

Abstract
Although incorporation of amino acid analogs provides a powerful means of producing new protein structures with interesting functions, many amino acid analogs cannot be incorporated easily by using the wild-type aminoacyl-tRNA synthetase (aaRS). To be able to incorporate specific amino acid analogs site-specifically, it is useful to build a mutant aaRS that preferentially activates the analog compared with the natural amino acids. Experimental combinatorial studies to find such mutant aaRSs have been successful but can easily become costly and time-consuming. In this article, we describe the clash opportunity progressive (COP) computational method for designing a mutant aaRS to preferentially take up the analog compared with the natural amino acids. To illustrate this COP procedure, we apply it to the design of mutant Methanococcus jannaschii tyrosyl-tRNA synthetase (M.jann-TyrRS). Because the three-dimensional structure for M.jann-TyrRS was not available, we used the STRUCTFAST homology modeling procedure plus molecular dynamics with continuum solvent forces to predict the structure of wild-type M.jann-TyrRS. We validate this structure by predicting the binding site for tyrosine and calculating the binding energies of the 20 natural amino acids, which shows that tyrosine binds the strongest. With the COP design algorithm we then designed a mutant tyrosyl tRNA synthetase to activate O-methyl-l-tyrosine preferentially compared with l-tyrosine. This mutant [Y32Q, D158A] is similar to the mutant designed with combinatorial experiments, [Y32Q, D158A, E107T, L162P], by Wang et al. [Wang, L., Brock, A., Herberich, B. & Schultz, P. G. (2001) Science 292, 498–500]. We predict that the new one will have much greater activity while retaining significant discrimination between O-methyl-l-tyrosine and tyrosine.
Proteins are synthesized with precise control over sequence, leading to the vast range of specific structures and functional properties observed in nature. Even so, the monomer pool for proteins is limited to the 20 natural amino acids. Increasing the monomer pool by incorporating new amino acid analogs would allow development of fascinating new bioderived polymers exhibiting novel but well controlled architectures (1, 2) and could lead to many interesting applications ranging from incorporating a fluorescence probe to elucidating specifics of protein structure and function (3), to incorporating selenium-substituted serine to facilitate crystallization processes in proteins (4).
The in vivo incorporation of amino acid analogs into proteins is controlled in large measure by aminoacyl-tRNA synthetases (aaRS), the class of enzymes that safeguards the fidelity of amino acid incorporation into proteins. It has been demonstrated that the wild-type translational apparatus can be used to incorporate some amino acid analogs into protein (5–11). However, few amino acid analogs have been incorporated in proteins in vivo and the functionalities of these analogs have been limited. To expand the range of amino acid analogs that can be incorporated in vivo, it is desirable to manipulate the activity of the aaRS (12, 13). There has been steady progress in developing the 21st aaRS-suppressor tRNA pairs in vivo (14, 15). A big success is the design of a novel orthogonal tRNA and tyrosyl-tRNA synthetase (TyrRS) from Methanococcus jannaschii TyrRS (hereafter denoted as M.jann-TyrRS) that incorporates O-methyl-l-tyrosine (OMe-Tyr) site-specifically in protein in response to an amber nonsense codon (16). Such procedures have tremendous potential to expand the genetic codes in living cells, but the current combinatorial experiments, which considered 520 mutation trials on 5 residues expected to be at the binding site of the tyrosine ligand, can become cumbersome. This article describes the clash opportunity progressive design algorithm (denoted as COP) and its application to redesign the binding site of M.jann-TyrRS for the preferential binding of OMe-Tyr over Tyr. COP design leads to three mutants expected to bind OMe-Tyr strongly while discriminating against Tyr. The best mutant [Y32Q, D158A] is similar to the one [Y32Q, D158A, E107T, L162P], designed by Wang et al. (16) by using combinatorial experiments. We predict that the new mutant will have much greater activity while retaining significant discrimination between OMe-Tyr and Tyr. The COP procedure should allow the efficient design of mutant aaRS for incorporation of other new analogs.
Because there is no crystal structure available for M.jann-TyrRS, we predicted the three-dimensional structure for wild-type M.jann-TyrRS, by combining the structfast sequence alignment and structure prediction algorithm with molecular dynamics (MD) including continuum solvent forces. [To select the 5 residues to modify in their experiments, Wang et al. (16) used a sequence alignment between M.jann-TyrRS and Bacillus stearothermophillus TyrRS.] To validate the predicted structure for M.jann-TyrRS, we use MD plus continuum solvent energies to demonstrate that Tyr is the preferred ligand over the 19 other natural amino acids.
Methods
Structure Prediction of Wild-Type M.jann-TyrRS.
No crystal structure is available for M.jann-TyrRS, but three crystal structures (2ts1, 3ts1, and 4ts1) have been reported for B. stearothermophillus TyrRS. The 2ts1 structure has no bound ligand (17), 3ts1 has the Tyr-AMP complex bound (17), and 4ts1 has just the Tyr bound (18). By using the sequence of wild-type M.jann-TyrRS from GenBank (accession no. Q57834), the three-dimensional structure of the main chain of M.jann-TyrRS was predicted with the structfast (D.D. and W.A.G., unpublished result) homology modeling technique, with the 4ts1 crystal structure as template. The sequence identity between the two sequences is 32.1%. The main chain atoms of the predicted M.jann-TyrRS structure agree with the corresponding residues of 4ts1 structure to 0.64 Å coordinates rms difference after aligning the two structures by using dali (19).
