


Abstract
Recent advances in ultra-low coverage whole-genome sequencing (WGS) of single cells have enabled detailed analysis of copy number variation at a throughput approaching that of single-cell RNA sequencing. However, downstream computational methods have not seen comparable advances and are largely adaptations of deep sequencing methodology with reduced precision. Here, we present ASCENT, a computational method built to take full advantage of modern direct tagmentation-based WGS at ultra-low depth. Using joint segmentation with high-resolution bins, we accurately detect small segments, achieving accurate copy number profiles even at 100 000 reads per cell. ASCENT implements true absolute copy state inference for single cells, based on statistical modeling of coverage rather than comparison to a reference, while taking variable segment copy state into account. Further, ASCENT implements per-segment copy-neutral loss of heterozygosity (LOH) calling without the need for non-tumor or bulk WGS reference. When applied to a pediatric B-ALL sample, ASCENT finds copy-neutral LOH in a small segment and a minor subclone defined by breakpoints missed in bulk WGS. Thus, by applying appropriate computational methods, single-cell WGS provides clear advantages over bulk, even at a relatively low cell number and sequencing depth.
Graphical Abstract
Graphical Abstract.

Introduction
Direct tagmentation is used in a collection of single-cell whole-genome sequencing (scWGS) methods, which provide high-quality data at ultra-low to low coverage [1–7]. The uniform genomic coverage and high throughput have enabled detailed insight into genetic differences between individual cells and how they evolve over time [1, 3–9]. Genetic heterogeneity is of particular significance in tumor biology, since it is the substrate of tumor evolution that will lead to subclones associated with progressive disease, e.g. by responding differently to treatment and eventually causing relapse [5, 10–12]. Subclones are readily identified by clone-specific copy number variation (CNV), which is why genomic segmentation and copy number calling, and the subsequent classification of individual cell genomes into subclones, are key steps in the analysis of scWGS data. Segmentation of scWGS data has been traditionally performed with algorithms based on Hidden Markov chains [13], Circular Binary segmentation [14], or joint segmentation [15], all initially developed for bulk DNA sequencing. To account for the lower depth of sequencing, resolution has been reduced to 200–500-kb bins [3, 5]. While this achieves some stabilization of variance on the bin-level measurements, it also results in imprecise segment boundaries and a de facto minimum segment size of above 1 Mb, with some variation depending on the segmentation method chosen.
Following segmentation, copy number (or ploidy) is estimated. Inferring single-cell copy numbers can be done in four ways. The first is to determine the lowest copy state that fits with an integer copy number [16]. This does not guarantee finding the correct absolute ploidy, but rather finds the lowest possible ploidy consistent with the data, which in practice will often be the correct one. The second option is to use nuclear staining to estimate DNA content, which requires index sorting of cells into wells to take full advantage of per cell estimates [4]. A third option is to utilize minor B allele frequencies to call integer copy numbers [17, 18]. However, this fails to detect a perfectly balanced duplication event such as an M-phase cell, and has limited statistical power at lower sequencing depth. The fourth approach, applicable to libraries with no preamplification, is to use a measure based on the degree of overlap between fragments to infer the absolute copy states of single cells [1, 19]. Since fragments from the same template cannot contribute to overlaps, the fractional overlap at a certain depth of sequencing will increase with ploidy.
Tumor evolution can be reconstructed from scWGS by tracking single-nucleotide variants (SNVs) or CNVs. SNV detection at the single-cell level requires whole-genome amplification, typically achieved by using methods such as multiple displacement amplification [20] or linear amplification via transposon insertion [21]. When sequenced deeply, these methods enable SNV detection, but accurate CNV analysis is difficult because of their highly uneven coverage. More recently, primary template-directed amplification [22] has been developed to support both CNV and SNV detection. However, its relatively low throughput and high price have limited its use in large-scale studies. Alternatively, direct tagmentation-based methods, which result in accurate CNV calling, can be supplemented with haplotyping to enable detection of mirrored subclonal CNVs and copy-neutral loss of heterozygosity (LOHs) [17, 18]. By incorporating these events, a more accurate evolutionary inference can be achieved than by CNVs alone. Effective haplotyping does require sufficient heterozygous single nucleotide polymorphisms (SNPs), which in turn necessitates deep sequencing, a matched normal sample, or a large number of cells.
