

Abstract
Deregulated expression of the c-Myc transcription factor is found in a wide variety of human tumors. Because of this significant role in oncogenesis, considerable effort has been devoted to elucidating the molecular program initiated by deregulated c-myc expression. The primary transforming activity of Myc is thought to arise through transcriptional regulation of numerous target genes. Thus far, Myc target genes involved in mitochondrial function have not been characterized in depth. Here, we describe a nuclear c-Myc target gene, PRDX3, which encodes a mitochondrial protein of the peroxiredoxin gene family. Expression of PRDX3 is induced by the mycER system and is reduced in c-myc−/− cells. Chromatin immunoprecipitation analysis spanning the entire PRDX3 genomic sequence reveals that Myc binds preferentially to a 930-bp region surrounding exon 1. We show that PRDX3 is required for Myc-mediated proliferation, transformation, and apoptosis after glucose withdrawal. Results using mitochondria-specific fluorescent probes demonstrate that PRDX3 is essential for maintaining mitochondrial mass and membrane potential in transformed rat and human cells. These data provide evidence that PRDX3 is a c-Myc target gene that is required to maintain normal mitochondrial function.
The c-Myc transcription factor has been implicated in the control of a variety of cellular processes, including cell growth, cell-cycle progression, and apoptosis (1). The c-Myc protein is a member of the basic helix-loop-helix leucine zipper family of transcription factors. In cooperation with its heterodimerization partner Max, Myc binds DNA in a sequence-specific manner and activates transcription at E box elements with the consensus sequence 5′-CACGTG-3′. In an effort to dissect the molecular pathways regulated by Myc, several recent studies have focused on the use of microarray technology to identify the transcriptional targets of c-Myc (2–4). A coherent picture is beginning to emerge whereby Myc functions to accelerate multiple metabolic pathways, including amino acid and nucleotide synthesis, lipid metabolism, and glycolysis. Whether Myc also affects mitochondrial metabolism remains unclear. Because mitochondria play a central role in energy production as well as the execution of cell death, they represent a potential site for the regulation of both proliferation and apoptosis. Therefore, Myc target genes encoding mitochondrial proteins could play a significant role in tumorigenesis.
PRDX3 was first identified as a putative c-Myc target gene by using representational difference analysis (RDA) to identify genes that were differentially expressed between Rat1a (R1a) fibroblasts and R1a fibroblasts stably overexpressing c-Myc (R1a-myc) under conditions of anchorage-independent growth (5). Originally cloned as a gene expressed during the differentiation of murine erythroleukemia cells (6), PRDX3 was subsequently shown to possess peroxide reductase activity (7). PRDX3 belongs to an expanding family of highly conserved proteins termed peroxiredoxins, which catalyze the reduction of peroxides in the presence of thioredoxin (8, 9). Members of this gene family have been shown to be involved in diverse cellular roles, including proliferation (10), apoptosis (11), and the response to oxidative stress (12). The bovine PRDX3 homolog, SP-22, localizes to mitochondria, and SP-22 expression is induced after exposure to peroxides or mitochondrial respiratory chain inhibitors (12). The potential role of PRDX3 in tumorigenesis has recently been examined in breast cancer, where elevated levels of PRDX3 protein were found in 79% of the cases examined (13).
In our present study, we sought to determine whether PRDX3 was a bona fide Myc target gene by using Northern analysis of PRDX3 in several model systems. By using chromatin immunoprecipitation (ChIP), we also have examined the occupancy of Myc at multiple sites within the PRDX3 genomic sequence during serum stimulation of 2091 primary human fibroblasts. Then, we evaluated whether PRDX3 has a functional role in Myc-mediated cellular phenotypes. Deregulated c-myc expression induces cell-cycle progression (14), cellular proliferation, anchorage-independent growth (15), and apoptosis after withdrawal of serum (16) or glucose (17). In an effort to establish whether PRDX3 expression affects Myc-induced transformation, we generated stable Rat1a-myc fibroblast cell lines expressing murine PRDX3 in either the sense or antisense (AS) conformation. These cell lines then were evaluated in proliferation and apoptosis assays. To apply our findings to other cell systems, we chose the MCF7/ADR human breast cancer epithelial cell line (18) for further study of PRDX3. Our results demonstrate that c-Myc directly activates expression of a mitochondrial peroxiredoxin that is required for Myc-mediated transformation.
Materials and Methods
Northern Blotting.
