

Abstract
Reactive oxygen species induce the expression of detoxification and repair genes critical for life in an aerobic environment. Bacterial factors that sense reactive oxygen species use either thiol-disulfide exchange reactions (OxyR, RsrA) or redox labile 2Fe–2S clusters (SoxR). We demonstrate that the reduced form of Bacillus subtilis OhrR binds cooperatively to two adjacent inverted repeat sequences in the ohrA control region and thereby represses transcription. In the presence of organic hydroperoxides, OhrR is inactivated by the reversible oxidation of a single conserved cysteine residue to the corresponding cysteine-sulfenic acid, and perhaps to higher oxidation states.
Keywords: peroxide stress|oxidative stress|peroxiredoxin|alkyl hydroperoxide reductase|Bacillus subtilis
Reactive oxygen species (ROS), including H2O2, superoxide anion (O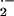 ), hydroxyl radical (⋅OH), and organic hydroperoxides (ROOH), are toxic to cells by virtue of their ability to damage DNA, protein, and membranes (1). To overcome oxidative stress, cells activate the expression of both detoxification and repair systems.
), hydroxyl radical (⋅OH), and organic hydroperoxides (ROOH), are toxic to cells by virtue of their ability to damage DNA, protein, and membranes (1). To overcome oxidative stress, cells activate the expression of both detoxification and repair systems.
In bacteria, the induction of oxidative stress stimulons is controlled by transcription factors that can directly sense the presence of ROS. One common mechanism is the reversible formation of a disulfide bond. For example, in Escherichia coli OxyR reacts with H2O2 to form an intramolecular disulfide bond, and the resulting conformational change activates the protein (2, 3). Similarly, in Streptomyces coelicolor oxidation of the RsrA anti-sigma factor by H2O2 or diamide leads to disulfide bond formation and results in release of σR and activation of appropriate target genes (4). The reaction of Bacillus subtilis PerR with H2O2 is modulated by the identity of the bound regulatory metal ion and may also involve disulfide bond formation (5). A similar mechanism has been proposed for the S. coelicolor PerR homolog CatR (6).
B. subtilis displays a complex adaptive response to peroxide stress coordinated by at least three transcription factors: PerR, σB, and OhrR. PerR, a metalloprotein of the ferric uptake regulator (Fur) family, represses the major vegetative catalase (KatA), alkyl hydroperoxide reductase (AhpCF), the MrgA DNA-binding protein, and other peroxide stress genes (7, 8). The general stress response, coordinated by the σB protein, includes paralogs of many of these stress proteins including two additional catalases (KatB and KatX) and the MrgA homolog, Dps. OhrR is representative of a recently identified class of peroxide sensors and acts as a repressor of the ohrA organic peroxide resistance gene (9, 10). Interestingly, a second organic peroxide resistance gene, ohrB, is under control of σB (11). The role of Ohr proteins in organic peroxide resistance has now been demonstrated in Xanthomonas campestris, Pseudomonas aeruginosa, Enterococcus faecalis, Actinobacillus pleuropneumoniae, and B. subtilis (10, 12–16). In many organisms, ohr genes are flanked by genes encoding OhrR homologs (9).
Here we characterize the interactions of B. subtilis OhrR with its cognate operator sequences and chemical inducers. Our results indicate that reduced OhrR binds to two adjacent inverted repeat sequences in the ohrA control region. Exposure to ROS leads to the reversible oxidation of a single conserved cysteine residue to cysteine-sulfenic acid, and perhaps higher oxidation states, and thereby leads to derepression of ohrA transcription.
Materials and Methods
OhrR Purification.
