

Abstract
In this study, a passively membrane‐permeable short peptide inhibitor targeting the measles virus fusion protein (MeV‐F) is reported. Measles virus (MeV) is highly contagious, yet no approved antiviral drugs are currently available. MeV‐F plays a crucial role in viral infection, making it an attractive target for drug development. The fusion inhibitor peptide (FIP) is a well‐known short peptide that binds to MeV‐F and prevents its structural rearrangement. However, improving both inhibitory activity and passive membrane permeability is essential for developing orally available MeV‐F inhibitors. Herein, FIP derivatives are explored through hydrogen‐to‐fluorine substitution and a derivative with enhanced inhibitory activity (IC50 = 90 nM) and passive membrane permeability (P e = 1.4 × 10–6 cm s−1) was identified. This study highlights the potential of the long‐studied fusion inhibitor peptide as a promising lead compound for the development of orally available drugs against measles infection.
Keywords: fluorination, fusion inhibitor peptides, measles virus, membrane permeability
Fusion inhibitor peptide (FIP) is a known small peptide capable of inhibiting measles virus infection. In this study, hydrogen‐to‐fluorine substitution is employed to derivatize FIP, leading to the discovery of a FIP derivative with enhanced inhibitory activity and passive membrane permeability. This molecule represents a promising lead compound for the development of an orally available therapeutic against measles infection.

1. Introduction
The measles virus (MeV) is a single‐stranded, negative‐sense RNA virus with a high basic reproduction number (R0) ranging from 12 to 18,[ 1 , 2 – 3 ] indicating significant contagiousness. Despite the availability of effective vaccines, measles remains a significant determinant of pediatric mortality in developing countries. Additionally, even with rigorous vaccination initiatives in developed nations, up to 5% of vaccine recipients fail to produce adequate immunity, and the acquired immunity by the vaccination gradually decreases,[ 4 ] leading to subsequent infections. The global pandemic triggered by SARS‐CoV‐2 has exacerbated this issue by further delaying the progression of measles vaccination programs across various countries.[ 5 , 6 ] To date, there remains no approved drug for measles. Given these challenges, the development of effective therapeutics for MeV is desirable.
The viral envelope proteins are essential for MeV infection and, therefore, attractive drug targets. Hemagglutinin (MeV‐H) and fusion protein (MeV‐F) are the two envelope proteins of MeV, which mediate its entry, a process crucial for MeV to infect cells (Figure 1a). Initially, MeV‐H interacts with specific receptors on the host cell surface, such as the signaling lymphocytic activation molecule (SLAM)[ 7 ] and nectin cell adhesion molecule 4 (nectin‐4),[ 8 , 9 ] which triggers a cascade of structural rearrangements in MeV‐F (Figure 1a, left). It leads to the exposure and subsequent insertion of the fusion peptide (FP) of MeV‐F into the host cell membrane (Figure 1a, middle). Subsequently, the heptad‐repeat domains N (HRN) and C (HRC) of MeV‐F align to form a six‐helix bundle structure, transitioning MeV‐F to a postfusion state.[ 10 ] These structural rearrangements of MeV‐F facilitate the fusion of the MeV envelope with the host cell membrane, enabling viral entry into the cell. Following entry, MeV replicates within the host cell, initiating budding and the infection of neighboring cells. Infection also induces the expression of MeV‐H and MeV‐F on the host cell's surface, leading to cell‐to‐cell fusion between the infected cell and adjacent cells expressing a receptor, resulting in the formation of syncytia. Inhibition of the structural changes of MeV‐F is an effective strategy to prevent MeV infection.
Figure 1.

a) A schematic illustration of MeV‐F transformation. MeV‐H is colored green. The domains of MeV‐F are colored as follows: HRN (cyan), HRC (purple), fusion peptide (black), transmembrane (gray), and other parts of prefusion state MeV‐F (pink). FIP and AS‐48 bind and stabilize the prefusion state of MeV‐F. b) Structures and IC50 values of MeV‐F inhibitor peptides (ref. [13]). c) The crystal structure of the prefusion state of MeV‐F bound with FIP (PDB ID: 5YZD).