In this work, we develop a set of methods directly aimed at direct tagmentation scWGS and implement them into a full computational pipeline called ASCENT (Absolute Single cell Copy number Evaluation and subcloNe Topology). We apply ASCENT to scWGS data from direct tagmentation-based protocols. We show that by segmenting at high resolution, we achieve a more accurate breakpoint detection than at lower resolution, despite the noise of ultra-low coverage data. To call absolute copy numbers, we develop a method based on fractional overlap and derive accurate log-odds ratios that are based on a closed-form solution of the expected overlap at absolute copy states to select the correct ploidy. Finally, we implement methods that allow per-clone haplotyping with ultra-low coverage scWGS data without a matched normal sample. Using these methods, we are able to call allele-specific absolute copy numbers in a majority of our segments and identify a sub-5-Mb copy-neutral LOH in one of our previously identified two-copy-state segments.
Materials and methods
Sample preparation
HCT116 cells were cultured for two passages in RPMI medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, then split 1 day before harvesting to achieve 40% confluency and ensure presence of cycling cells. Cells were collected, centrifuged, and resuspended in 1× phosphate buffered saline (PBS) with 2% FBS and stained with Hoechst 34580 at 1 μg/ml final concentration for 1 h at 37°C. Before sorting, cells were strained through a 40 μM cell strainer. Cells were sorted based on nuclei marker intensity as a proxy for cell cycle.
For the leukemia sample (ALL40), bone marrow was collected at diagnosis after written informed consent was received by the patient and/or guardians in accordance with the ethical standards of the Helsinki Declaration. This study was approved by the Regional Ethics Committee in Pirkanmaa, Tampere Finland (#R13109) and Regional Ethics Committee in Stockholm, Sweden (Dnr 2021-02718). Mononuclear cells were extracted by gradient separation and frozen viably in FBS supplemented with 10% dimethyl sulfoxide (DMSO). On the day of sorting the cells were thawed in culture medium, washed with 1× PBS, and depleted of dead cells. Cells were stained with antibodies for CD34, CD10, CD20, CD38, CD45, and CD19 according to the manufacturer’s protocol. Cells were washed, resuspended in 1× PBS with 2% FBS, and filtered by passing them through a 40 μM strainer before sorting. Gates were set to exclude debris and potential doublets and sorted (Sony SH800) into 384-well plates with a hypotonic lysis buffer, containing oligo(dT), RNase Inhibitor, Triton X-100, ERCC controls, and dNTP. After sorting, the plates were spun at 2000 × g at 4°C for 5 min, snap frozen on dry ice and kept at −80°C until processing.
Library preparation and sequencing
The WGS part of DNTR-seq was performed as previously described [1]. In short, each cell nucleus in 1 μl lysis buffer underwent proteinase K treatment, tagmentation, and sodium dodecyl sulfate (SDS) treatment before being barcoded and amplified with 18 cycles of PCR. Libraries were pooled and cleaned three times with 0.9× ratio of SPRI beads:solution. ALL40 library was sequenced on a NextSeq 550, with 2 × 37 bp paired-end sequencing to an average of 280 000 bam read pairs, and HCT cell libraries were sequenced on a NextSeq 550 2 × 36 bp paired-end (average of 1 231 000 bam read pairs) and a NovaSeq 6000 with 2 × 150 bp paired-end sequencing (average of 2 832 000 bam read pairs). All QC metrics are summarized per cell in Supplementary file S1. Data were delivered as demultiplexed FASTQ files.