The ohrR coding region was PCR-amplified by using primers 527 (5′-GGTGAACACCATGGAAAATAAATT-3′) and 528 (5′-CCGGATCCGTTGCTGAATAAATAAA-3′), digested with NcoI and BamHI (sites underlined), and ligated to the same sites in pET16x to create pMF5. The gene sequence was confirmed by sequencing. E. coli BL21(DE3)/pLysS (17) containing pMF5 was grown in 500 ml LB containing 100 μg/ml ampicillin and 10 μg/ml chloramphenicol until the OD600 reached 0.6, then OhrR expression was induced with 1 mM isopropyl β-d-thiogalactoside for 2 h at 37°C with aeration. Cells were harvested by centrifugation at 10,000 rpm for 5 min at 4°C, and the pellet was kept at −80°C overnight. The cell pellet was thawed on ice for 30 min and suspended in 10 ml resuspension solution (50 mM Tris⋅Cl, pH 8.0/2 mM EDTA, pH 8.0/0.1 mM DTT/1 mM β-mercaptoethanol/100 mM NaCl/1 mM PMSF/5% glycerol), and the cells were broken by French Pressure cell. The lysate was clarified by centrifugation at 10,000 rpm for 5 min at 4°C and applied to a heparin column. All further steps were done at 4°C. Bound proteins were eluted with gradient of NaCl (0.05–1 M) in elution buffer (50 mM Tris⋅Cl, pH 8.0/2 mM EDTA, pH 8.0/0.1 mM DTT/1 mM PMSF/5% glycerol). Samples were loaded on 15% SDS/PAGE to identify the fractions that contained OhrR. OhrR was concentrated by using a Centricon-10 microconcentrator (Amicon) and applied to a 25-ml Superdex75 FPLC column (Amersham Pharmacia) and eluted at a flow rate of 0.5 ml/min in elution buffer (50 mM Tris⋅Cl, pH 8.0/2 mM EDTA, pH 8.0/0.1 mM DTT/150 mM NaCl/5% glycerol). The protein concentration of purified OhrR was measured by BioRad protein assay. OhrR was estimated to be 99% pure. The molecular mass of OhrR in monomer form was as expected (about 17.0 kDa) as seen on SDS/PAGE gel. In solution OhrR is a dimer with an apparent molecular mass of 42.6 as determined by Superdex-75 FPLC (using chicken ovalbumin, 47 kDa; carbonic anhydrase, 29 kDa; cytochrome c, 12.4 kDa as standards).
DNaseI Footprinting.
A 211-bp ohrA promoter fragment was generated by PCR with wild-type B. subtilis chromosomal DNA (CU1065) and primers 539 (5′-GCAAATCCGTATACAGCGGG-3′) and 529 (5′-CGGGATCCAATGACCTTTCCTTCTCTTC-3′) containing AccI and BamHI sites (underlined). The PCR product was labeled by fill-in reaction by using [α-32P]dATP and Klenow fragment after digestion with either BamHI (top strand) or AccI (bottom strand). The ohrA* promoter fragment was prepared by using the same strategy with HB2031 (ohrA*-cat-lacZ) chromosomal DNA as template. The ohrA, ohrA1, and ohrA2 promoter fragments were generated by using pMF3, pMF15, or pMF16 (containing ohrA, ohrA1, or ohrA2 promoters in pJPM122) as templates. Primers used in these PCRs were 529 and 535 (5′-GTACATATTGTCGTTAGAAC-3′), which hybridizes upstream of the cloning site in the vector. Reaction mixtures (50 μl total volume) contained 1× binding buffer (20 mM Tris⋅Cl, pH 7.0/50 mM KCl/1 mM EDTA/5% glycerol/50 μg/ml BSA/5 μg/ml calf thymus DNA), labeled DNA fragment, and purified OhrR as indicated and were incubated at room temperature for 10 min. Fifty microliters of Ca/Mg solution (5 M CaCl2 and 10 mM MgCl2) was added, and then the reaction was digested with 0.06 unit DNaseI for between 1 and 3 min. Reactions were stopped by adding 700 μl stop solution (645 μl ethanol, 50 μl of 3 M sodium acetate, and 5 μl of 1 mg/ml yeast RNA), and the nucleic acids were recovered by centrifugation for 15 min at maximum speed. The DNA was resuspended in formamide loading buffer and loaded onto 6% sequencing gel. The G+A ladder used was generated by mixing approximately 20,000 cpm of labeled fragment with cleavage buffer (1 μl formic acid in 1 ml formamide loading buffer) and incubating at 104°C for 20 min (18). The image was obtained by exposing the dried gel to the Phosphor Screen (Molecular Dynamics). The amount of protein bound to DNA was quantified by using imagequant data analysis software. The unbound region of each reaction was used as an internal control.
ohrA Promoter Mutation.