Northern blot analysis was performed as described (5). Blots were analyzed and quantitated on a PhosphorImager (Molecular Dynamics). The murine PRDX3 cDNA probe was obtained from IMAGE clone 577524. For R1a and R1a-myc cells, RNA was collected from logarithmically growing cells (adherent) or from cells grown in suspension for 48 h over a layer 0.7% agarose in DMEM (nonadherent). The blot was hybridized simultaneously with probes for PRDX3 and rpL32 (5). For in vivo analysis of PRDX3 expression, total RNA was isolated from mouse liver at 3, 4, and 5 days after adenoviral injection, as described (19). Twenty μg of RNA was loaded for each sample. Analysis of PRDX3 expression in 2091 primary human fibroblasts was performed by placing 50% confluent 2091 cells (American Type Culture Collection) in media containing 0.1% serum. After 48 h, confluent cells were stimulated with DMEM containing 10% (vol/vol) serum, and RNA was collected at the indicated time points. Northern blots containing 10 μg of RNA were probed with either human c-myc or PRDX3. The PRDX3 and c-myc signals were normalized to the ethidium bromide-stained gel of 18S RNA, which was quantitated with LABWORKS image analysis software (Ultraviolet Products).
Chromatin Immunoprecipitation.
Quiescent human primary 2091 fibroblasts were serum stimulated for 0 or 2 h. ChIP was performed with α-Myc antibody (Santa Cruz Biotechnology, sc-764), as described (20). For PCR, 1/100th of the immunoprecipitate was used. PCR primers are given in Table 1, which is published as supporting information on the PNAS web site, www.pnas.org, and were designed by using the human PRDX3 genomic DNA sequence from the GenBank database (contig NT 008902). Real-time PCR was performed by using Sybr Green PCR core reagents (Applied Biosystems) according to the kit protocol (fragments D, F, G, I) or with 1× PCR buffer (Invitrogen), 2.5 mM MgCl2, 0.2 mM dNTPs, 1.25 units of Platinum Taq (Invitrogen), 0.5 μM primers, and 1× Sybr Green buffer (fragments A, B, C, E, H). Absolute quantitation of Myc-bound chromatin was performed by comparing the cycle threshold of each ChIP product to a standard curve generated with known amounts of total-input genomic DNA. Each reaction was analyzed within the linear range, and reactions were performed in triplicate.
Plasmids.
Murine PRDX3 cDNA was obtained from IMAGE consortium clone 577524. pSG5-PRDX3 and pSG5-PRDX3AS were created by cloning the Klenow-filled NotI-EcoRI fragment of 577524 into the Klenow-filled EcoRI site of pSG5 (Stratagene). Human PRDX3 cDNA was obtained from IMAGE consortium clone 50888. pSG5-PRDX3 and pSG5-PRDX3AS were created by NotI digestion of clone 50888 followed by partial digestion with HindIII. The 1.5-kb fragment corresponding to PRDX3 was filled with Klenow and cloned into the blunt Klenow-filled EcoRI site of pSG5. Constructs were screened for orientation and sequenced.
Stable Transfectants.
Stable pooled cell lines were generated by cotransfection of pSG5, pSG5-PRDX3, or pSG5-PRDX3AS with the puromycin resistance plasmid pBabe-puro (21) by using Lipofectamine (GIBCO) according to the manufacturer's instructions.
Immunoblotting.
Immunoblotting was performed as described (5). Polyclonal rabbit antipeptide antibodies to murine PRDX3 were generated against amino acids 80–95 of murine PRDX3 (Research Genetics, Huntsville, AL). Polyclonal rabbit antipeptide antibodies to human PRDX3 were generated against amino acids 241–256 of human PRDX3 (Zymed). Monoclonal β-actin antibody was from Sigma (A-5441).
Growth and Transformation Assays.
Growth curves were generated by plating triplicate samples for each cell line at an initial density of 5 × 103 cells per sample for R1a-myc cells or 1 × 104 cells per sample for MCF7/ADR cells. Live cells were counted by using a hemocytometer. The average cell number was plotted, curve fits were used to calculate doubling times, and R2 values were greater than 0.97 in each case. Methylcellulose assays consisted of four 35-mm dishes per cell line, at a density of 2 × 103 cells per dish, plated in 1 ml of 1.3% methylcellulose in DMEM. Photomicrographs were taken after 8 days (pSG5 and PRDX3) or 16 days (PRDX3AS). Colonies of all sizes from two experiments were counted after 7 days (pSG5 and PRDX3) or 14 days (PRDX3AS) to adjust for differences in doubling time.
