

Abstract
In the context of cancer diagnosis and treatment, we consider the problem of constructing an accurate prediction rule on the basis of a relatively small number of tumor tissue samples of known type containing the expression data on very many (possibly thousands) genes. Recently, results have been presented in the literature suggesting that it is possible to construct a prediction rule from only a few genes such that it has a negligible prediction error rate. However, in these results the test error or the leave-one-out cross-validated error is calculated without allowance for the selection bias. There is no allowance because the rule is either tested on tissue samples that were used in the first instance to select the genes being used in the rule or because the cross-validation of the rule is not external to the selection process; that is, gene selection is not performed in training the rule at each stage of the cross-validation process. We describe how in practice the selection bias can be assessed and corrected for by either performing a cross-validation or applying the bootstrap external to the selection process. We recommend using 10-fold rather than leave-one-out cross-validation, and concerning the bootstrap, we suggest using the so-called .632+ bootstrap error estimate designed to handle overfitted prediction rules. Using two published data sets, we demonstrate that when correction is made for the selection bias, the cross-validated error is no longer zero for a subset of only a few genes.
As explained by Xiong et al. (1), there is increasing interest in changing the emphasis of tumor classification from morphologic to molecular. In this context, the problem is to construct a classifier or a prediction (discriminant) rule R that can accurately predict the class of origin of a tumor tissue with feature vector x, which is unclassified with respect to a known number g (≥2) of distinct tissue types. Here the feature vector x contains the expression levels on a very large number p of genes (features). In many applications g = 2, corresponding to cancer and benign tumors. To train the rule R, there are available n tumor tissue samples, denoted by x1,… , xn, of known classification. These data are obtained from n microarrays, where the jth microarray experiment gives the expression levels of the p genes in the jth tissue sample xj of the training set. A useful account of microarray technology and the use therein of the latest discriminant analysis techniques may be found in Dudoit et al. (2). Also, Xiong et al. (3) and Zhang et al. (4) have provided a concise account of previous work on discriminant analysis for this technology, which has been considered also in refs. 5–9, among other papers. Note that we are considering here only the supervised classification of tumor tissue samples and not the unsupervised classification (cluster analysis) of them as, for example, is done in refs. 10–15.
In a standard discriminant analysis, the number of training observations n usually is much larger than the number of feature variables p, but in the present context of microarray data, the number of tissue samples n is typically between 10 and 100, and the number of genes p is in the thousands. This situation presents a number of problems. First, the prediction rule R may not be able to be formed by using all p available genes. For example, the pooled within-class sample covariance matrix required to form Fisher's linear discriminant function is singular if n < g + p. Second, even if all the genes can be used as, say, with a Euclidean-based rule or a support vector machine (SVM; refs. 9, 16, and 17), the use of all the genes allows the noise associated with genes of little or no discriminatory power, which inhibits and degrades the performance of the rule R in its application to unclassified tumors. That is, although the apparent error rate (AE) of the rule R (the proportion of the training tissues misallocated by R) will decrease as it is formed from more and more genes, its error rate in classifying tissues outside of the training set eventually will increase. That is, the generalization error of R will be increased if it is formed from a sufficiently large number of genes. Hence, in practice consideration has to be given to implementing some procedure of feature selection for reducing the number of genes to be used in constructing the rule R.
A number of approaches to feature-subset selection have been proposed in the literature (18). All these approaches involve searching for an optimal or near optimal subset of features that optimize a given criterion. Feature-subset selection can be classified into two categories based or whether the criterion depends on the learning algorithm used to construct the prediction rule (19). If the criterion is independent of the prediction rule, the method is said to follow a filter approach, and if the criterion depends on the rule, the method is said to follow a wrapper approach.
Regardless of how the performance of the rule is assessed during the feature-selection process, it is common to assess the performance of the rule R for a selected subset of genes by its leave-one-out cross-validated (CV) error. But, if it is calculated within the feature-selection process, there is a selection bias in it when it is used as an estimate of the prediction error. As to be made more precise in Correction For Bias Selection, an external cross-validation (20) should be undertaken subsequent to the feature-selection process to correct for this selection bias. Alternatively, the bootstrap (21–23) can be used.