To introduce mutations in the OhrR binding site we used mega-primer PCR mutagenesis taking advantage of unique flanking HindIII and HinfI sites. PCR fragment A, generated from CU1065 chromosomal DNA with primers 539 and 529, was digested with HinfI at a site downstream from the OhrR binding site. The digested fragments were used as a template for a second PCR using primers 539 and 621 (5′-TTAAATTCCCCACAATTAAA-3′). Single-stranded primers and nontemplated nucleotides were degraded by addition of 5 units Klenow fragment at 37°C for 45 min. This PCR product was the mega-primer used in another PCR with HindIII-cleaved PCR product A as template and primer 529. The final product contained the same length as the first PCR product with primer 529 and primer 539 except it contained the mutation that was introduced into primer 621 (underlined). After BamHI–HindIII digestion, this fragment was cloned into pJPM122 (19) at the BamHI–HindIII sites to generate pMF15. Primer 622 (5′-ACACAATTACCCCTGAAATG-3′) was used instead of primer 621 to generate pMF16. The resulting mutant promoter regions, designated ohrA1 and ohrA2, were fused to cat-lacZ reporter genes in pJPM122 and transferred into the SPβc2Δ2∷Tn917∷pBSK10Δ6 site of strain ZB307A (20) by double crossover recombination. The fusions were then transduced into CU1065 and HB2000 (ohrR∷kan) (10). The ohrA promoter fusion in wild type and ohrR mutant strains have been described (10). The pJPM122 cat-lacZ operon without an introduced promoter was used as a negative control.
PCR Mutagenesis of C15 of OhrR.
Plasmids pMF12 (C15S) and pMF13 (C15G) were produced by using PCR mutagenesis and CU1065 chromosomal DNA as template. The first PCR fragment was obtained by using primer 577, 5′-CGGGATCCCGAATGACAAAAAAGAGTTG-3′ (located upstream of ohrR coding sequence; BamHI site underlined), and a mutagenic antisense primer [either 574 (5′-CGCATATAGCAAAAAAGAAAGC-3′) for C15S or 576 (5′-CGCATATAGCAAAAAACCAAGC-3′) for C15G] in which the mutated bases are underlined. The second PCR fragment was obtained by using primer 578, 5′-CCCAAGCTTGTTGCTGAATAAATAAA-3′ (located downstream of ohrR coding sequence, HindIII site underlined) and the corresponding mutagenic sense primers [either 573 (5′-GCTTTCTTTTTTGCTATATGCG-3′) for C15S, or 575 (5′-GCTTGGTTTTTTGCTATATGCG-3′) for C15G], which are complementary to the mutagenic antisense primers. The two PCR products were denatured, reannealed, and extended by using Klenow fragment. PCR product generated from this extended fragment (using primers 577 and 578) was obtained, digested with BamHI and HindII, and cloned into pXT, a plasmid that integrates into the B. subtilis thrC locus and contains a xylose-inducible promoter of B. subtilis (T. Msadek, personal communication). The resulting plasmids, pMF12 and pMF13, were transformed into HB2014 [CU1065 SPβc2Δ2∷Tn917∷(ohrA-cat-lacZ)ohrR∷kan] (10). The sequences of the ohrR genes were verified by sequencing.
Transcription Fusion Analysis.
Cells were grown overnight in LB containing appropriate antibiotics, and then diluted 1:100 in the same medium. Cultures were grown at 37°C with aeration until an OD600 of 0.4. One-milliliter samples were treated with oxidants and incubated at 37°C with aeration for 15 min. Samples were harvested and assayed for β-galactosidase activities (21).
7-Chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl) Modification.
Purified OhrR in storage buffer was reduced with 2 mM DTT for 30 min at room temperature and concentrated and rediluted three times in modification buffer (50 mM potassium phosphate buffer, pH 7.0/1 mM EDTA) by using Microcon YM-10 (Amicon) ultrafiltration. Reduced protein (30 μM) was treated with a 2-fold molar excess of cumene hydroperoxide (CHP) for 30 s or 10 min, and then CHP was removed by ultrafiltration with Microcon YM-10. Sulfenic acid was trapped by addition of 1 mM NBD-Cl (Sigma) (22) and incubation for 1 h in the dark. NBD-Cl was removed by Microcon YM-10 ultrafiltration and then the absorbance at 300–600 nm was scanned with a microtiter plate reader. The reaction without OhrR was used as blank.