Nude Mouse Assays.
Cells (5 × 106) in 200 μl of sterile PBS were injected s.c. into the right flank of male homozygous nude mice at 6 weeks of age. Tumors were allowed to establish until the estimated tumor mass exceeded 1,500 mg. Experiments were approved by the Johns Hopkins School of Medicine Animal Care and Use Committee.
Flow Cytometric Analyses.
For apoptosis assays, cells were seeded at 5 × 105 per 10-cm2 plate and exposed to either media containing 0.1% serum or glucose-free media for 24 h. Cells were collected and stained with 5 μg/ml propidium iodide and FITC-conjugated annexin V (BioSource International, Camarillo, CA), followed by analysis using a Coulter EPICS 752 flow cytometer. All annexin V positive cells were included for statistical analysis.
For fluorescence activated cell sorter (FACS) analysis of reactive oxygen species, mitochondrial membrane potential, and mitochondrial mass, cells were seeded at 5 × 105 per 10-cm2 plate and incubated at 37°C in 5% CO2 for 30 min in the presence of 5 mg/ml 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), 20 nM DiOC6, or 100 nM NAO, respectively, all from Molecular Probes. Cells were washed with PBS, trypsinized, and resuspended. Cells incubated with DCFH-DA were resuspended in ice-cold media containing 5 mg/ml DCFH-DA and maintained on ice until analysis. Cells in DiOC6 were resuspended in 37°C media and analyzed immediately. NAO-labeled cells were resuspended in 37°C media containing 100 nM NAO and analyzed immediately. All analyses were performed with a Becton-Dickinson FACScan flow cytometer with a 488-nm argon laser. All analyses were performed at least three times, and a representative histogram is shown.
Electron Microscopy.
Adherent cells were embedded by using the Pelco Eponate 12 kit (Ted Pella, Inc., Redding, CA). Then, cells were sectioned, followed by staining with uranyl acetate and lead citrate. Analysis was performed by using transmission electron microscopy.
Results
Analysis of PRDX3 Expression in Response to c-Myc.
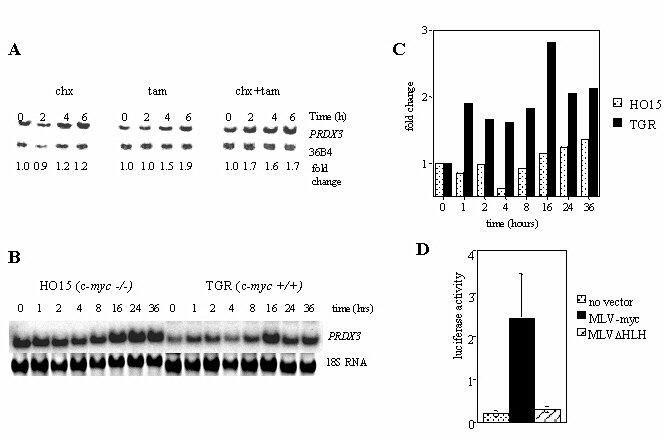
Northern analysis confirms the results of the original RDA screen, as shown in Fig. 1A. PRDX3 is two-fold more highly expressed in adherent R1a-myc cells relative to R1a cells, with the difference in expression becoming six-fold when the cells are nonadherent. Because R1a cells growth-arrest when they are not attached to a substrate while R1a-myc cells continue to proliferate (5), the original RDA screen cannot distinguish between direct c-Myc target genes and genes that are growth-related, non-Myc targets. Therefore, we used the Rat1MycER system (14, 22) to determine whether Myc directly activates PRDX3. This system utilizes a fusion of c-myc to the hormone-binding domain of the estrogen receptor. The fusion protein is retained in the cytosol until the addition of tamoxifen, an estrogen analog, whereupon the protein translocates to the nucleus and activates its biological targets. Cycloheximide is used to inhibit protein synthesis, thereby allowing identification of genes that are directly activated by Myc. Fig. 5A, which is published as supporting information on the PNAS web site, shows that PRDX3 expression increases in the presence of both cycloheximide and tamoxifen, suggesting that c-Myc directly activates transcription of PRDX3. Examination of logarithmically growing c-myc-null fibroblasts (23) indicates that PRDX3 expression is decreased by 50% in the absence of myc (Fig. 1B). PRDX3 expression is also induced during serum stimulation of quiescent c-myc+/+ cells (Fig. 5 B and C). PRDX3 expression increases after 1 h of serum stimulation and reaches a maximum of 2.8-fold after 16 h. However, only a 1.3-fold increase is seen after serum stimulation of c-myc−/− cells. These results indicate that PRDX3 is a c-Myc responsive gene and that PRDX3 expression is minimally induced by serum in the absence of Myc.