To place the Tyr ligand in the predicted structure, we matched the side chain conformation of the five conserved residues (Tyr-32, Tyr 151, Gln-155, Asp-158, and Gln-173) in the binding site of M.jann-TyrRS with those conformations from the 4ts1 crystal structure. The rest of the side chains for the predicted M.jann-TyrRS were added by using scwrl version 2.7 (20, 21) keeping the conformation of the binding site fixed. The resulting structure was then minimized with mpsim (22), allowing all side-chain atoms to move but keeping the main chain fixed. The MD and minimization calculations with mpsim used the cell multipole method (23) to calculate the nonbond interactions. The protein was described by using the dreiding force field (24) with CHARMM22 (25) charges.
For the Tyr ligand and the OMe-Tyr analog, we used Mulliken charges based on molecular orbitals from quantum mechanics (QM). The QM calculations were at the HF level with the 6-31G** basis set. Here the geometry was optimized by using forces calculated with Poisson Boltzmann continuum solvation (26). QM calculations were carried out with JAGUAR4.0 (Schrödinger, Portland, OR).
After optimizing the side-chain configurations in the protein, the energy of the whole protein was minimized with all atoms movable but with distance constraints on the hydrogen bonds between the phenolic OH group of the Tyr ligand and the Tyr-32 and Asp-158 side chains. This minimized structure was then used as the starting structure for annealing MD, where all constraints were relaxed. Each cycle of annealing MD involved heating the system from 50 to 600 K and cooling from 600 to 50 K in steps of 20 K for 0.5 ps. These annealing MD calculations included solvent forces from the surface-generalized born (SGB) continuum solvation method (27) with a dielectric constant of 80 and a solvent radius of 1.4 A. The final structure, shown in Fig. 1, was used to predict the binding of all 20 natural amino acids and to design the mutant protein.
Figure 1.

Comparison of predicted M.jann-TyrRS (in blue) and the crystal structure for B. stearothermophillus TyrRS (4ts1) (in yellow). The Tyr ligand is shown as a ball model.
Docking All 20 Amino Acids to M.jann-TyrRS.
In this article we assume that selectivity in the binding of the amino acid or analog to the aaRS is necessary for the activation of amino acid by aaRS. It is known that TyrRS shows selectivity binding tyrosine over 19 other amino acids (28, 29). It has also been shown that the calculated binding energies correlate well with the measured in vivo translational activities of a set of Phe analogs to PheRS (30).
Thus, to test over the predicted M.jann-TyrRS sructure 20 natural amino acids were docked into the Tyr binding site. The resulting 20 mutated protein structures were then minimized followed by annealing MD.
The binding energy of each amino acid is calculated as
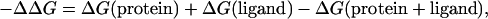 |
1 |
where ΔG(protein + ligand) is the free energy for the protein-ligand complex, ΔG(protein) is the free energy for the protein, and ΔG(ligand) is the free energy for the ligand alone. Because the structure optimizations included solvation forces with the SGB continuum solvent approximation with the experimental dielectric constant, we considered that the calculated energies were free energies (31).
Mutant Design for Preferential Binding of an Amino Acid Analog.
We next describe the COP procedure for structure-based rational redesign of a binding site. This procedure is useful in predicting which mutations in the binding site are essential for preferential binding to a specific ligand. We demonstrate the design strategy by designing aaRS mutants that activate a specific amino acid analog preferentially compared to all natural amino acids. The mutants are selected based on the calculated differential binding energy of the desired analog against any other potential competitor ligand that might bind to the mutant. For example, in redesigning TyrRS, we calculate the differential binding energy of the analog against Tyr and Phe. For cases in which the analog is much larger than Tyr, then we might consider Trp as a potential competitor for the redesigned mutant aaRS. The COP design procedure for designing mutant aaRS comprises the progressive sequence of steps.
Step 0: Conformation determination.
We first determine the favorable conformations of the analog. We generated the various rotamers of the ligand over a grid of dihedral angles and calculated their energies in solution with quantum mechanics.
Step 1: Clash identification.
The low energy rotamers from step 0 are then docked into the binding site. To do this the natural amino acid in the binding pocket was replaced with the energetically favorable rotamers of the analog while keeping the backbone of the ligand fixed (in order that the reaction center for the formation of the aminoacyl–AMP complex would be retained for the analog). Then the analog rotamer was matched onto the binding site, and the nonbond energy contributions (Ek) were calculated for each residue k in the binding pocket, using Eq. 2 [the functional forms for these Coulomb, Van der Waals, and hydrogen bond nonbond interactions are from the dreiding force field (24)]:
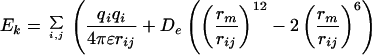 |
2 |
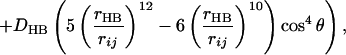 |
where i and j sum over all atoms in the ligand and protein residue k, of interest, qi and qj are partial charges on atoms i and j, respectively. rij is the distance between atoms i and j, rm and De are van der Waals distance and well depth of atoms i and j, and rHB and DHB are hydrogen bond distance and well depth, respectively. θ is the hydrogen bond angle between atoms i and j and their bridging hydrogen atom.