Mass Spectrometry.
Purified OhrR in storage buffer was reduced with 2 mM DTT for 30 min at room temperature and concentrated and rediluted three times in 1 mM ammonium bicarbonate (pH 7.0) by using Microcon YM-10 (Amicon) ultrafiltration. Electrospray MS was carried out on a Bruker Esquire-LC ion trap mass spectrometer (Breman, Germany). The protein sample (20 μM) with and without CHP treatment was injected at a flow rate of 70 μl/hr. The mass spectrometer was calibrated with myoglobin before protein sample analysis and operated in the m/z range 1,050 to 1,800. Each MS spectrum was recorded by averaging 40 spectra. The protein mass spectra were deconvoluted with Bruker Daltonics dataanalysis software.
Results and Discussion
OhrR represses ohrA by cooperative binding to two inverted repeat elements. To define the mechanism of OhrR-mediated regulation it is necessary to understand the interactions of OhrR both with its DNA operator site and with chemical inducers. Previously, we selected a mutant strain that had elevated transcription of ohrA and thereby identified a region required for OhrR-mediated regulation (10). The resulting derepressed mutant (Fig. 1A, ohrA*) contains a 15-bp deletion in the ohrA promoter/operator region that regenerates a constitutively active promoter. Inspection of the ohrA regulatory region reveals one perfect inverted repeat (TACAATT–AATTGTA) and an adjacent imperfect repeat with three mismatches (Fig. 1A). Alternatively, this regulatory region can be viewed as a single direct repeat (AATTTAATTGT–AATTTAATTGT). Because the deletion in the ohrA* mutant destroys the direct repeat and the perfect inverted repeat the determinants necessary for OhrR recognition are not immediately apparent.
Figure 1.

Binding of OhrR to the ohrA and ohrA* promoters. (A) The ohrA promoter and variants used in this study. The wild-type promoter (ohrA) contains two inverted repeats shown by thick arrows. Bases matching the perfect inverted repeat are identified by a vertical line. The thin arrows indicate the 11-bp direct repeats. The −10 and −35 regions are shown in bold letters, and the transcription start site (+1 position) is in italics. ohrA* is a previously described 15-bp deletion in the promoter region (10) as indicated with a dashed line. The ohrA1 mutation (uppercase bold letters) destroys the perfect inverted repeat (but not the direct repeat). The ohrA2 mutation (uppercase bold letters) destroys the direct repeat as well as the imperfect inverted repeat. −10* is a new −10 region in the ohrA* promoter. (B) DNase I footprinting analysis of OhrR binding to the ohrA (lanes 1–9) and ohrA* (lanes 10–18) promoters. Both promoter regions (2 nM DNA) were labeled on the top strand, and the amount of OhrR (nM) is shown above the lane number. Note that at high concentrations the protein binding site extends in the downstream direction. DTT (1 mM) was present in all binding reactions. The −10, −35, and +1 positions are shown next to the G+A ladders. The inverted repeats are shown with arrows.
To define the nature of the interaction between OhrR and this control region we purified OhrR after overexpression in E. coli and analyzed its interaction with the DNA operator region by DNase I footprinting. OhrR binds with high affinity (Kd = 5 nM) to a region spanning both the perfect and imperfect inverted repeat elements (from +14 to −28 relative to the start site of transcription; Fig. 1B). OhrR also binds to the remaining imperfect inverted repeat in the ohrA* promoter (10) but with significantly lower affinity (Kd = 30 nM) (Fig. 1B). This finding is consistent with the small extent of derepression of ohrA* in an ohrR strain (10). Because OhrR is a dimer in solution (see Materials and Methods) this finding is consistent with the interaction with the inverted repeat sequences noted above.