ASCENT
ASCENT (available on GitHub: https://github.com/EngeLab/ASCENT) was implemented as an end-to-end pipeline using snakemake within conda environments. For an overview of the pipeline, see Supplementary Fig. S1. The pipeline can be run either as a DNA- or RNA-only pipeline, or combined, whereby cell phase information can be implemented to eliminate S and G2M phase cells from segmentation and clustering. ASCENT performs standard quality control and clustering of RNA data, enabling direct comparison between expression and copy state. Total runtime for the full ASCENT pipeline is ∼7 CPU minutes per cell. This estimate includes all pipeline features, concluding in a phylogenetic tree, and is based on typical non-ideal circumstances—1024 FASTQ files (one per cell), sequenced to a relatively high depth of 2.5 million reads per cell finish in 10 h when run on 12 cores on a workstation with a 16-core (32-thread) processor and 128 GB of RAM, running a 64-bit Linux operating system.
Filtering of raw sequencing data
Adapter sequences and low-quality bases were removed with trim-galore (https://zenodo.org/records/7598955) with error rate set to maximum 0.1. Paired DNA reads were aligned with bwa [23] to GRCh38, minimum seed length at 25 and -M for marking shorter split hits as secondary, and sorted with samtools [24]. Picard’s (https://github.com/broadinstitute/picard) MarkDuplicates was used to remove duplicates from bam files. Bam files were filtered by minimum mapq 20 and max insert size 1000. Two different fragment files were created, one with stricter filtering, which was used for overlap statistics (see below) and another that was used for copy number calling. Fragment files were created with bedtools [25] bamtobed, and only autosomal and sex chromosomes were kept. When aligning to GRCh38, we removed fragments that fell within Encode’s exclusion list [26], and when aligning to T2T (Telomere-to-Telomere reference) [27], we removed fragments that fell within a list from the R package excluderanges (https://doi.org/10.18129/B9.bioc.excluderanges). Reads were piled up in fixed bins (sizes 10, 40, 200, and 500 kb) using bedtools intersect.
Filter bin-level data
An exclusion list was created to filter out problematic regions of the genome. We first filtered the fixed-size bins to exclude mappability under 0.8 and GC fraction under 0.2. At 10 kb resolution, this removes 43 339 out of 308 837 bins. Further, we used a curated list of diploid cells sequenced on a NextSeq 550 to extend the list to genomic regions where the coverage significantly deviates from expected. We modeled the expected counts with a Poisson distribution, adjusting for GC content and mapping biases, flagging bins with significant deviations as “bad bins.” This removed 5209 more bins, leaving 260 289 bins at 10 kb resolution.
Raw bin counts that fall in “good bins” were summed up, and cells with bin counts under a certain threshold (default: 60 000) were removed from further analysis. If mRNA has been prepared for the same cells, we recommend removing replicating cells for less noise during breakpoint detection.
Hypothesis-free segmentation
The hypothesis-free segmentation method was based on determining the log-likelihood ratio for a given genomic bin position and a window extending from each side of this position, between two competing hypotheses: (i) copy state is the same on each side (ii) copy state is different on the two sides. Counts [X] were modeled as a poisson process, where the mean was determined by a scaling factor [S], copy state [p] effect of gc [Q(g)], and mappability [m]. For bin i: 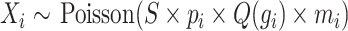
We use this equation to calculate the log-likelihood ratio between hypothesis 1 and 2:
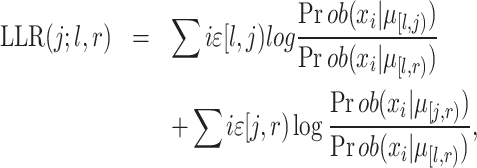 |
where j is the bin index that is evaluated, l is the lower bound of the window and r is the upper bound of the window (l < j < r). 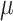 [l,j) is the average copy state in the bin interval [l,j), and Prob(X, |
[l,j) is the average copy state in the bin interval [l,j), and Prob(X, | 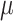 [l,j)) is the poisson point probability of observing X counts in bin i. Raw bin counts were collected as explained earlier. Bin counts were normalized with a quadratic model for GC bias and mappability, followed by mean centering. Window size is determined dynamically based on the average number of bin counts for each cell, and is set to the average number of bins that encompasses 200 reads, with a minimum size of 100 and maximum size of 10 000. Window sizes larger than half the chromosome arm being segmented were rounded to half the chromosome arm length. The LLR was calculated for each bin position across the genome, and each region of consecutive bins with LLR > 5 constitutes a candidate breakpoint. The precise breakpoint position within each such region was chosen based on the bin position with the highest LLR (Supplementary Fig. S2A). Subsequently, segments <10 bins were excluded from further analysis, regardless of window size.