Previous Results
For three published data sets including the two considered in this paper, Xiong et al. (1) reported that Fisher's linear discriminant rule R formed with only two or three genes via sequential forward selection can achieve 90% or more accuracy of classification. During the selection process, the set S of all available tissue samples was used to carry out the feature selection in the training of R. To evaluate the accuracy of R for the chosen subset of genes, they randomly partitioned the tissue samples into two sets: a training set (S1) and a test set (S2). The rule R then was trained on S1 and tested on S2. The test error rate of R thus formed then was averaged over 200 such partitions (24). But this evaluation does not take into account the selection bias, because the test error is based on S2, which as a subset of S is used in the selection of the genes for R. Thus this test error gives too optimistic an assessment of the prediction error, although this bias is offset to some extent by the fact that the training subset S1 is smaller in size than the full training set S. For example, for the selected subset of three genes, they reported that for a 95/5% split of the data set into training/test sets, Fisher's rule had an (average) test error of 10.7 and 0% for the colon and leukemia data sets, respectively (see table 3 in ref. 1). This result contrasts with an estimated prediction error of ≈15 and ≈5% for the colon and leukemia data sets, respectively, as given by cross-validation and the bootstrap when correction for the selection bias is undertaken (discussed in Correction For Bias Selection).
In other work, Zhang et al. (4) demonstrated on the colon data (11) how a classification tree based on three genes had an error of only 2% after a localized validation. However, if these genes had been selected from the p = 2,000 available genes on the basis of some or all of the tissue samples used to train the classification tree, then this estimated error still would have a selection bias present in it.
Guyon et al. (9) noted for the leukemia data set that a support vector with recursive feature elimination (RFE) discovers two genes that yield a zero leave-one-out error. However, they relied on the internal CV error, which does not take into account the selection bias. In our results, we shall demonstrate that a selection bias is incurred with the RFE procedure in selecting genes for an SVM.
Before we proceed to present our results, we briefly describe two approaches for assessing the selection bias in practice.
Correction for Bias Selection
As explained above, the CV error of the prediction rule R obtained during the selection of the genes provides a too-optimistic estimate of the prediction error rate of R. To correct for this selection bias, it is essential that cross-validation or the bootstrap be used external to the gene-selection process. Of course a straightforward method of estimating the error rate of a prediction rule R is to apply it to a test sample that has been chosen randomly from the training tissue samples that are available. However, because the tissue samples in the test set must not be used in the training of the rule R, it means that R has to be formed from a reduced set of tissue samples. Given that the entire set of available tissue samples is relatively small, in practice one would like to make full use of all available tissue samples in the gene selection and training of the prediction rule R.
Cross-Validation.
With M-fold cross-validation of a prediction rule R, the training set is divided into M nonoverlapping subsets of roughly equal size. The rule is trained on M − 1 of these subsets combined together and then applied to the remaining subset to obtain an estimate of the prediction error. This process is repeated in turn for each of the M subsets, and the CV error is given by the average of the M estimates of the prediction error thus obtained. In the case of M = n, where each observation (tissue sample) in the training set is deleted in turn before it is allocated by the rule formed from the remaining (n − 1) observations, we obtain the so-called leave-one-out CV error.
In the present context where feature selection is used in training the prediction rule R from the full training set, the same feature-selection method must be implemented in training the rule on the M − 1 subsets combined at each stage of an (external) cross-validation of R for the selected subset of genes. Of course, there is no guarantee that the same subset of genes will be obtained as during the original training of the rule (on all the training observations). Indeed, with the huge number of genes available, it generally will yield a subset of genes that has at most only a few genes in common with the subset selected during the original training of the rule.
The leave-one-out CV error is nearly unbiased, but it can be highly variable. This proved to be the case in the present context where the prediction rule is formed from a relatively small subset of the available genes. Hence we considered 10-fold (M = 10) cross-validation, which gives a more biased but less variable estimate than the leave-one-out CV error. Leave-one-out cross-validation for bias correction due to gene selection has been considered in refs. 25 and 26 and by D. V. Nguyen and D. M. Rocke (personal communication).
Bootstrap Approach.
Suitably defined bootstrap procedures (22, 23) can reduce the variability of the leave-one-out error in addition to providing a direct assessment of variability for estimated parameters in the prediction rule. Also, if we take the number of bootstrap replications K to be less than n, it will result in some saving in computation time relative to leave-one-out cross-validation.