Small molecule inhibitors have the advantages of low production costs and the potential for oral administration. Two small molecule inhibitors that bind to the MeV‐F prefusion state and prevent its conformational change have been reported: AS‐48 (IC50 = 3.0 µM)[ 11 ] and FIP (fusion inhibitor peptide)[ 12 ] (IC50 = 1.3 µM)[ 13 ] (Figure 1b). However, their inhibitory activity is relatively weak, particularly compared to antibodies[ 14 , 15 ] and large peptide inhibitors,[ 16 , 17 ] necessitating further modification.
Recently, we discovered an FIP derivative with an enhanced inhibitory activity through a structure‐guided derivatization study.[ 13 ] We first analyzed the interaction between MeV‐F and FIP based on the crystal structure of MeV‐F bound with FIP (PDB ID: 5YZD),[ 18 ] revealing that the C‐terminus of FIP does not interact with MeV‐F[ 13 ] (Figure 1c). Consequently, we chemically modified the C‐terminus of FIP to achieve new interactions without affecting the existing interactions, aiming to enhance the inhibitory activity. As a result, we successfully discovered an FIP derivative with an enhanced inhibitory activity (IC50 = 210 nM) named FIP‐G3r‐NH2 (Figure 1b). Although improvement of inhibitory activity was achieved, the introduction of the D‐Arg side chain increases the molecular weight and hydrophilicity of the peptide, which is generally detrimental to oral bioavailability.
Here, we report a new derivatization study of FIP using small, noncharged modifications. Specifically, we employ hydrogen‐to‐fluorine substitution as a derivatization strategy. This modification minimally alters the structure and size of the molecule while potentially introducing new interactions and enhancing lipophilicity. We evaluate the inhibitory activity and membrane permeability of the derivatives to identify potent, membrane‐permeable fusion inhibitor peptides.
2. Results and Discussion
2.1. Design and Evaluation of FIP Derivatives
In our previous study, we observed that modifying the carboxylic acid terminus of FIP to an amide terminus, resulting in FIP‐NH2, led to a slight improvement in inhibitory activity compared to the original FIP. Moreover, the neutral amide structure is more favorable for passive membrane permeation than the negatively charged carboxylate terminus. Considering this result, we recruited FIP‐NH2 as the parent structure for derivatization in the current study.
According to the crystal structure of MeV‐F bound with FIP, all the phenyl rings of FIP have close contact with MeV‐F, and there is not much space for introducing additional functional groups to increase the interaction (Figure 1c). Therefore, we recruited hydrogen‐to‐fluorine substitution as a strategy to increase the interaction.[ 19 , 20 ] The introduction of fluorine atoms can enhance the binding affinity to the target protein via electrostatic, hydrophobic, and van der Waals interactions. Moreover, the substitution can increase the lipophilicity of the compound, which is favorable for passive membrane permeation.
We synthesized FIP‐NH2 derivatives with a hydrogen‐to‐fluorine substitution at ortho‐, meta‐, and para‐positions of each phenyl ring. In addition, we synthesized derivatives with a phenyl‐to‐pentafluorophenyl substitution at each phenyl ring of FIP‐NH2.
The inhibitory activities of these fluorinated FIP derivatives were then evaluated using a cell‐to‐cell fusion assay (Figure 2 ). Vero cells stably expressing hSLAM (Vero/hSLAM cells)[ 21 ] were transfected with plasmids encoding MeV‐F, MeV‐H, and EGFP, and incubated with inhibitors for 24 h. The transfected Vero/hSLAM cells form syncytia due to the fusiogenic activity of MeV‐F. FIP and the FIP derivatives can inhibit the syncytia formation by preventing the transformation of MeV‐F. The inhibition of syncytia formation was qualitatively analyzed by assessing the size and area of the EGFP‐emitting cells. First, as a control, FIP was examined, and the peptide showed negligible inhibition of syncytia formation at 1 µM (Figure 2a, FIP). In comparison, the previously reported derivative FIP‐G3r‐NH2 almost completely inhibited the syncytia formation at 1 µM (Figure 2a, FIP‐G3r‐NH2). These results align with the results reported in our previous study.[ 13 ]
Figure 2.