Those residues in the wild-type protein having bad clashes with the analog are marked for mutation. Because the protein backbone is fixed in step 2, we require that the analog rotamer should not clash with the backbone of the protein. Analog rotamers having a severe clash with the protein backbone are discarded. Table 3 shows an example of the bad clashes of the analog in the wild-type protein.
Table 3.
Interaction energies of OMe-Tyr ligand (both rotamers) and of Tyr ligand with the predicted structure (wild type) of M.jann-TyrRS
| Residue | OMe-Tyr (rotamer 1) | OMe-Tyr (rotamer 2) | Tyr | Diff‡ |
|---|---|---|---|---|
| Gln-155 | −11.15 | −10.65 | −10.66 | −0.48 |
| Met-154 | −0.93 | −0.60 | −0.60 | −0.32 |
| Ala-67 | −1.69 | −1.43 | −1.42 | −0.27 |
| Gln-109 | −0.99 | −0.91 | −0.91 | −0.08 |
| Leu-66 | −0.21 | −0.13 | −0.13 | −0.08 |
| Asn-157 | 0.13 | 0.20 | 0.20 | −0.08 |
| Val-156 | −0.24 | −0.17 | −0.17 | −0.07 |
| Phe-108 | −0.16 | −0.10 | −0.10 | −0.06 |
| Leu-65 | −1.28 | 265.94* | −1.23 | −0.05 |
| His-160 | −0.25 | −0.21 | −0.21 | −0.04 |
| Gly-105 | −0.08 | −0.03 | −0.03 | −0.04 |
| Phe-35 | −1.52 | −1.50 | −1.49 | −0.04 |
| Pro-152 | −0.32 | −0.28 | −0.28 | −0.04 |
| Ile-159 | −0.05 | −0.02 | −0.02 | −0.03 |
| Ile-33 | −0.23 | −0.21 | −0.20 | −0.03 |
| His-70 | −2.89 | −2.87 | −2.88 | −0.01 |
| Tyr-161 | −0.08 | −0.06 | −0.07 | −0.01 |
| His-177 | −0.38 | −0.39 | −0.37 | −0.01 |
| Leu-69 | 0.02 | 0.02 | 0.02 | 0.00 |
| Gly-34 | −2.04 | 302.84† | −2.06 | 0.02 |
| Gln-173 | −11.15 | −11.20 | −11.17 | 0.03 |
| Asp-68 | −0.94 | −0.98 | −0.97 | 0.03 |
| Tyr-151 | −9.44 | −9.52 | −9.66 | 0.21 |
| Glu-36 | −1.14 | −1.27 | −1.36 | 0.22 |
| Tyr-32 | −13.45 | 12745.3* | −15.69 | 2.25 |
| Asp-158 | 2450.97* | −0.80 | −15.44 | 2466.41 |
The interactions are shown for all ligands with any atom within 6 Å of the Tyr (referred to as the binding site).
Large van der Waals energy showing steric clashes of protein side chain with OMe-Tyr ligand.
Steric clash with main chain.
Difference between OMe-Tyr rotamer 1 and Tyr.
Step 2: Relieving clashes with point mutations.
Those residues having bad clashes with the analog are mutated sequentially to all of the other 19 amino acids. These point mutations use a backbone-dependent side-chain rotamer library (20). The backbone of the protein is held fixed in this stage. After each point mutation, the side chain alone is energy minimized while keeping the rest of the protein fixed. Then the contribution from this mutated side chain to the binding energy of both the analog and the wild-type amino acid are calculated by using Eq. 2. Also, the clashes of this mutated residue with neighboring residues in the protein are calculated. The best mutations are selected with a scoring energy function consisting of a weighted sum of the differential nonbond interaction energy of the mutated residue with the ligand and the analog, and the nonbond interaction energy of the mutated residue with the rest of the residues in the protein. Generally the weights are 0.75–1.0 for ligand–protein interaction, and 0.0–0.25 for protein–protein interactions. We also add desolvation penalty to the energy of the mutated residue, calculated with the SGB continuum solvent method with mpsim. Here we calculate the differential binding energies for all 20 possible amino acid mutations, as in Table 4. This procedure is repeated for all of the residues showing a clash in the binding pocket of the analog (step 1). The mutation candidates for further consideration are selected based on the scoring energy between the analog and the natural ligand.
Table 4.
Binding scores of the best 6 mutations for Tyr-32 and Asp-158 in M.jann-TyrRS
| Tyr-32 | Tyr | OMe-Tyr | Diff |
|---|---|---|---|
| Glu | 0.13 | −0.28 | −0.41 |
| Asp | −0.14 | −0.37 | −0.23 |
| Gln | −0.10 | −0.28 | −0.18 |
| Met | −0.32 | −0.37 | −0.05 |
| Phe | −0.45 | −0.49 | −0.04 |
| Ser | −0.08 | −0.07 | 0.01 |
| Asp-158 | |||
| Ala | −0.41 | −0.92 | −0.51 |
| Gly | −0.26 | −0.08 | 0.18 |
| Ser | −0.52 | 2.68 | 3.20 |
| Cys | −0.99 | 4.88 | 5.87 |
| Asp | −1.70 | 4.36 | 6.06 |
| Asn | −0.64 | 10.54 | 11.18 |
Scores (in kcal/mol) are nonbond interaction energies of the mutated residue with the OMe-Tyr or Tyr. Based on these results, we selected the 5 mutations with negative difference for Tyr-32, and the one case for Asp-158.