[l,j)) is the poisson point probability of observing X counts in bin i. Raw bin counts were collected as explained earlier. Bin counts were normalized with a quadratic model for GC bias and mappability, followed by mean centering. Window size is determined dynamically based on the average number of bin counts for each cell, and is set to the average number of bins that encompasses 200 reads, with a minimum size of 100 and maximum size of 10 000. Window sizes larger than half the chromosome arm being segmented were rounded to half the chromosome arm length. The LLR was calculated for each bin position across the genome, and each region of consecutive bins with LLR > 5 constitutes a candidate breakpoint. The precise breakpoint position within each such region was chosen based on the bin position with the highest LLR (Supplementary Fig. S2A). Subsequently, segments <10 bins were excluded from further analysis, regardless of window size.
Joint segmentation
The joint segmentation (mpcf [multiple piecewise constant fitting] from the copynumber package [15]) is based on a piecewise constant fit (pcf) to the data through penalized least squares minimization. The penalty applied for each discontinuity or breakpoint in the segmentation curve is set by the gamma parameter.
The input to mpcf needs to be normalized. We first applied a Freeman-Tukey (FT) transformation followed by a lowess smoothing on bin counts falling in good bins for variance stabilization. In addition, we normalized (in the same way) a curated list of normal cells [available on GitHub for NovaSeq 6000 (2 × 150 bp) and NextSeq 550 (2 × 37 bp)], matching the sequencer of the current experiment. We averaged normalized normal-cell bin counts per bin and used that factor as a normalization factor for our data. This normalization is optional, but in our experience, it can be helpful to remove noise. If this normalization is not performed, gamma values need to be 2–10 times higher to achieve a similar result. mpcf was run with setting mpcf_fast = FALSE and was run per chromosome to enable parallelization. For each sample, we ran multiple gammas ranging from 0.5–5 for cell numbers between 100 and 1000, and bin sizes between 10 and 40 kb. Larger bin sizes and higher cell numbers require lower gamma values. This resulted in candidate breakpoints to define segments.
Scale factor estimation for initial integer copy number conversion
To convert read counts to copy number integers, we estimated a scale factor for each cell based on the fact that genomic segments can only exist in whole numbers (integers). While FT transformation (see earlier) helps detect breakpoints, its nonlinear compression of count values disrupts the proportional relationship between counts and copy numbers needed in this context. Instead, we applied a quadratic model to correct for GC bias and mappability, followed by depth normalization. For segments identified through either hypothesis-free or joint segmentation, we calculated the mean normalized bin count per segment and assigned this value to all bins within that segment. We then used a maximum likelihood (ML) approach to determine the integer copy numbers that best fit these normalized segment values. We found the scale factor S, which minimized the objective function based on the Manhattan distance between integer and real-valued copy values c: 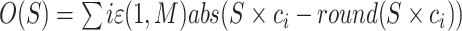 , across the set of all M bins. For each cell, factors between 1.2 and 4.5 were tested (Supplementary Fig. S2B). This factor was used to call initial copy numbers, which will be corrected during absolute copy number calling.
, across the set of all M bins. For each cell, factors between 1.2 and 4.5 were tested (Supplementary Fig. S2B). This factor was used to call initial copy numbers, which will be corrected during absolute copy number calling.