Evaluation of the inhibitory activity of fluorinated FIP derivatives at 1 µM using a cell‐to‐cell fusion assay. a) Control results. “w/o peptide” refers to control cells incubated without peptides. “EGFP only” refers to cells transfected with the EGFP plasmid but not with the MeV‐F and MeV‐H plasmids. b) Results of fluorinated FIP derivatives. Experiments were performed in triplicate, and one of the images is shown. The full set of triplicate images is presented in Figure S1, Supporting Information.
Among the synthesized fluorinated FIP derivatives, some derivatives exhibited improved inhibitory activity compared to FIP. First, among the derivatives fluorinated at the N‐terminal benzyloxycarbonyl (Z) group, the ortho‐fluorinated derivative (FIP‐ZoF‐NH2) exhibited stronger inhibitory activity than FIP, whereas other derivatives did not show improved inhibitory activity (Figure 2b, top). Second, none of the derivatives with fluorination at first D‐phenylalanine residue of FIP exhibited stronger inhibitory activity than FIP (Figure 2b, middle). Lastly, among the derivatives fluorinated at the second phenylalanine residue, the para‐fluorinated derivative (FIP‐F2pF‐NH2) exhibited stronger inhibitory activity than FIP, while other derivatives did not (Figure 2b, bottom). Altogether, FIP‐ZoF‐NH2 and FIP‐F2pF‐NH2 exhibited stronger inhibitory activity compared to FIP. The inhibitory activities of the two compounds were compared at 0.5 µM (Figure S2, Supporting Information). The result suggested that FIP‐F2pF‐NH2 is a stronger inhibitor than FIP‐ZoF‐NH2. Based on these results, we selected FIP‐F2pF‐NH2, the most potent inhibitor among the derivatives, for further study. Note that FIP and FIP‐F2pF‐NH2 as well as the previously reported FIP‐G3r‐NH2 did not exhibit cytotoxicity to Vero/SLAM cells in CCK‐8 assay (Table S1, Supporting Information), demonstrating that the fusion inhibition by these compounds is not derived from nonspecific cytotoxicity.
To quantitatively evaluate the inhibitory activity of FIP‐F2pF‐NH2 against MeV infection in cells, we conducted a viral entry inhibition assay. Vero/hSLAM cells were infected with EGFP‐recombinant MeV (MeV‐EGFP) in the presence of the inhibitors for 1 h. The virus‐containing medium was removed, and the cells were further incubated in a medium containing 100 µM FIP for 48 h. After the incubation, the number of virus‐infected cells was counted based on the fluorescence signal from the infected cells. FIP and FIP‐G3r‐NH2 were also tested under the same conditions as controls. The IC50 values for FIP, FIP‐G3r‐NH2, and FIP‐F2pF‐NH2 were 2.1, 0.25, and 0.090 µM, respectively (Table 1 and Figure S3, Supporting Information). The IC50 values of FIP and FIP‐G3r‐NH2 are consistent with our previous report.[ 13 ] FIP‐F2pF‐NH2 exhibited a 23‐fold enhancement in inhibitory activity compared to FIP. Compared to the previously reported strong inhibitor FIP‐G3r‐NH2, FIP‐F2pF‐NH2 exhibited comparable or even stronger inhibitory activity.
Table 1.
Virus entry inhibitory activity of FIP and its derivatives.
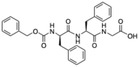


The values were determined by viral cell infection inhibition assay. The experiments were conducted in triplicate.
The values were determined by surface plasmon resonance assay.
The values are from the previous report (ref. [13]).
2.2. Investigating the Interaction between FIP‐F2pF‐NH2 and MeV‐F
To investigate whether the enhanced inhibitory activity of FIP‐F2pF‐NH2 is derived from improved binding affinity to MeV‐F, we used surface plasmon resonance (SPR) to determine the dissociation constants (K D) between FIP‐F2pF‐NH2 and MeV‐F. To immobilize the peptide as a ligand on the CM5 chip, we synthesized a derivative of FIP‐F2pF‐NH2, namely FIP‐F2pF‐NH‐EG‐K, which contains an ethylene glycol spacer (EG) and a lysine (Lys) residue at the C‐terminus (the structure is shown in Figure S4, Supporting Information). The amino group of Lys was used for immobilizing the peptide onto the CM5 chip. For the SPR measurement, the ectodomain trimer of MeV‐F, which is stabilized in its prefusion state by introducing cysteine mutations in the stalk region,[ 18 ] was utilized.