Figure 1.

PRDX3 is regulated by c-myc expression. (A) RNA from Rat1a (R1a) fibroblasts or Rat1a fibroblasts expressing ectopic c-Myc (R1a-myc). rpL32 is shown as a loading control. RNA was isolated from adherent cells (A) or nonadherent cells grown over a layer of agar (N). (B) PRDX3 expression in logarithmically growing c-myc+/+, +/−, or −/− Rat1 fibroblasts. PRDX3 expression was calculated relative to vimentin. (C) Hepatic RNA from mice injected with either adenoviral LacZ or c-myc. Numbers represent days after injection with adenovirus. 18S RNA is shown as a loading control. (D) Schematic representation of the PRDX3 genomic locus. Exons are indicated by black boxes. Fragments analyzed for Myc binding are indicated by lettered black bars. The sole canonical E box is indicated in bold, and noncanonical E boxes (38, 39) in fragments C and D are also shown. (E) Ethidium bromide-stained gels of PCR products. (F) Sybr green analysis of PCR fragments evaluated for Myc binding. The absolute amount of DNA in each sample was calculated, and the average was plotted ± SD. (G) Relative mRNA levels for c-myc and PRDX3 during serum stimulation. Signals were normalized to the level of 18S RNA and plotted relative to the 0 h time point for each series.
A recently described in vivo model of transient c-Myc overexpression (19) also indicates that c-Myc regulates PRDX3. Mice injected with adenoviral c-myc show a dramatic increase in hepatic PRDX3 expression, whereas mice injected with control LacZ adenovirus show a minimal increase in PRDX3 (Fig. 1C). The increase in PRDX3 expression parallels that of c-myc. To determine whether Myc binds directly to PRDX3 in vivo, we performed chromatin immunoprecipitation during serum stimulation of primary human fibroblasts. Scanning analysis of the 11-kb genomic PRDX3 sequence (Fig. 1D) indicates that Myc binds to a region containing the sole canonical E box 179 bp upstream from the translational start site, as well as two noncanonical E boxes within the first intron of PRDX3 (Fig. 1E). Quantitative real-time PCR analysis of PRDX3 when Myc levels are low, at 0 h, indicates that most fragments exhibit a similar level of binding (Fig. 1F, white bars). At 2 h, Myc binds fragments B, C, and D preferentially, with fragment C showing a 22-fold increase in binding relative to negative distal sites F, H, and I (Fig. 1F, black bars). Despite the presence of multiple noncanonical E boxes located throughout the genomic PRDX3 sequence, Myc binds specifically within a 930-bp region, spanning fragments B, C, and D at the 5′ end of PRDX3. Northern blot analysis during serum stimulation of 2091 fibroblasts indicates that myc expression is maximal between 1–2 h (Fig. 1G). Expression of PRDX3 is induced after 2 h and reaches a maximum at 12 h. Taken together, our results establish that Myc binds directly to PRDX3 in vivo and activates transcription.
Effect of PRDX3 on Proliferation and Apoptosis in Rat1a-myc Cells.