Absolute copy number calling
Absolute copy number calling depends on accurate segmentation of the genome and initial scaling of copy numbers. The input was copy numbers per segment per single cell and fragment files. Fragment files for absolute copy number calling were stringently filtered; we performed additional deduplication of possibly index hopped or mispriming events by removing all fragments that had a start site within 1 bp of each other. The fragments were additionally filtered to remove aberrant insert sizes, so that only fragments that fall within the 5th and 95th percentile of fragment size (per cell) were used. Note that while this procedure removes some accurate data along with artifactual reads, the resulting overlap is not altered since sampling is done at a set depth (see below). Five base pairs were removed on each side of the fragment to ensure removal of the 9 bp overhangs inherent with tagmentation with Tn5. To gather overlap statistics per cell per estimated copy number, the filtered fragments were grouped so that all fragments originating from segments with the same estimated copy number were calculated separately. For these calculations, we excluded segments smaller than a hundred 10 kb bins, and segments where the copy number did not exist in at least a thousand 10 kb bins. Any cells where the remaining segments covered less than half the genome were removed from further absolute copy number analysis. This was rare in our dataset but will happen at very low read depth or very short insert sizes combined with low read depth. To calculate a standardized overlap estimate, copy number states per cell were randomly sampled from corresponding fragments to reach 0.5% coverage, using the coverage function from the R package GenomicRanges [28] to calculate the number of base positions with coverage of 2. Subsampling to a specified fixed coverage irrespective of original sequencing depth makes the subsequent overlap measurements independent of original depth and thereby comparable between samples. The average of over 20 independent random samplings was used. These initial calculations resulted in standardized overlaps per copy state per cell, e.g. a diploid male cell would have one score for all autosomal chromosomes (copy 2) and another score for sex chromosomes (copy 1). To adjust initial copy numbers, we used information from all separate copy states per cell to make a combined statistic to decide if copy numbers should be adjusted as follows.
We first calculated the probability of picking the same base pair twice at different copy numbers (1 through 4), at a given fraction of genome covered (0.5%) and a noise level (10%, tunable parameter). To get the probability of the current ploidy being right, we calculated:
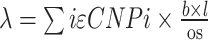 where p indicated the copy number specific probability, b = bins, l = bin size and OS = overlap size, and the poisson log probability was calculated as
where p indicated the copy number specific probability, b = bins, l = bin size and OS = overlap size, and the poisson log probability was calculated as 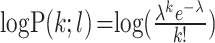 , where k was the sum of overlaps. Since fractional copy state differences are accurately detected using the scale factor calculation (see above), we need only consider the special cases of doubling (2× ploidy) or half (1/2× ploidy). We calculated log-odds ratios of doubling/estimated or halving/estimated copy numbers and used a P-value of 1% (log-odds = 4.6) as the cutoff to adjust the copy states.
, where k was the sum of overlaps. Since fractional copy state differences are accurately detected using the scale factor calculation (see above), we need only consider the special cases of doubling (2× ploidy) or half (1/2× ploidy). We calculated log-odds ratios of doubling/estimated or halving/estimated copy numbers and used a P-value of 1% (log-odds = 4.6) as the cutoff to adjust the copy states.
Initial clone assignment
Clone assignment is performed on samples that have undergone mpcf segmentation. If hypothesis-free segmentation was selected, the pipeline assumes that no clones should exist, and the absolute copy numbers per cell are the final output.
Absolute integer copy numbers per bin were winsorized to max 8, and segments smaller than a threshold (10 bins) were masked. A UMAP dimensionality reduction was run on the copy number matrix (including all good bins) with 30 dimensions and 20 nearest neighbors, then the density-based clustering algorithm, hdbscan [29], was used to identify clones. If the sample had too few unique copy states, counted as how many unique combinations of copy number per bin x cells (based on a threshold, n = 32), we group cells into clones based on copy number per bin. The clones underwent a series of quality checks. First, we calculated the distance per cell from the median copy number of its clone on both bin and segment level. The differences were z-scaled by clone (mean/standard deviation) and cells above a threshold (3, tunable) as well as cells that exhibited copy number differences in >20% of bins compared to the clone median were moved to clone zero (which consists of cells that did not fit any clones, by hdbscan).