As a result of the SPR measurements, the K D value for FIP‐F2pF‐NH‐EG‐Lys binding to MeV‐F was determined to be 16 nM (Table 1 and Figure S4, Supporting Information). We previously showed that the K D value for FIP‐NH2 binding to MeV‐F was 81 nM. This indicates that the binding affinity was enhanced approximately fivefold by the hydrogen‐to‐fluorine substitution. This result is consistent with the enhanced inhibitory activity of the derivative compared to FIP.
It is worth noting that the binding affinity of FIP‐F2pF‐NH2 (K D = 16 nM) is slightly weaker than that of FIP‐G3r‐NH2 (K D = 6.6 nM), whereas the inhibitory activity of FIP‐F2pF‐NH2 was stronger than that of FIP‐G3r‐NH2 (Table 1). This suggests that the binding affinity of these peptides to the artificially isolated and stabilized ectodomain MeV‐F may not perfectly reflect their binding affinity to MeV‐F on the cell surface. Otherwise, the binding affinity to the protein may not directly correlate with the inhibitory activity against the conformational transition of MeV‐F from the prefusion state to the fusion state.
The improvement of the binding affinity indicates that the improved inhibitory activity of FIP‐F2pF‐NH2 is attributed to the formation of new interactions with MeV‐F. To investigate the plausible new interactions formed by the fluorine atom in the derivative, we generated a model complex structure using the reported crystal structure of FIP binding to MeV‐F (PDB ID 5YZD).
Assuming the small modification by the hydrogen‐to‐fluorine substitution does not affect the binding conformation of FIP, the model complex structure was generated by simply substituting the hydrogen to fluorine at the para‐position of the phenyl group in the second phenylalanine residue of FIP in the crystal structure (Figure 3 ). There are no obvious MeV‐F residues that can form electrostatic interactions with the fluorine. In contrast, the increased volume of the phenyl group by the hydrogen‐to‐fluorine substitution is suggested to more effectively fill the space in the binding pocket (Figure 3b), leading to a greater number of van der Waals contacts with MeV‐F. More specifically, while the hydrogen atom in FIP contacts only α‐ and β‐carbons of the A466 residue of MeV‐F, the model structure suggests that the fluorine atom in FIP‐F2pF‐NH2 can additionally contact a δ‐carbon of the L470 residue of MeV‐F (Figure S5, Supporting Information).
Figure 3.

Investigation of new interactions formed by the hydrogen‐to‐fluorine substitution. a) FIP structure and a close‐up view of the crystal structure of FIP bound to MeV‐F (PDB ID: 5YZD). b) FIP‐F2pF‐NH2 structure and a close‐up view of a modeled structure of FIP‐F2pF‐NH2 bound to MeV‐F. Hydrogen, carbon, nitrogen, oxygen, and fluorine atoms are shown in white, beige, blue, red, and green, respectively. The MeV‐F surface is shown in blue with 50% transparentcy. Since hydrogen atoms are not visible in the crystal structure, they were added in appropriate orientations using ChimeraX.[ 22 ]
2.3. Passive Membrane Permeability Measurements
Finally, we assessed the oral bioavailability of FIP and FIP‐F2pF‐NH2 by measuring their passive membrane permeability. Compounds that passively permeate lipid membranes exhibit high gastrointestinal adsorption and are effectively transported into systemic circulation after oral administration. Therefore, passive membrane permeability serves as an indicator of oral bioavailability. We evaluated the passive membrane permeability of the peptides using a parallel artificial membrane permeability assay (PAMPA). As a result, FIP exhibited a negligible level of permeability, while FIP‐F2pF‐NH2 exhibited passive membrane permeability (P e = 1.4 × 10–6 cm s−1) (Table 2 ). The enhanced membrane permeability can be attributed to the following two structural alterations: 1) Removal of the negative charge of the peptide by the amidation of the C‐terminal carboxylate of FIP; 2) Enhanced hydrophobicity by the hydrogen‐to‐fluorine substitution.
Table 2.
