Interstitial lung disease in non-sicca onset primary Sjögren’s syndrome: a large-scale case-control study. Int J Rheum Dis. 2018;21(7):1423–1429. doi: 10.1111/1756-185X.13334 [DOI] [PubMed] [Google Scholar]
- 27.He SH, He YJ, Guo KJ, Liang X, Li SS, Li TF. Risk factors for progression of interstitial lung disease in Sjögren’s syndrome: a single-centered, retrospective study. Clin Rheumatol. 2022;41(4):1153–1161. doi: 10.1007/s10067-021-05984-1 [DOI] [PubMed] [Google Scholar]
- 28.Dong X, Gao Y, Li M, Wang D, Li J, Zhang Y. The characteristics of chest HRCT and pulmonary function tests in lung-onset primary sjogren’s syndrome. Immun Inflamm Dis. 2023;11(8):e957. doi: 10.1002/iid3.957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta S, Ferrada MA, Hasni SA. Pulmonary manifestations of primary Sjögren’s syndrome: underlying immunological mechanisms, clinical presentation, and management. Front Immunol. 2019;10:1327. doi: 10.3389/fimmu.2019.01327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luppi F, Sebastiani M, Sverzellati N, Cavazza A, Salvarani C, Manfredi A. Lung complications of Sjogren syndrome. Eur Res Rev. 2020;29(157):200021. doi: 10.1183/16000617.0021-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang M, Wu Y, Liu X, et al. Efficacy and safety of antifibrotic agents in the treatment of CTD-ILD and RA-ILD: a systematic review and meta-analysis. Respir Med. 2023;216:107329. doi: 10.1016/j.rmed.2023.107329 [DOI] [PubMed] [Google Scholar]
- 32.Matteson EL, Kelly C, Distler JHW, et al. Nintedanib in patients with autoimmune disease-related progressive fibrosing interstitial lung diseases: subgroup analysis of the INBUILD trial. Arthritis Rheumatol. 2022;74(6):1039–1047. doi: 10.1002/art.42075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oldham JM, Lee CT, Wu Z, et al. Lung function trajectory in progressive fibrosing interstitial lung disease. Eur Respir J. 2022;59(6):2101396. doi: 10.1183/13993003.01396-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, Dong Xu, upon reasonable request.