Next, we determined if sub-clustering was possible by k-means clustering. First, we found the maximum k that could be used, based on the number of cells in each clone. Then we smoothed the copy number data per clone and ran k-means clustering with all different k’s (as determined by cell number or the max k parameter). We found an elbow point in the measured within-clusters sums of squares to estimate the best k, if that k fell above a threshold (0.4), we used that k to split the clones further. We made revised median profiles of the clone’s copy numbers, merged identical clones if any, and removed clones that had no cells after filtering.
Clone and breakpoint refinement
Initial clones derived from the gamma that visual inspection revealed to correspond to a good segmentation and clustering, were further refined, mainly in regard to breakpoint detection. Pseudobulk profiles were obtained per clone. Mpcf was run on the pseudobulked counts in 10 kb bins, normalized the same way as the input to single cell mpcf. Here it is recommended to use 10 kb bins, regardless of which bin size was used for initial clones. To determine the correct gamma parameter for mpcf, values ranging from 0.1–5 were tested. A script is provided (on https://github.com/EngeLab/ASCENT ) that allows optimization of the gamma parameter by performing segmentation across different closely spaced gamma parameters and finding the inflexion point where the number of breaks resulting from the segmentation flatten out. This approach is highly CPU intensive, and in practice a range of gamma parameters will give an identical result. The correct gamma parameter can generally be determined by visually inspecting the resulting clone heatmap at a much lower number of gamma values. In general, a lower gamma is required for more complex samples, and higher if normal cell normalization was not performed. Small segments were merged with the larger of two neighbouring segments (minimum segment size set to 10 bins), and segments that were at chromosome boundaries were merged with their adjacent segment, if they were <40 bins. If any segments had high residuals (threshold 0.3), splitting of mixed clones was performed if the splitting decreased the residual at previously high residual segments, as well as keeping the residuals at all other segments low. This process was done iteratively with tunable thresholds. Some segments did not improve by this and were masked if they had high residuals in multiple clones. Clones with a high fraction (30%) of high residual segments were removed, and the cells from those clones were moved to clone zero. At this stage, duplicate clones might have been created, and those were merged. In some cases, manual masking of segments could be preferable (e.g. at TCR regions where variability might not be tumor cell specific). If the segments that distinguished clones were high residual, fuzzy merging of those clones was performed. To finalize the clones, bad cells were removed by removing cells that had more than a thousand 10 kb bins different from the median of the clone. The assignment of cells into clones at that stage was considered to be final, and final CNVs had been achieved.
Allele specific copy numbers
Aligned reads were aggregated across all cells per patient and genotyped for common SNPs (1KG phase 3 [30], population allele frequency >0.5%) using cellsnp-lite [31]. Positions covered with at least five reads, including at least two supporting the alternate allele, were kept as likely germline heterozygous SNPs. These were phased with Eagle2 [32] with a reference population panel from the harmonized Human Genome Diversity Project (HGDP) and the 1000 Genome Project (1kp) [33]. Reads supporting each allele were summarized per cell with bcftools mpileup. First, we identified fully homozygous segments by calculating the ratio between heterozygous and homozygous SNPs per segment over all cells. This allowed us to detect LOH when no heterozygous alleles existed (i.e. when every subclone is LOH and no normal cells were sequenced). Fully homozygous segments appeared as outliers, characterized by their very low ratio. We defined a cutoff as the 90th quantile of ratios × 0.1. Segments falling under this cutoff were labeled LOH for all cells in the sample. Leveraging the fact that reference-backed phasing is correct to a high probability within short segments [17], A and B alleles were summarized per 50 kb genomic blocks per clone. To assign a major/minor allele per segment, we followed three rules. First, if the CN was 1, we set the alleles to major. Second, if the CN was >1 and significantly skewed in abs(BAF-0.5), indicating likely cnLOH, we filtered for minimum SNPs/block and minimum SNPs/segment (tunable parameter), and then set the major allele. Third, if the CN was >2, then BAF > 0.5 after filtering determined the major allele. Allele-specific copy numbers were not called in low confidence segments, identified as low genotype probability, <2 SNPs per block or <20 SNPs per segment. Genotype probability by copy state was modeled from the BAF as a binomial distribution, including an empirical error rate, and the scaled log likelihood of the most likely genotype was required to be >0.97 for a reliable call (Supplementary Fig. S3). Medicc2 [34] was run with default settings, indicating a diploid clone, on the allele specific copy number matrix.