As discussed by Efron and Tibshirani (22), a bootstrap smoothing of leave-one-out cross-validation is given by the leave-one-out bootstrap error B1, which predicts the error at a point xj only from bootstrap samples that do not contain the point xj. To define B1 more precisely, suppose that K bootstrap samples of size n are obtained by resampling with replacement from the original set of n classified tissue samples. We let R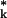 be the bootstrap version of the rule R formed from the kth bootstrap sample in exactly the same manner that R was formed from the original training set using the same gene-selection procedure. Then the (Monte Carlo) estimate of B1 on the basis of the K bootstrap samples is given by
be the bootstrap version of the rule R formed from the kth bootstrap sample in exactly the same manner that R was formed from the original training set using the same gene-selection procedure. Then the (Monte Carlo) estimate of B1 on the basis of the K bootstrap samples is given by
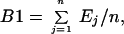 |
where
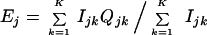 |
and Ijk is one if xj is not contained in the kth bootstrap sample and is zero otherwise, and Qjk is one if R misallocates xj and is zero otherwise.
misallocates xj and is zero otherwise.
Typically, B1 is based on ≈.632n of the original data points, and it was confirmed in the case where there is no feature selection that it closely agrees with half-sample cross-validation (23). Thus B1 is upwardly biased and Efron (23) proposed the .632 estimator,
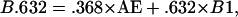 |
for correcting the upward bias in B1 with the downwardly biased AE.
For the present problem where the prediction rule is an overfit as a consequence of its being formed from a very large number of genes relative to the number of tissues, we need to use the so-called .632+ estimator, B.632+, which puts relatively more weight on the leave-one-out bootstrap error B1. It was proposed by Efron and Tibshirani (22) for highly overfit rules such as nearest-neighbors, where the AE is zero. The .632+ estimate is defined as
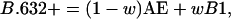 |
1 |
where the weight w is given by
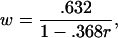 |
and where
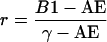 |
2 |
is the relative overfitting rate and γ is the no-information error rate that would apply if the distribution of the class-membership label of the jth tissue sample did not depend on its feature vector xj. It is estimated by
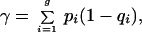 |
3 |
where pi is the proportion of the (original) tissue samples from the ith class, and qi is the proportion of them assigned to the ith class by R. The rate r may have to be truncated to ensure that it does not fall outside the unit interval [0,1] (22). The weight w ranges from .632 when r = 0 (yielding B.632) to 1 when r = 1 (yielding B1).
The B.632+ estimate hence puts more weight on the bootstrap leave-one-out error B1 in situations where the amount of overfitting as measured by B1 − AE is relatively large. It thus is applicable also in the present context where the prediction rule R is overfitted because of feature selection.
Backward Elimination with SVM
We consider two well known data sets in the microarray literature: the colon data analyzed initially by Alon et al. (11) and the leukemia data first analyzed in Golub et al. (5). Both data sets consist of absolute measurements from Affymetrix oligonucleotide arrays: the first contains n = 62 tissue samples on p = 2,000 human gene expressions (40 tumors and 22 normal tissues), and the second contains n = 72 tissue samples on p = 7,129 genes (47 cases of acute lymphoblastic leukemia and 25 cases of acute myeloid leukemia).
We first illustrate the selection bias incurred for an SVM with linear kernel. We considered the selection procedure of Guyon et al. (9), who used a backward selection procedure that they termed RFE. The selection considers all the available genes, because subset selection can be undertaken very quickly for an SVM using the vector weights as selection criteria.
We let CV1IE denote the CV (leave-one-out) internal error that does not correct for the selection bias. We let CV10E denote the CV 10-fold error obtained by an external cross-validation, which thus corrects also for the selection bias. The .632+ bootstrap error estimate, B.632+, was formed by using K = 30 bootstrap replications for each of the 50 splits of a full training set.