To determine the role of PRDX3 in Myc-mediated transformation, we generated pooled R1a-myc fibroblast cell lines stably expressing murine PRDX3 in either the sense or AS conformation (Fig. 2A). Characterization of the growth rate of these cells shows a decrease in the growth rate of R1a-myc-PRDX3AS cells, whereas control and R1a-myc-PRDX3 cells display similar doubling times (Fig. 2B). This decrease in growth rate is not caused by increased apoptosis, as staining with annexin V is nearly identical among the three cell lines (data not shown). Because PRDX3 was originally identified in a screen under conditions of anchorage-independent growth, we hypothesized that PRDX3 would affect colony formation in semisolid media. Fig. 2C demonstrates that R1a-myc-PRDX3 cells form colonies at a higher frequency than pSG5 control cells, whereas cells with PRDX3AS form very few colonies. To determine whether our observations applied in vivo, we injected these same cells into nude mice (Fig. 2D). R1a-myc cells expressing AS PRDX3 did not readily form tumors and were only slightly more tumorigenic than R1a cells expressing control vectors alone (R1a pSG5 MLV). In contrast, R1a-myc cells overexpressing PRDX3 formed larger tumors than R1a-myc cells, suggesting that elevated PRDX3 expression confers a growth advantage in vivo. These results indicate that PRDX3 affects both growth rate and transformation in R1a-myc cells. We also used these cell lines to examine Myc-induced apoptosis after serum or glucose deprivation. We found that PRDX3 expression does not affect apoptosis after serum deprivation (Fig. 2E, light bars). However, PRDX3 does affect apoptosis after glucose withdrawal (Fig. 2E, dark bars). Cells expressing AS PRDX3 are resistant to apoptosis after removal of glucose, whereas cells with increased PRDX3 remain sensitive to glucose deprivation-induced apoptosis.
Figure 2.

Effect of PRDX3 expression on doubling time, transformation, and apoptosis in R1a-myc cells. (A) Immunoblot analysis of cell lysates from R1a-myc cells transfected with pSG5 empty vector, pSG5-PRDX3, or pSG5-PRDX3AS. (B) Growth curves of R1a-myc transfectants: pSG5 (□), PRDX3 (▵), and PRDX3AS (○). Doubling times were 10.4, 10.9, and 19.0 h, respectively. (C) Photomicrographs of methylcellulose colonies. (Bar = 500 μM.) The bar graph represents the average colony number per 35-mm dish ± SD. (E) Tumor formation in nude mice. The average estimated tumor mass was plotted at 2, 3, and 4 weeks after injection ± SD (n = 8). (D) Percentage of apoptotic cells 24 h after serum deprivation (light bars) or glucose deprivation (dark bars). The average ± SD of three experiments is shown.
Effect of PRDX3 on Proliferation and Apoptosis in MCF7/ADR Cells.
To demonstrate that our findings were not specific to R1a-myc cells, we chose the MCF7/ADR human breast cancer epithelial cell line (18). This cell line undergoes extensive apoptosis after glucose withdrawal, and apoptosis can be inhibited by reduction of c-Myc expression with AS oligonucleotides (24). Apoptosis depends on the formation of oxygen radicals, as inhibition of oxygen radical formation using the free radical scavenger sodium pyruvate is sufficient to inhibit apoptosis (25). By using full-length human PRDX3 cDNA, we generated stable pooled cell lines that either overexpress or show decreased levels of human PRDX3 protein (Fig. 3A). Analysis of the growth rate of these cells shows that PRDX3AS cells show a decreased growth rate relative to control cells, although the result is less dramatic than that for R1a-myc cells (Fig. 3B). We also assayed apoptosis after glucose deprivation for 24 h. Cells that overexpress PRDX3 show a reproducible increase in apoptosis, whereas cells with diminished PRDX3 are resistant to apoptosis (Fig. 3C). These results confirm that PRDX3 is required for proliferation in transformed cells, and that AS PRDX3 inhibits apoptosis after glucose deprivation.
Figure 3.

Effect of PRDX3 expression on doubling time and apoptosis in MCF7/ADR cells. (A) Immunoblot analysis of cells lysates from MCF7/ADR cells transfected with pSG5, pSG5-PRDX3, or pSG5-PRDX3AS. (B) Growth curves of MCF7/ADR transfectants: pSG5 (□), PRDX3 (▵), and PRDX3AS (○). Doubling times were 43.0, 37.6, and 60.2 h, respectively. (C) Percentage of apoptotic cells 24 h after glucose withdrawal. The average ± SD of three separate experiments is shown.
Effect of PRDX3 on Mitochondrial Function and Structure.