Membrane permeability of FIP and FIP derivatives.
| Chemical structurea) | P e [ × 10–6 cm s–1]b) | |
|---|---|---|
| FIP |

|
<0.013 |
| FIP‐F2pF‐NH2 |

|
1.4 ± 0.1 |
| FIP‐NH2 |

|
1.2 ± 0.2 |
| FIP‐G3r‐NH2 |

|
<0.013 |
The structures are shown in their charged states at physiological pH.
The values were determined by PAMPA. The experiments were conducted in triplicate.
To examine which is the dominant factor for the enhanced membrane permeability, we also examined the membrane permeability of FIP‐NH2. FIP‐NH2 exhibited passive membrane permeability, P e = 1.2 × 10–6 cm s−1 (Table 2), which is only slightly lower than that of FIP‐F2pF‐NH2. This result suggests that the C‐terminal amidation majorly contributed to the enhanced membrane permeability, while the hydrogen‐to‐fluorine substitution might contribute a little to the permeability enhancement.
As a comparison, we also examined the membrane permeability of FIP‐G3r‐NH2. This peptide did not passively permeate the lipid membrane, probably due to the positively charged D‐Arg residue (Table 2). This result confirms that the neutral modification by hydrogen‐to‐fluorine substitution is more favorable for improving the membrane permeability of FIP than introducing the charged Arg residue in FIP‐G3r‐NH2 for developing orally available derivatives.
3. Conclusion
In this study, we aimed to develop potent inhibitors of MeV infection with enhanced activity and passive membrane permeability by derivatizing FIP at their phenyl rings using a hydrogen‐to‐fluorine substitution strategy. Among the synthesized derivatives, FIP‐F2pF‐NH2 demonstrated significant improvements, showing higher inhibitory activity (IC50 = 90 nM) and membrane permeability (P e = 1.4 × 10–6 cm s−1) compared to FIP (IC50 = 2.1 µM, P e < 0.013 × 10–6 cm s−1), making FIP‐F2pF‐NH2 as a promising candidate for further development.
This study demonstrated the utility of the long‐studied fusion inhibitor peptide for developing potent and passively membrane‐permeable MeV infection inhibitors. The membrane permeable inhibitor, FIP‐F2pF‐NH2, is potentially useful as a lead for developing an orally available therapeutic for treating MeV infection.
FIP derivatives are not expected to broadly inhibit other paramyxoviruses or unrelated viruses, considering that the original FIP does not inhibit even the fusion protein of canine distemper virus,[ 18 ] a virus closely related to MeV. However, since fusion proteins of other viruses also contain hydrophobic pockets in their prefusion states, it may be possible to develop similar small‐peptidic hydrophobic inhibitors for other paramyxoviruses, which could potentially be orally available.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supplementary Material
Acknowledgements
This research was supported by AMED under grant nos. (JP24wm0325069) (T.H. and J.M.), (JP24jf0126002) (T.H.), The Uehara Memorial Foundation (J.M.), JST PRESTO (JPMJPR21AF) (J.M.), JSPS KAKENHI under grant nos. (JP23H02725 and JP20K20596) (T.H.), The Naito Foundation (T.H.), and CREST (JPMJCR21N5), Japan Science and Technology Agency (S.S.). This research was partially supported by JSPS Core‐to‐Core Program (A. Advanced Research Networks) grant no. (JPJSCCA20240006) (T.H.). The authors thank Mao Yamaguchi, Motoki Sugano, and Ayumi Inayoshi for supporting the measurements of passive membrane permeability. The authors also thank Tomoaki Suzuki for his preliminary assessment of fluorinated FIP derivatives. The authors thank One‐stop Sharing Facility Center for Future Drug Discovery (The University of Tokyo) for the use of Biacore T100 for surface plasmon resonance analysis. Molecular graphics and analyses were performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health grant no. R01‐GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
Contributor Information
Shinsuke Sando, Email: ssando@chembio.t.u-tokyo.ac.jp.
Takao Hashiguchi, Email: hashiguchi.takao.1a@kyoto-u.ac.jp.