Step 3: Stabilizing point mutations (opportunities).
After identifying candidate mutation for relieving clashes, we then look for opportunities for mutations in the binding pocket that would stabilize the analog ligand or disrupt the bonding with the natural ligand. Thus, we consider residues near the ligand (within 6 Å) and look for residues that might, for example, take advantage of hydrogen bond donor or acceptor atoms that are different between the analog and the natural ligand. These opportunity mutations are selected with the same procedure as for clash mutations.
Step 4: Combined mutations.
Steps 2 and 3 lead to a subset of mutation candidates that are expected to either relieve clashes or provide stabilization opportunities to the binding of the analog in preference to the wild-type ligand. In step 4, we generate simultaneous mutations from each of the chosen subsets of mutations. For example, if the clash analysis (step 2) leads to 3 residues with 2, 3, and 4 candidates and the opportunity analysis leads to 1 residue with 5 possible mutation candidates (say for making hydrogen bonds with the analog), then we would consider 2 × 3 × 4 × 5 = 120 possible protein mutants. We generate the best possible rotamer combination for each of these 120 mutant proteins (optimizing the side chains by conjugate gradient minimization). Then, after selecting optimum side chains for all 120 cases, the structure of the whole mutant protein is minimized both with the natural ligand and with the analog. Finally, the differential binding energy of the analog to the natural amino acid in the mutant is calculated by using Eq. 1 with dreiding force field and including SGB solvation.
Step 5: Relaxation of the free mutant protein without the ligand.
In step 4, we considered mutations and side-chain conformations that enhance binding of the protein to the analog. However, it is possible that some mutations would disrupt the folding of the free protein when the ligand is not present. Thus, for the best mutants selected from step 4, we reoptimize the side chains without the ligand in the binding site. The optimization allows the side chain of each mutation to go into the part of the binding site normally occupied by the ligand. In this step, we first reselect the side-chain conformation from the side-chain library and then optimize the structure of the full protein with the SGB continuum solvent procedure. Once the mutant structure is optimized without ligand, the ligand is then matched on to the binding site and the potential energy of the resulting structure is minimized with SGB solvation. This procedure is done for both the analog and the natural ligand (and any other ligands that might bind to the mutated site). For the analog ligand this will generally lead to a weaker binding energy than step 4, because we now include the penalty paid to push the side chains away from the binding site as the ligand binds. However, the natural ligand may have a stronger binding energy for step 5. Thus, the differential binding energy in step 5 will generally be smaller than in step 4. We denote this differential binding energy as the “relaxed protein binding energy,” because the mutants were optimized with no ligand in the binding pocket.
Step 6: Selection.
From steps 4 and 5 we select candidate mutants with both good binding energies to the analog and high differential binding energies to the natural amino acid. While redesigning aaRS for binding to a specific analog, it is important that the mutant aaRS activates only the analog and not any other natural amino acid. Thus, the best candidate mutants are tested further for binding to other natural amino acids. To do this testing we dock likely natural amino acid competitors into both the relaxed and optimized binding sites, using the procedures described in step1. The binding energy is calculated for each ligand/mutant pair. The mutants are finally ranked by the difference in binding energies between the analog and its competitors. The better binding energy is taken either from the relaxed or the optimized mutant cases.
Steps 1–6 are repeated for other low energy rotamers of the analog from step 0.
Results and Discussion
Assessment of the Quality of the Predicted Structure for M.jann-TyrRS.
Fig. 1 superimposes the experimental crystal structure of B. stearothermophillus TyrRS (4ts1) with the predicted structure of M.jann-TyrRS with the Tyr ligand shown in the binding site. Although the sequence identity is 32.1%, the general folding is very similar for the two proteins. The backbone structure, predicted with structfast, led to an initial coordinate rms (Crms) difference in the main chain atoms between the two superimposed structures of 0.64 Å before structure optimization. After the side-chain optimization, the Crms (main chain) increases to 1.75 Å for the 139 residues aligned structurally. However, the conserved residues (Tyr-32, Tyr 151, Gln-155, Asp-158, and Gln-173) in the binding sites have a Crms difference of 0.62 Å (all atoms except hydrogens). These comparisons were made with the dali structural-alignment program (19) to superimpose structures.
The main difference between the two structures is that M.jann-TyrRS lacks the α-helical domain present in 4ts1 (residues 246–317). This domain is involved only in tRNA binding, which is consistent with the observation that M.jann exhibits only a minimalist tRNA anticodon loop-binding domain (32).
For M.jann-TyrRS, the tyrosine ligand binds in the deep cleft formed by several α-helices and β-strands in the α/β domain. The phenolic hydroxyl group of the Tyr ligand makes hydrogen bonds with Tyr-32-Oη and Asp-158-Oδ1, both with a hydrogen bond distance of 2.87 Å. The amino group of the ligand tyrosine makes three hydrogen bonds with Tyr-151-Oη, Gln-155-Oγ1, and Gln-173-Oγ1. Table 1 lists the hydrogen bond distances that the Tyr ligand makes in the binding pocket. Comparison of these distances with the 4ts1 structure (see Table 1) shows that the hydrogen bonds made by the Tyr ligand are very similar. After publication, the PDB coordinates for the predicted M.jann-TyrRS structure can be downloaded from the Molecular Simulation Center publication section at http://www.wag.caltech.edu/.