To illustrate the size of the selection bias for the colon data set, we split it into a training and a test set each of size 31 by sampling without replacement from the 40 tumor and 22 normal tissues separately such that each set contained 20 tumor and 11 normal tissues. The training set is used to carry out gene selection and form the error rate estimates AE, CV1IE, and CV10E for a selected subset of genes. An unbiased error rate estimate is given by the test error, equal to the proportion of tissues in the test set misallocated by the rule. We calculated these quantities for 50 such splits of the colon data into training and test sets. For the leukemia data set, we divided the set of 72 tissues into a training set of 38 tissues (25 acute lymphoblastic leukemia and 13 acute myeloid leukemia) and a test set of 34 tissues (22 acute lymphoblastic leukemia and 12 acute myeloid leukemia) for each of the 50 splits. The average values of the error rate estimates are plotted in Fig. 1, and the corresponding averages for the leukemia data are plotted in Fig. 2. The error bars on the test error refer to the 95% confidence limits.
Figure 1.

Error rates of the SVM rule with RFE procedure averaged over 50 random splits of the 62 colon tissue samples into training and test subsets of 31 samples each. TE, test error.
Figure 2.

Error rates of the SVM rule with RFE procedure averaged over 50 random splits of the 72 leukemia tissue samples into training and test subsets of 38 and 34 samples, respectively. TE, test error.
It can be seen from Fig. 1 that the true prediction error rate as estimated by the test error is not negligible, being above 15% for all selected subsets. The lowest value of 17.5% occurs for a subset of 26 genes. Similarly, for the leukemia data set, it can be seen from Fig. 2 that the selection bias cannot be ignored when estimating the true prediction error, although it is smaller (≈5%) for this second set as the leukemia classes acute lymphoblastic leukemia and acute myeloid leukemia are separated more easily.
Concerning the estimation of the prediction error by 10-fold cross-validation and the bootstrap, it can be seen that CV10E has little bias for both data sets. For the colon data set, the bootstrap estimate B.632+ is more biased. However, B.632+ was found to have a slightly smaller root mean squared error than CV10E for the selected subsets of both data sets. Efron and Tibshirani (22) comment that future research might succeed in producing a better compromise between the unbiasedness of cross-validation and the reduced variance of the leave-one-out bootstrap. It would be of interest to consider this for the present problem, where there is also feature-selection bias to be corrected for.
We observed comparable behavior of the SVM rule formed using all the available tissue samples (62 and 72 tissue samples for the colon and leukemia data sets, respectively). As to be expected for training samples of twice or near twice the size, the (estimated) bias was smaller: between 10 and 15% for the colon data set and between 2 and 3% for the leukemia data set.
It can be seen from Figs. 1 and 2 that the estimated prediction rate according to B.632+ and CV10E remains essentially constant as genes are deleted in the SVM until ≈64 or so genes when these estimates start to rise sharply. The CV1IE error, which is uncorrected for selection bias, also starts to rise then. Hence feature selection provides essentially little improvement in the performance of the SVM rule for the two considered data sets, but it does show that the number of genes can be reduced greatly without increasing the prediction error.
It is of interest to note that the plot by Guyon et al. (9) for their leave-one-out CV error is very similar to our plot of CV1IE in Fig. 1. However, for this data set, Guyon et al. transformed the normalized (logged) data further by a squashing function to “diminish the importance of outliers,” which could have an effect on the selection bias. For example, for this data set there is some doubt (7) as to the validity of the labels of some of the tissues, in particular, tumor tissues on patients 30, 33, and 36 and normal tissues on patients 8, 34, and 36 as labeled in ref. 11. When we deleted these latter tissue samples, the selection bias of the rule was estimated to be almost zero. But in a sense this is to be expected, for if all tissue samples that are difficult to classify are deleted, then the rule should have a prediction error that is close to zero regardless of the selected subset of genes.
Forward Selection with Fisher's Rule
We now consider the selection bias incurred with a sequential forward selection procedure for the rule based on Fisher's linear discriminant function. For the selection of genes for Fisher's rule, we first reduced the set of available genes to 400 for each data set to reduce the computation time. This exercise was undertaken by selecting the top 400 genes as ranked in terms of increasing order of the average of the maximum (estimated) posterior probabilities of class membership. This initial selection will incur some (small) bias, which we shall ignore here in our illustrative examples. The forward selection procedure was applied with the decision to add a feature (gene) based on the leave-one-out CV error. Here we used all the available tissue samples (62 for the colon and 72 for the leukemia data sets) to train the Fisher rule, and thus there was no test set available in each case. The cross-validation and bootstrap estimates of the prediction error of Fisher's rule formed via forward selection of the genes are plotted in Figs. 3 and 4 for the colon and leukemia data sets, respectively.