Jumpei Morimoto, Email: jmorimoto@chembio.t.u-tokyo.ac.jp.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Anderson R. M., May R. M., Science 1982, 215, 1053. [DOI] [PubMed] [Google Scholar]
- 2. Anderson R. M., May R. M., J. Hyg. 1985, 94, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guerra F. M., Bolotin S., Lim G., Heffernan J., Deeks S. L., Li Y., Crowcroft N. S., Lancet Infect. Dis. 2017, 17, e420. [DOI] [PubMed] [Google Scholar]
- 4. Robert A., Suffel A. M., Kucharski A. J., Lancet Public Health 2024, 9, e766. [DOI] [PubMed] [Google Scholar]
- 5. Local Burden of Disease Vaccine Coverage Collaborators , Nature 2021, 589, 415.33328634 [Google Scholar]
- 6. Ho L. L., Gurung S., Mirza I., Nicolas H. D., Steulet C., Burman A. L., Danovaro‐Holliday M. C., Sodha S. V., Kretsinger K., Int. J. Infect. Dis. 2022, 119, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tatsuo H., Ono N., Tanaka K., Yanagi Y., Nature 2000, 406, 893. [DOI] [PubMed] [Google Scholar]
- 8. Mühlebach M. D., Mateo M., Sinn P. L., Prüfer S., Uhlig K. M., Leonard V. H. J., Navaratnarajah C. K., Frenzke M., Wong X. X., Sawatsky B., Ramachandran S., McCray P. B., Cichutek K., Von Messling V., Lopez M., Cattaneo R., Nature 2011, 480, 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Noyce R. S., Bondre D. G., Ha M. N., Lin L. T., Sisson G., Tsao M. S., Richardson C. D., PLoS Pathog. 2011, 7, e1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Plattet P., Alves L., Herren M., Aguilar H. C., Viruses 2016, 8, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Plemper R. K., Doyle J., Sun A., Prussia A., Cheng L. T., Rota P. A., Liotta D. C., Snyder J. P., Compans R. W., Antimicrob. Agents Chemother. 2005, 49, 3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kelsey D. R., Flanagan T. D., Young J., Yeagle P. L., J. Biol. Chem. 1990, 265, 12178. [PubMed] [Google Scholar]
- 13. Gao Z., Sasaki J., Suzuki T., Suzuki T., Miwa Y., Sando S., Hashiguchi T., Morimoto J., RSC Med. Chem. 2025, 16, 1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mathieu C., Ferren M., Harder O., Bovier F. T., Marcink T. C., Predella C., Angius F., Drew‐Bear J., Dorrello N. V., Greninger A. L., Moscona A., Niewiesk S., Horvat B., Porotto M., Cell. Mol. Immunol. 2021, 18, 1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zyla D. S., Marca R. D., Niemeyer G., Zipursky G., Stearns K., Leedale C., Sobolik E. B., Callaway H. M., Hariharan C., Peng W., Parekh D., Marcink T. C., Avalos R. D., Horvat B., Mathieu C., Snijder J., Greninger A. L., Hastie K. M., Niewiesk S., Moscona A., Porotto M., Saphire E. O., Science 2024, 384, eadm8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Welsch J. C., Talekar A., Mathieu C., Pessi A., Moscona A., Horvat B., Porotto M., J. Virol. 2013, 87, 13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Figueira T. N., Palermo L. M., Veiga A. S., Huey D., Alabi C. A., Santos N. C., Welsch J. C., Mathieu C., Horvat B., Niewiesk S., Moscona A., Castanho M. A. R. B., Porotto M., J. Virol. 2017, 91, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hashiguchi T., Fukuda Y., Matsuoka R., Kuroda D., Kubota M., Shirogane Y., Watanabe S., Tsumoto K., Kohda D., Plemper R. K., Yanagi Y., Proc. Natl. Acad. Sci. 2018, 115, 2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swallow S., Prog. Med. Chem. 2015, 54, 65. [DOI] [PubMed] [Google Scholar]
- 20. Gillis E. P., Eastman K. J., Hill M. D., Donnelly D. J., Meanwell N. A., J. Med. Chem. 2015, 58, 8315. [DOI] [PubMed] [Google Scholar]
- 21. Ono N., Tatsuo H., Hidaka Y., Aoki T., Minagawa H., Yanagi Y., J. Virol. 2001, 75, 4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meng E. C., Goddard T. D., Pettersen E. F., Couch G. S., Pearson Z. J., Morris J. H., Ferrin T. E., Protein Sci. 2023, 32, e4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.