Table 1.
Hydrogen bonds in the binding site of the predicted M.jann-TyrRS structure, compared with the hydrogen bonds in B.sther-TyrRS crystal structure (PDB ID code 4ts1)
|
M.jann-TyrRS
|
B.sther-TyrRS (4ts1)
|
|||
|---|---|---|---|---|
| Ligand atoms | Protein atom | HB distance, Å | Protein atom | HB distance, Å* |
| Oη | Tyr-32 Oη | 2.80 | Tyr-34 Oη | 2.93 (2.87) |
| Oη | Asp-158 Oδ1 | 3.02 | Asp-176 Oδ1 | 2.27 (2.83) |
| N | Gln-75 Oɛ1 | 3.14 | Asp-78 Oδ1 | 2.91 (2.87) |
| N | Tyr-151 Oη | 2.83 | Tyr-169 Oη | 2.78 (2.94) |
| N | Gln-173 Oɛ1 | 3.12 | Gln-173 Oɛ1 | 3.13 (3.28) |
| OXT | Gln-75 Nɛ2 | 5.56† | Lys-82 Nɛ | 4.83 (4.97)† |
The values quoted here are from the crystal structure for B.sther-TyrRS (PDB ID code 4ts1). The values in parentheses are after minimization with the dreiding force field.
Water mediated hydrogen bonds (HB).
Docking of the 20 Amino Acids to M.jann-TyrRS.
Table 2 shows the binding energies of all 20 amino acids when docked into the predicted M.jann-TyrRS structure. Here we first build the amino acid into the structure as in step 1. Further, we optimize the ligand with protein fixed and then optimize the neighboring ligands with the more distant residues fixed. Finally, all atoms of the protein were optimized, without constrain. All these calculations included the forces due to solvation by means of the SGB continuum solvent method. As expected, the wild-type ligand Tyr has a much higher binding energy (16 kcal/mol) than any other natural amino acid.
Table 2.
Binding energies (including solvation) for the 20 natural amino acids docked to the binding site of the predicted structure for M.jann-TyrRS
| Amino acid | Binding energy, kcal/mol | Amino acid | Binding energy, kcal/mol |
|---|---|---|---|
| Tyr | 43.8 | Val | 16.2 |
| Ala | 27.2 | Ile | 14.1 |
| Asn | 27.2 | Leu | 12.0 |
| His | 27.1 | Gln | 9.7 |
| Thr | 26.8 | Arg | 2.3 |
| Phe | 26.6 | Pro | 1.3 |
| Ser | 25.6 | Glu | −3.5 |
| Gly | 24.1 | Met | −13.8 |
| Cys | 22.9 | Trp | −20.6 |
| Asp | 16.4 | Lys | −56.9 |
Because several steps are involved in the activation of aaRS (29), of which selective binding of the amino acid is only one, we cannot be certain that only the natural ligand is bound strongly. However, TyrRS is known to activate only Tyr and exhibits high selectivity even in the binding of the amino acid (28, 29). Hence, it is plausible that only the Tyr is bound strongly, as predicted in Table 2. Thus, we consider that the results in Table 2 validate the accuracy of the predicted structure for M.jann-TyrRS.
Design of Mutant M.jann-TyrRS for OMe-Tyr.
Starting with the predicted structure for M.jann-TyrRS, we used the COP algorithm to design mutants of M.jann-TyrRS for selective binding of OMe-Tyr (see Scheme S1).
Scheme 1.

Tyr and the analog OMe-Tyr (rotamer 1) for which the mutant TyrRS was designed.
OMe-Tyr has two equally favorable rotamers, one shown in Scheme S1 (denoted 1 and the other with the −OMe group pointed down (denoted 2). Both rotamers were matched in the binding site of Tyr in wild-type M.jann-TyrRS, keeping the zwitterion end fixed in the structure. Component analysis of the energy contribution of each residue in the binding site to the binding of OMe-Tyr was calculated with Eq. 2. The binding site was defined as the entire residue for all atoms of the protein within 6 Å of any atom of the ligand, leading to the 26 residues in Table 3. Because rotamer 2 had severe clashes with protein backbone of Gly-34 and rotamer 1 had none, we considered only rotamer 1 further.
The nonbond interaction energies between OMe-Tyr and residues within 6 Å of the ligand are summarized in Table 3. We find that Asp-158 has a very severe clash with the OMe-Tyr, and hence this residue was selected to be mutated to favor OMe-Tyr. In addition, Tyr-32 has a strong binding contribution to Tyr over OMe-Tyr and hence we targeted Tyr-32 for mutations to find a residue favorable for OMe-Tyr.
We considered all 20 amino acids as possible mutations for both Tyr-32 and Asp-158, and selected for further study all those that favor OMe-Tyr over Tyr. The results for the six most favorable mutations are shown in Table 4. The following choices minimize clashes (stage 1): for Tyr-32: Glu, Asp, Gln, Phe, and Met; for Asp-158: Ala.