To further clarify whether OhrR binding is determined by the inverted or the direct repeat sequences we constructed two additional mutant ohrA operator regions (Fig. 1A). The ohrA1 mutation affects the perfect inverted repeat, but not the direct repeat, whereas ohrA2 affects the direct repeat and the imperfect inverted repeat. In vitro, OhrR binding is significantly reduced by the ohrA1 mutation and, to a lesser extent, by the ohrA2 mutation (Fig. 2). The results with the ohrA1 mutant indicate that the direct repeat is not sufficient for high affinity binding. Together, these results indicate that OhrR can bind independently to either inverted repeat element, but that cooperative binding to both inverted repeats leads to a high affinity interaction. This finding is consistent with studies of in vivo repression of the ohrA* mutant (10) and the ohrA2 mutant (Fig. 2B). The ohrA2 promoter is still partially repressed by OhrR in vivo (9-fold vs. 130-fold for the wild type) and is still inducible by organic hydroperoxides. Note that we cannot observe in vivo activity of ohrA1 because of the destruction of the −10 region. Because the OhrR binding site covers the −10 promoter region, transcriptional repression is likely caused by direct inhibition of RNA polymerase binding (23).
Figure 2.

Binding of OhrR to mutant ohrA operators in vitro and in vivo. (A) The binding of OhrR (% DNA bound) to ohrA (⧫), ohrA1 (▪), and ohrA2 (▴) promoter DNA as determined from DNaseI footprinting data (promoter DNA at 1 nM). (B) In vivo expression from reporter fusions for the ohrA and ohrA2 promoters. Log phase cultures of each strain were induced with 100 μM CHP (gray bar) or 100 μM H2O2 (black bar) for 15 min at 37°C with aeration before β-galactosidase assay. Empty bars are nontreated cultures. Results are means ± SD from three independent experiments performed in duplicate. Note that ohrA1 promoter activity, which is not shown here, gave less than 1 Miller unit.
Sensing of Oxidants by OhrR Requires Cys-15.
Because the expression of OhrR itself is not affected by organic hydroperoxides (10), we hypothesized that ROS modulate the activity of OhrR. We measured the effects of organic hydroperoxides on the binding activity of OhrR by using DNaseI footprinting. As predicted, the binding activity of OhrR is lost upon exposure to CHP or tert-butyl hydroperoxide (Fig. 3). Significantly, binding activity is recovered upon addition of the thiol reductant, DTT. OhrR binding activity is also lost in the presence of diamide, a thiol oxidizing agent that causes the formation of disulfides between sulfhydryl groups within proteins or mixed disulfides with low molecular weight thiols. These results support the hypothesis that OhrR is directly and reversibly modified by peroxides.
Figure 3.

Effects of oxidants on binding of OhrR to the ohrA promoter. The fragment (0.1 nM DNA) used in this experiment was the same as in Fig. 2 except that AccI digestion was used to obtain the fragment for bottom-strand labeling using α-[32P]dATP. Either 1 mM CHP, 1 mM tert-butyl hydroperoxide (t-BuOOH), 1 mM H2O2, or 10 mM diamide was added to the binding reaction and incubated for 10 min at room temperature. When indicated, 100 mM DTT was then added into the reactions and incubation continued at room temperature for 30 min before DNaseI treatment. Lane 1 contained no protein; lanes 2–12 contained 2 nM OhrR.
Unexpectedly, H2O2 also caused a loss of OhrR binding activity in vitro (Fig. 3), although in vivo derepression of ohrA upon exposure to H2O2 has not been observed (10). In B. subtilis, there are many enzymes that can detoxify H2O2, including the major vegetative catalase, KatA (24), and alkyl hydroperoxide reductase (25, 26). Both of these enzymes are induced by low levels of H2O2, and we therefore hypothesized that levels sufficient for modification of OhrR may not be easily achieved. Consistent with this idea, in the katA and katA ahpC mutant strains we do observe derepression of ohrA in response to high levels (>100 μM) of H2O2 (Fig. 4). Thus, the failure to observe induction by H2O2 in wild-type cells is caused by the efficient enzymatic removal of this inducer.
Figure 4.