Because PRDX3 localizes to mitochondria, we examined several parameters that reflect mitochondrial integrity and function. Fig. 4A demonstrates that MCF7/ADR cells expressing PRDX3AS show increased levels of reactive oxygen species as measured by the redox-sensitive dye DCFH-DA (26), which is oxidized to fluorescent DCF. However, R1a-myc-PRDX3AS cells show a minimal increase in reactive oxygen species. Analysis of mitochondrial mass with 10-N-nonyl-acridine orange (NAO) (27) reveals that MCF7/ADR-PRDX3AS cells show decreased mitochondrial mass, whereas R1a-myc-PRDX3AS cells also show a small percentage of cells with reduced mitochondrial mass. Reduction of PRDX3 in both cell lines results in a decrease in mitochondrial membrane potential, indicated by the reduced uptake of 3,3′-dihexyloxacarbocyanine iodide (DiOC6), as shown in Fig. 4A. Although PRDX3AS cells have a lower mitochondrial mass and would, therefore, be expected to show reduced uptake of DiOC6, the membrane potential is diminished even after the mitochondrial mass is taken into account (data not shown). In addition to functional defects, we also observed severe morphological defects when using electron microscopy. R1a-myc-PRDX3AS cells show distorted mitochondrial architecture, with elongated mitochondria displaying branched or circular lobes (Fig. 4B). Analysis of 10 individual R1a-myc-pSG5 cells indicates that although some cells also had longer mitochondria, none of the cells showed branched or looped mitochondria. In contrast, 9 of 10 R1a-myc-PRDX3AS cells showed branched mitochondria, and 3 of 9 also showed looped mitochondria.
Figure 4.

PRDX3 affects mitochondrial membrane integrity and morphology. (A) Histograms generated by FACS analysis of cells incubated with dye specific for cellular reactive oxygen species (DCF), mitochondrial mass (NAO), or mitochondrial membrane potential (DiOC6): pSG5 (solid black line), PRDX3 (solid gray line), PRDX3AS (dotted line). (B) Transmission electron microscopy of R1a-myc-pSG5 and R1a-myc-PRDX3AS cells. (Bar = 1 μM.) (C) Analysis of ROS after glucose deprivation. Cells were exposed to glucose-free media for 1.5 h before incubation with DCFH-DA.
We hypothesized that the reduced mitochondrial membrane potential could prevent the generation of reactive oxygen species (ROS) during glucose deprivation-induced apoptosis. Previously, it had been shown that the mitochondrial membrane potential is required for generating ROS in bovine aortic endothelial cells after exposure to hyperglycemia (28). Analysis of MCF7/ADR cells after glucose withdrawal demonstrates that PRDX3AS cells show a minimal increase in ROS, whereas control cells show a dramatic increase in ROS (Fig. 4C).
Discussion
Our data suggest that one of the primary defects in PRDX3AS cells is a reduced mitochondrial membrane potential. The reduction in membrane potential may be a result of oxidative damage to components of the respiratory chain complexes (29), which, in turn, would disrupt the proton gradient across the inner mitochondrial membrane. A recent report attributing peroxynitrite reductase activity to bacterial peroxiredoxins (30) suggests peroxynitrite as a possible mediator of inhibition of respiratory chain activity and reduction of mitochondrial membrane potential (31). Our results are consistent with the observation that reduced levels of another mitochondrial antioxidant, MnSOD, also result in mitochondrial dysfunction and reduced mitochondrial membrane potential (32).
Several reports underscore the potential significance of nuclear c-Myc target genes whose protein products localize to mitochondria. In one case, the mycER system was used to analyze thousands of genes on microarrays (4). Three genes with protein products that localize to mitochondria, peptidyl-prolyl cis-trans isomerase F, heat shock protein 60, and the chaperone grpE were identified by using this technique. Another study focused on genes regulated by myc-induced lymphomagenesis in the bursa of Fabricius (33). Several mitochondrial genes, including matrix nucleoside diphosphate kinase and matrix protein P1, were identified. Additionally, recent evidence suggests that the response of Myc to diverse apoptotic stimuli converges at a common mitochondrial signaling element (34).
Microarray analysis comparing c-myc+/+ and c-myc−/− cells supports our conclusion that PRDX3 is a c-Myc target gene (3). Rat PRDX3, termed thioredoxin peroxidase, was identified as a gene that was more highly expressed in both wild-type fibroblasts and myc−/− fibroblasts with reconstituted c-myc as compared with c-myc−/− cells. This same study also found that PRDX3 expression was increased 2.4-fold upon expression of ectopic myc in normal c-myc+/+ fibroblasts. This finding indicates that deregulated overexpression of myc, which mimics conditions found in cancer cells, induces PRDX3, and it suggests that at least some of the target genes that are regulated by myc under physiological conditions are also activated when myc is overexpressed. We hypothesize that Myc is not the sole regulator of PRDX3 expression, as PRDX3 is still expressed in myc−/− fibroblasts. Rather, PRDX3 likely belongs to a class of genes that facilitates accelerated cellular growth and metabolism induced by c-Myc.