In stage 2, we used scwrl to generate the side-chain configurations for the two mutated residues for each of the five cases of simultaneous mutations selected from stage 1 (with five choices for Tyr-32 and 1 for Asp-158). These side chains were optimized separately for the analog and for Tyr in the binding site. This optimization led to 5 full protein structures for each ligand. We then carried out energy minimization of the new side chains with all other atoms fixed, followed by energy minimization of the whole protein with either OMe-Tyr or Tyr bound for all five mutants. The binding energies (including solvation) for both OMe-Tyr and Tyr were then calculated for all mutants. This calculation led to the binding energies in the top half of Table 5 for OMe-Tyr and Tyr bound to the five mutants.
Table 5.
Binding energies of OMe-Tyr and Tyr to the wild-type M.jann-TyrRS and mutants
| Y32 | D158 | E107 | L162 | OMe-Tyr, kcal/mol | Tyr, kcal/mol | Differential, OMe-Tyr−Tyr |
|---|---|---|---|---|---|---|
| Y | D | E | L | −12.34 | 43.83 | −56.17* |
| E | A | 37.11 | 37.64 | −0.54 | ||
| D | A | 43.85 | 38.24 | 5.60† | ||
| Q | A | 48.93 | 42.30 | 6.62† | ||
| F | A | 39.06 | 39.81 | −0.76 | ||
| M | A | 44.17 | 39.00 | 5.16† | ||
| E | A | T | P | 27.48 | 34.98 | −7.51 |
| D | A | T | P | 31.65 | 25.58 | 6.06 |
| Q | A | T | P | 27.06 | 17.72 | 9.33‡ |
| F | A | T | P | 31.54 | 34.06 | −2.53 |
| M | A | T | P | 27.20 | 28.69 | −1.50 |
The first row is for the wild type, which binds Tyr well but not OMe-Tyr. The next five rows consider the mutations for Y32 and D158 identified in Table 4. The three cases denoted as † are considered to be promising cases worth testing. The last five rows consider these same five mutations, but with the E107T and L162P mutations observed in the experiments. The case denoted as ‡ is the one determined experimentally.
Wild-type M.jann-TyrRS.
Chosen designed mutant M.jann-TyrRS.
Mutant M.jann-TyrRS found experimentally (ref. 16).
The results for the five mutant proteins can be compared with the wild type (given in the top row of numbers in Table 5), which leads to 44 kcal/mol for Tyr but −12 kcal/mol for OMe-Tyr. All five mutants bind Tyr less strongly (38–42 kcal/mol) whereas these five mutants bind OMe-Tyr by 37–49 kcal/mol. Of the five mutants, three favor binding of OMe-Tyr over Tyr by at least 5 kcal/mol. These are [Y32Q, D158A], [Y32M, D158A], and [Y32D, D158A], which lead to total binding energy for OMe-Tyr of 49, 44, and 44 and differential binding energies of 7, 5, and 6 kcal/mol. The other two cases both lead to weak binding and favor Tyr over OMe-Tyr, hence we will ignore them.
Fig. 2 shows the predicted binding site of OMe-Tyr for the best case, [Y32Q, D158A]. We see that residues Ala-67, Ala-158, and Leu-65 form a hydrophobic pocket for the methyl group. The amide Nɛ2 of Gln-32 has close contact with the oxygen atom of the OMe group (3.79 Å), whereas the Oɛ1 atom of Gln-32 is stabilized by forming a weak hydrogen bond (3.58 Å) with the main chain NH of Leu-65. These H-bonds might be stabilized further by an intervening water.
Figure 2.

The predicted binding site surrounding the OMe-Tyr in the COP-designed mutant [Y32Q, D158A] M.jann-TyrRS. The mutated residues (Gln-32 and Ala-158) are labeled in blue. Ala-67, Ala-158, and Leu-65 form a hydrophobic pocket for the methyl group. The amide Nɛ2 of Gln-32 has close contact with the oxygen of OMe, whereas the Oɛ1 atom of Gln-32 is stabilized by forming a weak hydrogen bond (3.58 Å) with the main chain NH of Leu-65. (Both may have intervening water molecules.)
The mutant [Y32M, D158A] is also a favorable candidate. However, for [Y32D, D158A], the charged group of the Asp does not seem to have a favorable stabilization of the charged group, which may lead to problems with folding.
Comparison to Experimental Results.
Wang et al. (16) carried out a combinatorial experiment to find a mutant M.jann-TyrRS optimum for OMe-Tyr. Because no three-dimensional structure was available they used a sequence alignment with 4ts1. Their alignment suggested that the five residues—Y32, D158, E107, L162, and I159—are in the active site. They then considered all 520 mutations and selected the best ones. This selection was carried out by first screening for binding any amino acid followed by screening against natural amino acids to find the mutants least able to bind Tyr and any other natural amino acids. Of these the ones that were most selective toward OMe-Tyr were considered. These studies led to [Y32Q, D158A, E107T, L162P, I159I].
Thus, the experiments identify the [Y32Q, D158A] replacements found from COP to be best.
However, Wang et al. (16) found their optimum mutant to also include E107T and L162P (they found that I159 did not change). We did not consider mutations of E107 or L162 because our predicted structure for M.jann-TyrRS places both residues far from the binding site (see Fig. 3). Thus we find that Glu-107 is on the surface of the protein, 12.9 Å from the Tyr ligand (from the Cα of Glu-107 to the Oη of Tyr ligand), and Leu-162 is 14.5 Å from the Tyr ligand. In the Wang et al. alignment, Leu-162 and Glu-107 in M.jann-TyrRS correspond to Leu-180 and Asn-123 in the 4ts1 structure. Leu-180 is in the middle of a β-strand on the bottom of the binding site in 4ts1. The Leu → Pro mutation at this position would be expected to disrupt the secondary structure. Because Asn-123 is close to the core of the protein in 4ts1, it seems unlikely that a charged Glu would fold into this structure.