Figure 3.

Error rates of Fisher's rule with stepwise forward selection procedure using all the colon data (62 samples).
Figure 4.

Error rates of Fisher's rule with stepwise forward selection procedure using all the leukemia data (72 samples).
Considering the colon data set, it can be seen in Fig. 3 that the leave-one-out CV error, CV1IE (with the selection bias still present), is optimized (6.5%) for only three genes. However, the CV error CV10E, which has the selection bias removed, is approximately equal to 15% for more than seven genes. The fact that the estimates B.632+ and CV10E are ≈15% for eight or so genes for the colon data set would appear reasonable given that this set has six tissues, the class of origin of which is in some doubt.
Similarly, it can be seen in Fig. 4 that the selection bias incurred with forward selection of genes for Fisher's rule on the leukemia data set is not negligible. As a consequence, although the leave-one-out error CV1IE is zero for only three selected genes, the 10-fold CV error CV10E and the bootstrap B.632+ error estimate are ≈5%.
The results described above for CV1IE for Fisher's rule formed via forward selection of the genes are in agreement with the results of Xiong et al. (1). Their error rate corresponding to our CV1IE was reported to be equal to 6.5 and 0% for the colon and leukemia data sets, respectively (see table 2 in ref. 1).
Comparing the performance of forward selection with Fisher's rule to backward elimination with SVM, we found that the latter procedure leads to slightly better results, with a 2 or 3% improved error rate for both data sets. When trying forward selection with SVM, we found very similar results to that obtained with Fisher's rule with a comparable number of genes. Hence it seems that the selection method and the number of selected genes are more important than the classification method for constructing a reliable prediction rule.
Discussion
For two data sets commonly analyzed in the microarray literature, we have demonstrated that it is important to correct for the selection bias in estimating the prediction error for a rule formed by using a subset of genes selected from a very large set of available genes. To illustrate this selection bias further, we generated a no-information training set by randomly permuting the class labels of the colon tissue samples. For each of these 20 no-information sets, an SVM rule was formed by selecting genes by the RFE method, and the AE and the leave-one-out CV error CV1IE were calculated. The average values of these two error rates and the no-information error γ (Eq. 3) over the 20 sets are plotted in Fig. 5, where we also plotted the average value of the CV10E and B.632+ error estimates that correct for the selection bias. It can be seen that although the feature vectors have been generated independently of the class labels, we can form an SVM rule that has not only an average zero AE but also an average CV1IE error close to zero for a subset of 128 genes and ≈10% for only eight genes in the selected subset. It is reassuring to see that the error estimates CV10E and B.632+, which correct for the selection bias, are between 0.40 and 0.45, consistent with the fact that we are forming a prediction rule on the basis of a no-information training set.
Figure 5.

Error rates of the SVM rule averaged over 20 noninformative samples generated by random permutations of the class labels of the colon tumor tissues.
From the three examples presented, it can be seen that it is important to recognize that a correction for the selection bias be made in estimating the prediction error of a rule formed by using genes selected from a very large set of available genes. It is important also to note that if a test set is used to estimate the prediction error, then there will be a selection bias if this test set was used also in the gene-selection process. Thus the test set must play no role in the feature-selection process for an unbiased estimate to be obtained.
Given that there is usually only a limited number of tissue samples available for the training of the prediction rule, it is not practical for a subset of tissue samples to be put aside for testing purposes. However, we can correct for the selection bias either by cross-validation or the bootstrap as implemented above in the examples. Concerning the former approach, an internal cross-validation does not suffice. That is, an external cross-validation must be performed whereby at each stage of the validation process with the deletion of a subset of the observations for testing, the rule must be trained on the retained subset of observations by performing the same feature-selection procedure used to train the rule in the first instance on the full training set.