Induction of ohrA by H2O2. β-Galactosidase activity of strains containing the SPβc2Δ2∷Tn917∷φ(ohrA-cat-lacZ) reporter fusion were determined in wild type (HB2012), an ahpC∷Tn10 mutant (HB2038), a catalase mutant strain [HB2043; CU1065 katA∷pJH101 (32)], and the double mutant (HB2077). Samples were taken 15 min after treatment with H2O2 to a final concentration of (left to right) 0, 100, 200, 400, or 800 μM.
Unlike OxyR, which directly senses peroxides by formation of a disulfide bond between two conserved cysteine residues (2, 3), OhrR contains only one cysteine, and this residue is conserved in other OhrR homologs (10). To determine whether Cys-15 plays a role in redox sensing, we used site-directed mutagenesis to generate OhrRC15G and C15S mutant proteins. The OhrR C15G and C15S mutant proteins allow complementation of an ohrR mutant and efficiently repress an ohrA-cat-lacZ transcriptional fusion. However, only the wild-type OhrR can be induced upon exposure to CHP (Fig. 5). Thus, Cys-15 is critical for the redox-sensing mechanism of OhrR, but is not required for DNA binding.
Figure 5.

Essential role for OhrR Cys-15 in peroxide sensing. β-Galactosidase activity of strains containing ohrA-cat lacZ, ohrR∷kan complemented with OhrR, OhrR C15S, or OhrR C15G. Log-phase cultures of each strain were induced with 100 μM CHP for 15 min at 37°C with aeration before β-galactosidase assay. The empty bars are nontreated cultures, and the filled bars are CHP-treated cultures. Results are means ± SD from three independent experiments performed in duplicate.
OhrR senses oxidants by the formation of cysteine sulfenic acid (Cys-15SOH). If Cys-15 is involved in disulfide bond formation, we hypothesized that the dimeric OhrR protein might form an intermolecular disulfide bond that could be visualized with nonreducing SDS/PAGE analysis. However, oxidation of OhrR by CHP, tert-butyl hydroperoxide, H2O2, or diamide does not lead to formation of covalent OhrR dimers (data not shown).
Oxidation of Cys residues by peroxides is also known to lead to the formation of cysteine-sulfenic acids (reviewed in refs. 27 and 28). Indeed, some enzymes, including the AhpC peroxiredoxin (22) and methionine sulfoxide reductase (29), form Cys-sulfenic acid during their catalytic cycle. We therefore hypothesized that OhrR might react with peroxides to form a Cys-sulfenic acid derivative (Cys-15SOH). To examine this hypothesis, we chemically modified OhrR with NBD-Cl, an electrophile that reacts with sulfhydryls and sulfenic acids to form distinct products: the sulfenate ester (λmax = 347 nm) and the thioether (λmax = 420 nm), respectively (22). As expected, the reduced form of OhrR reacted with NBD-Cl to yield the thioether with a maximum absorption at 420 nm (Fig. 6A). In contrast, OhrR treated for 30 sec with CHP reacted with NBD-Cl to give a product with maximum absorbance near 347 nm, consistent with the rapid formation of Cys-15SOH. The amount of sulfenic acid decreases as a function of time of exposure to CHP (Fig. 6A). This finding is suggestive of further oxidation of the labile CysSOH species (e.g., to Cys-15 sulfinic and/or sulfonic acids) or migration of the NBD group (L. Poole, personal communication). Further evidence for formation of Cys-15SOH was provided by electrospray ionization–MS. Exposure of OhrR to a molar excess of CHP leads to the rapid formation of oxidized protein, which has incorporated from one to as many as three additional oxygen atoms (Fig. 6B). This finding is suggestive of rapid formation of a Cys-15SOH derivative followed by further oxidation of the labile Cys-15SOH either before or during the electrospray ionization–MS experiment.
Figure 6.

Sulfenic acid formation at C15 upon oxidative stress. (A) NBD-Cl modification of purified OhrR. Reduced OhrR was treated with CHP for 30 s (dotted line) and 10 min (dashed line). The thiol group and sulfenic acid were trapped by addition of NBD-Cl as described (22). The absorbance at 300–600 mm was scanned with a microtiter plate reader. The reaction without OhrR was used as blank. Solid line is reduced OhrR reacted with NBD-Cl. (B) Detection of OhrR oxidation by electrospray ionization–MS. Protein was incubated with CHP and immediately (within 30 s) infused into a Bruker Esquire electrospray ionization–MS instrument for analysis. The deconvoluted spectra were derived as described in Materials and Methods and peak heights were normalized to the most abundant species (set to 100%). Peak a corresponds in mass to the fully reduced protein, and peaks b, c, and d indicate the incorporation of 1, 2, or 3 oxygen atoms, respectively.