Figure 3.

The predicted structure for M.jann-TyrRS with explicit side chains for the Tyr-32, Asp-158 residues involved in the design. The Tyr ligand is also shown (labeled in red). The predicted position of residues Glu-107 and Leu-62 (labeled in blue) M.jann-TyrRS are shown.
Because our predicted structure puts the residues Glu-107 and Leu-162 well outside the binding site for M.jann-TyrRS, COP did not find these residues as mutation targets.
To understand why the combinatorial experiments led to a different result than the COP design, we calculated the effect of including the [E107T, L162P] mutations along with the five cases from the COP analysis. The L162P mutation requires a change in the main chain conformation, and therefore we carried out annealing MD to optimize the backbone structure. Hence, the energy minimization was followed by one cycle of annealing MD with SGB solvation method, using mpsim. The resulting best-energy annealed protein structure was used to calculate binding energies.
The calculated binding energies of both OMe-Tyr and Tyr to the mutants with the five mutations are shown in the bottom half of Table 5. We find that the experimental optimum mutation, [Y32Q, D158A, E107T, L162P], leads to a dramatically weak binding (18 kcal/mol) to Tyr. Because the experiments conducted negative selections against any mutant TyrRS that binds Tyr, our favored mutants, which have higher affinity to Tyr, would be screened out in this process. We find also that the experimental mutant leads to a differential preference for OMe-Tyr over Tyr of 9 kcal/mol. This is by far the best differential we have seen and much better than for our designed case. However, the net binding of OMe-Tyr to the mutant is calculated to be only 27 kcal/mol. This finding could explain the observation that the mutant led to an incorporation rate much poorer than for the natural amino acid. Thus, the calculations do seem consistent with the experiment given how the experiment was carried out.
Because our predicted mutants all would have been in the experimental screen, it would be interesting to reexamine the three cases predicted by COP to determine how effective they are. We suspect that the predicted differentials of 5–7 kcal/mol may be sufficient to obtain selectivity. In addition, the total binding energies of 44–49 kcal/mol for OMe-Tyr suggest that these new mutants would be much more active.
Summary
We describe a generic structure-based rational functional site-design procedure (COP) to design mutant proteins that would bind optimally and preferentially to new ligands and we apply this procedure to predict mutant M.jann-TyrRS proteins optimal for binding OMe-Tyr. This procedure leads to a mutant, [Y32Q-D158A] (and two others), predicted to have a good preferential binding over Tyr and an excellent absolute binding energy to OMe-Tyr.
We compare our predictions with the experimentally optimized mutant [Y32Q-D158A-E107T-L162]. Experiment and theory agree with the [Y32Q-D158A] mutations. However, we predict that residues E107 and L162 are remote from the binding sites. We find that these mutations lead to extremely poor binding of Tyr but to an extremely high differential binding of OMe-Tyr over Tyr, which is what the experiments were designed to do. However, we calculate that the activity of the experimental mutant is far lower than the designed one. Because our predicted mutation is a subset of the ones considered in the experiments, it would be interesting to test the selectivity and incorporation efficiency for our three predicted cases.
Because no experimental three-dimensional structure was available for wild-type M.jann-TyrRS, we used structfast to predict the alignment and backbone folding. We then used a series of energy minimization and annealing calculations to obtain the structure for the full protein. We found that this predicted protein structure binds Tyr much stronger then the other 19 natural amino acids, which we consider as validation of the predicted structure. In addition, the success in predicting mutants that compare consistently with experiment provides additional evidence in favor of the predicted structure for M.jann-TyrRS
The experimental comparison of our predicted mutations with observed incorporation would provide additional tests of the validity of these new structure-prediction tools.
Acknowledgments
We thank Dr. Peter G. Schultz, Mr. Lei Wang, and Mr. Sheng Ding for suggesting this project to us. This research was supported by National Institutes of Health/BioEngineering Research Group Grant GM62523. The computing facilities for this project were provided by an IBM Shared University Research grant. The facilities of the Materials and Process Simulation Center used in this project are supported also by the Department of Energy/Accelerated Strategic Computing Initiative/Academic Strategic Alliances Program, National Science Foundation Grants CHE9985574 and MRI99–77872), the National Institutes of Health, the Army Research Office-Multidisciplinary University Research Initiative, Chevron Corp., the Defense Advanced Research Planning Agency, 3M, Seiko–Epson, Avery–Dennison Corp., Kellogg's, General Motors, Beckman Institute, Asahi Chemical, and the Office of Naval Research.