Abbreviations
- AE
apparent error rate
- CV
cross-validated
- RFE
recursive feature elimination
- SVM
support vector machine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Xiong M, Li W, Zhao J, Jin L, Boerwinkle E. Mol Genet Metab. 2001;73:239–247. doi: 10.1006/mgme.2001.3193. [DOI] [PubMed] [Google Scholar]
- 2.Dudoit S, Fridlyand J, Speed T P. J Am Stat Assoc. 2002;97:77–87. [Google Scholar]
- 3.Xiong M, Fang X, Zhao J. Genome Res. 2001;11:1878–1887. doi: 10.1101/gr.190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H, Yu C-Y, Singer B, Xiong M. Proc Natl Acad Sci USA. 2001;98:6730–6735. doi: 10.1073/pnas.111153698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golub T R, Slonim D K, Tamayo P, Huard C, Gassenbeck M, Mesirov J P, Coller H, Loh M L, Downing J R, Caligiuri M A, et al. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 6.Moler E J, Chow M L, Mian I S. Physiol Genomics. 2000;4:109–126. doi: 10.1152/physiolgenomics.2000.4.2.109. [DOI] [PubMed] [Google Scholar]
- 7.Chow M L, Moler E J, Mian I S. Physiol Genomics. 2001;5:99–111. doi: 10.1152/physiolgenomics.2001.5.2.99. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen D V, Rocke D M. In: Methods of Microarray Data Analysis. Lin S M, Johnson K F, editors. Dordrecht, The Netherlands: Kluwer; 2001. pp. 109–124. [Google Scholar]
- 9.Guyon I, Weston J, Barnhill S, Vapnik V. Mach Learn. 2002;46:389–422. [Google Scholar]
- 10.Eisen M B, Spellmann P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;195:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alon U, Barkai N, Notterman D A, Gish K, Ybarra S, Mack D, Levine A J. Proc Natl Acad Sci USA. 1999;96:6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Dor A, Bruhn L, Friedman N, Nachman I, Schummer M, Yakhini Z. J Comp Biol. 2000;7:559–584. doi: 10.1089/106652700750050943. [DOI] [PubMed] [Google Scholar]
- 13.Getz G, Levine E, Domany E. Proc Natl Acad Sci USA. 2000;97:12079–12084. doi: 10.1073/pnas.210134797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sorlie T, Perou C M, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen M B, van de Rijn M, Jeffrey S S, et al. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLachlan G J, Bean R W, Peel D. Bioinformatics. 2002;18:413–422. doi: 10.1093/bioinformatics/18.3.413. [DOI] [PubMed] [Google Scholar]
- 16.Furey T S, Cristianini N, Duffy N, Bednarski D W, Schummer M, Haussler D. Bioinformatics. 2000;16:906–914. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- 17.Brown M P S, Grundy W N, Lin D, Cristianini N, Sugnet C W, Furey T S, Ares M, Jr, Haussler D. Proc Natl Acad Sci USA. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kudo M, Sklansky J. Pattern Recognit. 2000;33:25–41. [Google Scholar]
- 19.Kohavi R, John G H. Artif Intell. 1996;97:273–324. [Google Scholar]
- 20.McLachlan G J. Discriminant Analysis and Statistical Pattern Recognition. New York: Wiley; 1992. [Google Scholar]
- 21.Efron B. Ann Statist. 1979;7:1–26. [Google Scholar]
- 22.Efron B, Tibshirani R. J Am Stat Assoc. 1997;92:548–560. [Google Scholar]
- 23.Efron B. J Am Stat Assoc. 1983;78:316–331. [Google Scholar]
- 24.Roth F P. Genome Res. 2001;11:1801–1802. doi: 10.1101/gr.215501. [DOI] [PubMed] [Google Scholar]
- 25.Spang R, Blanchette C, Zuzan H, Marks J R, Nevins J R, West M. In Silico Biol. 2001. www.bioinfo.de/isb/gcb01/talks/spang/ , www.bioinfo.de/isb/gcb01/talks/spang/. . [PubMed] [Google Scholar]
- 26.West M, Blanchette C, Dressman H, Huang E, Ishida S, Spang R, Zuzan H, Olsen J A, Jr, Marks J R, Nevins J R, et al. Proc Natl Acad Sci USA. 2001;98:11462–11467. doi: 10.1073/pnas.201162998. [DOI] [PMC free article] [PubMed] [Google Scholar]