Our results demonstrate that the ability of OhrR to sense peroxidative stress requires a single conserved Cys residue that is reversibly oxidized to the sulfenic acid, and possibly to higher oxidation states. In vivo, sulfenic acids are reactive with free thiols, leading to the formation of either intramolecular or intermolecular disulfide bonds. Indeed, a sulfenic acid has been proposed as an intermediate in the formation of the disulfide bond in OxyR (2). B. subtilis does not contain glutathione, and the major free intracellular thiols are cysteine and CoASH (30). Experiments to measure the formation, and fate, of OhrR Cys-15SOH in vivo will require new techniques currently under development (L. Poole, personal communication). It is possible that the OhrR-Cys-15SOH reacts, via S-thiolation, to form a mixed disulfide with either or both of these thiols in vivo. The S-thiolation of proteins, usually involving formation of adducts with glutathione, has emerged as a major response of eukaryotic cells to oxidative stress. S-thiolation appears to regulate the activity of several enzymes and, intriguingly, may account for the regulation of DNA binding by the c-Jun transcription factor (31).
In summary, we have described a biochemical mechanism of peroxide sensing for B. subtilis OhrR, a member of a conserved family of organic peroxide-sensing transcription factors. OhrR represses transcription of the ohrA resistance gene by cooperative binding to two inverted repeat elements overlapping the promoter. Induction by peroxides requires a conserved Cys residue that is reversibly oxidized by peroxides to a sulfenic acid. Formation of higher oxidation states in vitro may reflect the known propensity of cysteine sulfenic acids to undergo further oxidation. However, oxidation does not lead to disulfide bond formation as noted for OxyR (2) and several other peroxide-sensing regulators. Thus, Cys-sulfenic acids, which have been shown to participate in the catalytic cycle of several enzymes (27, 28), have also been adapted by the cell to sense peroxidative stress in this family of transcription factors.
Acknowledgments
We thank Dr. Leslie Poole for her insightful comments on this manuscript. This work was supported by a grant from the National Science Foundation (MCB-9983656).
Abbreviations
- ROS
reactive oxygen species
- CHP
cumene hydroperoxide
- NBD-Cl
7-chloro-4-nitrobenzo-2-oxa-1,3-diazole
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Storz G, Imlay J A. Curr Opin Microbiol. 1999;2:188–194. doi: 10.1016/s1369-5274(99)80033-2. [DOI] [PubMed] [Google Scholar]
- 2.Zheng M, Aslund F, Storz G. Science. 1998;279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 3.Choi H, Kim S, Mukhopadhyay P, Cho S, Woo J, Storz G, Ryu S. Cell. 2001;105:103–113. doi: 10.1016/s0092-8674(01)00300-2. [DOI] [PubMed] [Google Scholar]
- 4.Kang J G, Paget M S, Seok Y J, Hahn M Y, Bae J B, Hahn J S, Kleanthous C, Buttner M J, Roe J H. EMBO J. 1999;18:4292–4298. doi: 10.1093/emboj/18.15.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herbig A, Helmann J D. Mol Microbiol. 2001;41:849–859. doi: 10.1046/j.1365-2958.2001.02543.x. [DOI] [PubMed] [Google Scholar]
- 6.Hahn J S, Oh S Y, Chater K F, Cho Y H, Roe J H. J Biol Chem. 2000;275:38254–38260. doi: 10.1074/jbc.M006079200. [DOI] [PubMed] [Google Scholar]
- 7.Bsat N, Herbig A, Casillas-Martinez L, Setlow P, Helmann J D. Mol Microbiol. 1998;29:189–198. doi: 10.1046/j.1365-2958.1998.00921.x. [DOI] [PubMed] [Google Scholar]
- 8.Herbig A, Helmann J D. In: Bacillus Subtilis and Its Closest Relatives. Sonenshein A L, Hoch J A, Losick R, editors. Washington, DC: Am. Soc. Microbiol.; 2002. pp. 405–414. [Google Scholar]