Abbreviations
- aaRS
aminoacyl-tRNA synthetase
- OMe-Tyr
O-methyl-l-tyrosine
- M.jann-TyrRS
Methanococcus jannaschii tyrosyl tRNA synthetase
- COP
clash opportunity progressive design
- MD
molecular dynamics
- SGB
surface-generalized born
References
- 1.Petka W A, Harden J L, McGrath K P, Wirtz D, Tirrell D A. Science. 1998;281:389–392. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 2.Tang Y, Ghirlanda G, Petka W A, Nakajima T, DeGrado W F, Tirrell D A. Angew Chem Int Ed Engl. 2001;40:1494–1498. [PubMed] [Google Scholar]
- 3.Noren C J, Anthony-Cahill S J, Griffith M C, Schultz P G. Science. 1989;244:182–188. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- 4.Hendrickson W A, Horton J R, LeMaster D M. EMBO J. 1990;9:1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richmond M H. J Mol Biol. 1963;6:284–294. doi: 10.1016/s0022-2836(63)80089-3. [DOI] [PubMed] [Google Scholar]
- 6.Yoshikawa E, Fournier M J, Mason T L, Tirrell D A. Macromolecules. 1994;27:5471–5475. [Google Scholar]
- 7.van Hest J C M, Kiick K L, Tirrell D A. J Am Chem Soc. 2000;122:1282–1288. [Google Scholar]
- 8.Kothakota S, Mason T L, Tirrell D A, Fournier M J. J Am Chem Soc. 1995;117:536–537. [Google Scholar]
- 9.Cowie D B, Cohen G N. Biochim Biophys Acta. 1957;26:252–261. doi: 10.1016/0006-3002(57)90003-3. [DOI] [PubMed] [Google Scholar]
- 10.Budisa N, Steipe B, Demange P, Eckerskorn C, Kellermann J, Huber R. Eur J Biochem. 1995;230:788–796. doi: 10.1111/j.1432-1033.1995.tb20622.x. [DOI] [PubMed] [Google Scholar]
- 11.Duewel H, Daub E, Robinson V, Honek J F. Biochemistry. 1997;36:3404–3416. doi: 10.1021/bi9617973. [DOI] [PubMed] [Google Scholar]
- 12.Kast P, Hennecke H. J Mol Biol. 1991;222:99–124. doi: 10.1016/0022-2836(91)90740-w. [DOI] [PubMed] [Google Scholar]
- 13.Ibba M, Hennecke H. FEBS Lett. 1995;364:272–275. doi: 10.1016/0014-5793(95)00408-2. [DOI] [PubMed] [Google Scholar]
- 14.Liu D R, Magliery T J, Pastrnak M, Schultz P G. Proc Natl Acad Sci USA. 1997;94:10092–10097. doi: 10.1073/pnas.94.19.10092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowal A K, Kohrer C, RajBhandary U L. Proc Natl Acad Sci USA. 2001;98:2268–2273. doi: 10.1073/pnas.031488298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Brock A, Herberich B, Schultz P G. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 17.Brick P, Bhat T N, Blow D M. J Mol Biol. 1989;208:83–98. doi: 10.1016/0022-2836(89)90090-9. [DOI] [PubMed] [Google Scholar]
- 18.Brick P, Blow D M. J Mol Biol. 1987;194:287–297. doi: 10.1016/0022-2836(87)90376-7. [DOI] [PubMed] [Google Scholar]
- 19.Holm L, Sander C. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 20.Bower M J, Cohen F E, Dunbrack R L., Jr J Mol Biol. 1997;267:1268–1282. doi: 10.1006/jmbi.1997.0926. [DOI] [PubMed] [Google Scholar]
- 21.Dunbrack R L., Jr Proteins. 1999;Suppl. 3:81–87. doi: 10.1002/(sici)1097-0134(1999)37:3+<81::aid-prot12>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 22.Lim K T, Iotov M, McClurg R B, Vaidehi N, Dasgupta S, Taylor S, Goddard W A., III J Comput Chem. 1997;18:501–521. [Google Scholar]
- 23.Ding H-Q, Karasawa N, Goddard W A, II. J Chem Phys. 1992;97:4309–4315. [Google Scholar]
- 24.Mayo S L, Olafson B D, Goddard W A., III J Phys Chem. 1990;94:8897–8909. [Google Scholar]
- 25.MacKerell A D, Bashford D, Bellott M, Dunbrack R L, Evanseck J D, Field M J, Fischer S, Gao J, Guo H, Ha S, et al. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 26.Tannor D J, Marten B, Murphy R, Friesner R A, Sitkoff D, Nicholls A, Ringnalda M, Goddard W A, III, Honig B. J Am Chem Soc. 1994;116:11875–11882. [Google Scholar]
- 27.Ghosh A, Rapp C S, Friesner R A. J Phys Chem B. 1998;102:10983–10990. [Google Scholar]
- 28.Fersht A R, Shindler J S, Tsui W C. Biochemistry. 1980;19:5520–5524. doi: 10.1021/bi00565a009. [DOI] [PubMed] [Google Scholar]
- 29.Freist W, Sternbach H, Pardowitz I, Cramer F. J Theor Biol. 1998;193:19–38. doi: 10.1006/jtbi.1998.0672. [DOI] [PubMed] [Google Scholar]
- 30. Wang, P., Vaidehi, N., Tirrell, D. A. & Goddard, W. A., III (2002) J. Am. Chem. Soc., in press. [DOI] [PubMed]
- 31.Hendsch Z S, Tidor B. Protein Sci. 1999;8:1381–1392. doi: 10.1110/ps.8.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steer B A, Schimmel P. J Biol Chem. 1999;274:35601–35606. doi: 10.1074/jbc.274.50.35601. [DOI] [PubMed] [Google Scholar]