The mechanism by which c-Myc regulates both proliferation and apoptosis remains unclear. The c-Myc target gene ODC has been found to affect both processes, in that overexpression stimulates apoptosis, whereas inhibiting ODC activity blocks cell-cycle progression (35). This observation led to the multiple-effector model, whereby c-Myc regulates targets that overlap in function. In support of this model, our data indicate that Myc regulates a mitochondrial peroxiredoxin that is required for proliferation as well as apoptosis in transformed cells. Additionally, our results suggest that reduced mitochondrial function affects Myc-mediated transformation. Although is has been reported that the loss of mitochondrial membrane potential is a downstream event in Myc-mediated apoptosis (36), it is not known how the mitochondrial membrane potential affects proliferation and the apoptotic signaling cascade. Recently, it has been suggested that the mitochondrial membrane potential could be an integrator of growth, maturation, and apoptotic pathways (37). The observation that both transformation and apoptosis are affected in PRDX3AS cells supports this hypothesis.
Supplementary Material
Acknowledgments
We thank members of the Dang lab for assistance. This work was supported by National Institutes of Health Grants CA51497, T32HL07525, and T32GM07819.
Abbreviations
- AS
antisense
- ROS
reactive oxygen species
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Henriksson M, Luscher B. Adv Cancer Res. 1996;68:109–182. doi: 10.1016/s0065-230x(08)60353-x. [DOI] [PubMed] [Google Scholar]
- 2.Schuhmacher M, Kohlhuber F, Holzel M, Kaiser C, Burtscher H, Jarsch M, Bornkamm G W, Laux G, Polack A, Weidle U H, Eick D. Nucleic Acids Res. 2001;29:397–406. doi: 10.1093/nar/29.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo Q M, Malek R L, Kim S, Chiao C, He M, Ruffy M, Sanka K, Lee N H, Dang C V, Liu E T. Cancer Res. 2000;60:5922–5928. [PubMed] [Google Scholar]
- 4.Coller H A, Grandori C, Tamayo P, Colbert T, Lander E S, Eisenman R N, Golub T R. Proc Natl Acad Sci USA. 2000;97:3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis B C, Shim H, Li Q, Wu C S, Lee L A, Maity A, Dang C V. Mol Cell Biol. 1997;17:4967–4978. doi: 10.1128/mcb.17.9.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto T, Matsui Y, Natori S, Obinata M. Gene. 1989;80:337–343. doi: 10.1016/0378-1119(89)90297-7. [DOI] [PubMed] [Google Scholar]
- 7.Tsuji K, Copeland N G, Jenkins N A, Obinata M. Biochem J. 1995;307:377–381. doi: 10.1042/bj3070377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chae H Z, Chung S J, Rhee S G. J Biol Chem. 1994;269:27670–27678. [PubMed] [Google Scholar]
- 9.Kang S W, Chae H Z, Seo M S, Kim K, Baines I C, Rhee S G. J Biol Chem. 1998;273:6297–6302. doi: 10.1074/jbc.273.11.6297. [DOI] [PubMed] [Google Scholar]
- 10.Prosperi M T, Ferbus D, Karczinski I, Goubin G. J Biol Chem. 1993;268:11050–11056. [PubMed] [Google Scholar]
- 11.Zhang P, Liu B, Kang S W, Seo M S, Rhee S G, Obeid L M. J Biol Chem. 1997;272:30615–30618. doi: 10.1074/jbc.272.49.30615. [DOI] [PubMed] [Google Scholar]
- 12.Araki M, Nanri H, Ejima K, Murasato Y, Fujiwara T, Nakashima Y, Ikeda M. J Biol Chem. 1999;274:2271–2278. doi: 10.1074/jbc.274.4.2271. [DOI] [PubMed] [Google Scholar]
- 13.Noh D Y, Ahn S J, Lee R A, Kim S W, Park I A, Chae H Z. Anticancer Res. 2001;21:2085–2090. [PubMed] [Google Scholar]
- 14.Eilers M, Picard D, Yamamoto K R, Bishop J M. Nature (London) 1989;340:66–68. doi: 10.1038/340066a0. [DOI] [PubMed] [Google Scholar]