- 9.Sukchawalit R, Loprasert S, Atichartpongkul S, Mongkolsuk S. J Bacteriol. 2001;183:4405–4412. doi: 10.1128/JB.183.15.4405-4412.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuangthong M, Atichartpongkul S, Mongkolsuk S, Helmann J D. J Bacteriol. 2001;183:4134–4141. doi: 10.1128/JB.183.14.4134-4141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volker U, Andersen K K, Antelmann H, Devine K M, Hecker M. J Bacteriol. 1998;180:4212–4218. doi: 10.1128/jb.180.16.4212-4218.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mongkolsuk S, Praituan W, Loprasert S, Fuangthong M, Chamnongpol S. J Bacteriol. 1998;180:2636–2643. doi: 10.1128/jb.180.10.2636-2643.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ochsner U A, Hassett D J, Vasil M L. J Bacteriol. 2001;183:773–778. doi: 10.1128/JB.183.2.773-778.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rince A, Giard J C, Pichereau V, Flahaut S, Auffray Y. J Bacteriol. 2001;183:1482–1488. doi: 10.1128/JB.183.4.1482-1488.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atichartpongkul S, Loprasert S, Vattanaviboon P, Whangsuk W, Helmann J D, Mongkolsuk S. Microbiology. 2001;147:1775–1782. doi: 10.1099/00221287-147-7-1775. [DOI] [PubMed] [Google Scholar]
- 16.Shea R J, Mulks M H. Infect Immun. 2002;70:794–802. doi: 10.1128/iai.70.2.794-802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Studier F W, Rosenberg A H, Dunn J J, Dubendorff J W. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 18.Liu S T, Hong G F. Anal Biochem. 1998;255:158–159. doi: 10.1006/abio.1997.2457. [DOI] [PubMed] [Google Scholar]
- 19.Slack F J, Mueller J P, Sonenshein A L. J Bacteriol. 1993;175:4605–4614. doi: 10.1128/jb.175.15.4605-4614.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuber P, Losick R. J Bacteriol. 1987;169:2223–2230. doi: 10.1128/jb.169.5.2223-2230.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller J H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 22.Ellis H R, Poole L B. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 23.Rojo F. Curr Opin Microbiol. 2001;4:145–151. doi: 10.1016/s1369-5274(00)00180-6. [DOI] [PubMed] [Google Scholar]
- 24.Bol D K, Yasbin R E. J Bacteriol. 1994;176:6744–6748. doi: 10.1128/jb.176.21.6744-6748.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costa Seaver L, Imlay J A. J Bacteriol. 2001;183:7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bsat N, Chen L, Helmann J D. J Bacteriol. 1996;178:6579–6586. doi: 10.1128/jb.178.22.6579-6586.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Claiborne A, Yeh J I, Mallett T C, Luba J, Crane E J, 3rd, Charrier V, Parsonage D. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 28.Claiborne A, Mallett T C, Yeh J I, Luba J, Parsonage D. Adv Protein Chem. 2001;58:215–276. doi: 10.1016/s0065-3233(01)58006-7. [DOI] [PubMed] [Google Scholar]
- 29.Boschi-Muller S, Azza S, Sanglier-Cianferani S, Talfournier F, Van Dorsselear A, Branlant G. J Biol Chem. 2000;275:35908–35913. doi: 10.1074/jbc.M006137200. [DOI] [PubMed] [Google Scholar]
- 30.Newton G L, Arnold K, Price M S, Sherrill C, Delcardayre S B, Aharonowitz Y, Cohen G, Davies J, Fahey R C, Davis C. J Bacteriol. 1996;178:1990–1995. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klatt P, Molina E P, De Lacoba M G, Padilla C A, Martinez-Galesteo E, Barcena J A, Lamas S. FASEB J. 1999;13:1481–1490. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 32.Casillas-Martinez L, Setlow P. J Bacteriol. 1997;179:7420–7425. doi: 10.1128/jb.179.23.7420-7425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]