- 15.Stone J, de Lange T, Ramsay G, Jakobovits E, Bishop J M, Varmus H, Lee W. Mol Cell Biol. 1987;7:1697–1709. doi: 10.1128/mcb.7.5.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evan G I, Wyllie A H, Gilbert C S, Littlewood T D, Land H, Brooks M, Waters C M, Penn L Z, Hancock D C. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 17.Shim H, Chun Y S, Lewis B C, Dang C V. Proc Natl Acad Sci USA. 1998;95:1511–1516. doi: 10.1073/pnas.95.4.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fairchild C R, Ivy S P, Kao-Shan C S, Whang-Peng J, Rosen N, Israel M A, Melera P W, Cowan K H, Goldsmith M E. Cancer Res. 1987;47:5141–5148. [PubMed] [Google Scholar]
- 19.Kim S, Li Q, Dang C V, Lee L A. Proc Natl Acad Sci USA. 2000;97:11198–11202. doi: 10.1073/pnas.200372597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyd K E, Wells J, Gutman J, Bartley S M, Farnham P J. Proc Natl Acad Sci USA. 1998;95:13887–13892. doi: 10.1073/pnas.95.23.13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egan S E, Giddings B W, Brooks M W, Buday L, Sizeland A M, Weinberg R A. Nature (London) 1993;363:45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- 22.Littlewood T D, Hancock D C, Danielian P S, Parker M G, Evan G I. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mateyak M K, Obaya A J, Adachi S, Sedivy J M. Cell Growth Differ. 1997;8:1039–1048. [PubMed] [Google Scholar]
- 24.Lee Y J, Galoforo S S, Berns C M, Tong W P, Kim H R, Corry P M. J Cell Sci. 1997;110:681–686. doi: 10.1242/jcs.110.5.681. [DOI] [PubMed] [Google Scholar]
- 25.Lee Y J, Galoforo S S, Berns C M, Chen J C, Davis B H, Sim J E, Corry P M, Spitz D R. J Biol Chem. 1998;273:5294–5299. doi: 10.1074/jbc.273.9.5294. [DOI] [PubMed] [Google Scholar]
- 26.Hockenbery D M, Oltvai Z N, Yin X M, Milliman C L, Korsmeyer S J. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 27.Petit J M, Maftah A, Ratinaud M H, Julien R. Eur J Biochem. 1992;209:267–273. doi: 10.1111/j.1432-1033.1992.tb17285.x. [DOI] [PubMed] [Google Scholar]
- 28.Nishikawa T, Edelstein D, Du X L, Yamagishi S, Matsumura T, Kaneda Y, Yorek M A, Beebe D, Oates P J, Hammes H P, et al. Nature (London) 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies K J. J Biol Chem. 1990;265:16330–16336. [PubMed] [Google Scholar]
- 30.Bryk R, Griffin P, Nathan C. Nature (London) 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 31.Brown G C. Biochim Biophys Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- 32.Kokoszka J E, Coskun P, Esposito L A, Wallace D C. Proc Natl Acad Sci USA. 2001;98:2278–2283. doi: 10.1073/pnas.051627098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neiman P E, Ruddell A, Jasoni C, Loring G, Thomas S J, Brandvold K A, Lee R, Burnside J, Delrow J. Proc Natl Acad Sci USA. 2001;98:6378–6383. doi: 10.1073/pnas.111144898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soucie E L, Annis M G, Sedivy J, Filmus J, Leber B, Andrews D W, Penn L Z. Mol Cell Biol. 2001;21:4725–4736. doi: 10.1128/MCB.21.14.4725-4736.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Packham G, Porter C W, Cleveland J L. Oncogene. 1996;13:461–469. [PubMed] [Google Scholar]
- 36.Hotti A, Jarvinen K, Siivola P, Holtta E. Oncogene. 2000;19:2354–2362. doi: 10.1038/sj.onc.1203567. [DOI] [PubMed] [Google Scholar]
- 37.Augenlicht L H, Heerdt B G. Nat Genet. 2001;28:104–105. doi: 10.1038/88800. [DOI] [PubMed] [Google Scholar]
- 38.Grandori C, Mac J, Siebelt F, Ayer D E, Eisenman R N. EMBO J. 1996;15:4344–4357. [PMC free article] [PubMed] [Google Scholar]
- 39.Blackwell T K, Huang J, Ma A, Kretzner L, Alt F W, Eisenman R N, Weintraub H. Mol Cell Biol. 1993;13:5216–5224. doi: 10.1128/mcb.13.9.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}