Downsampling and different resolution analysis
Downsampling was performed with the DownsampleMatrix function from the scater [35] library with by_cols = TRUE on filtered bin counts of cells from ALL40 that had >400 000 counts in good bins and were confirmed to be in G1 by expression (n = 56). For joint segmentation with mpcf, gammas were scaled according to bin number and three different gammas were tested per resolution. The gamma values where most conditions were stable with low bin counts was chosen for the main figure (Supplementary Fig. S4A). Segments from joint segmentation were filtered based on the number of bins (probes) before clustering. At resolutions 10, 40, 80, 200, and 500 kb, a minimum of 10, 5, 3, 1, and 1 bins were required, respectively. For hypothesis-free segmentation based on log-likelihood ratios all segments <10 bins at each resolution were discarded.
Preparation of reference (mpcf post refinement)
ALL40 was run through the pipeline at resolution 10 kb, with minimum bin number 10 and using only cells with at least 60 000 counts. A gamma of 10 was picked as optimal for joint segmentation, with a minimum of six cells per cluster and k-means max 12 and k-means threshold 0.4. To refine the clones, pseudobulk profiles were created per clone and run with gamma 0.5 through multipcf. Segments <10 bins (100 kb) were merged with their larger neighbor. Segments that were on chromosome edges and <40 bins were merged with their neighbor. The function split_mixed_clones was run with residual_threshold = 0.3 and improvement_threshold = 0.8, but no clones were split. High residual segments were masked with function mask_high_residual, with settings max_residual = 0.3 and clone_filter_fraction = 0.3. Remove_bad_clones was run, but no clone was removed. Bad cells were removed (remove_bad_cells) with max_diff_bins = 1000. Single-cell copy numbers were re-calculated per refined segment in the refined clones.
Simulations
Depth of two was simulated for different ploidy states at different noise levels by sampling genomic ranges without replacement and calculating the coverage at two. This was done for ploidies one through four, with artificial noise level added. We added noise by introducing duplicated genomic ranges at different percentages, 5, 10, 15, and 20 shown in Supplementary Fig. S5.
WGS
ALL40 was sequenced with paired-end sequencing (150 bp) and analyzed with the BALSAMIC workflow v8.2.10 in tumor-normal mode, and breakpoints analyzed in this paper were detected with AscatNGS within the BALSAMIC workflow. ASCAT output is available in Supplementary file S2.
Benchmarking
ASCENT, Alleloscope [18], CHISEL [17], and scAbsolute [19] were run on ALL40 in order to benchmark the accuracy against ASCAT output from WGS analysis. ASCENT was run as previously described. Alleloscope was run using heterozygous SNPs from ASCENT. Filtering settings in the function Matrix_filter were cell_filter = 1000 and SNP_filter = 3, in the function Segments_filter nSNP = 100, and in the function Est_regions max_SNP = 30 000. Normal cells sequenced on the same sequencer were used as a reference during segmentation. CHISEL was run using phased SNPs by ASCENT at 5 Mb resolution in –no-normal mode. scAbsolute was run at 1 Mb resolution in order to reach their recommended threshold of reads per bin. The scAbsolute source code had several hard-coded genome parameters that tied it exclusively on the outdated hg19 genome build. Minor modifications were therefore made to allow us to obtain comparable results from the GRCh38 build (available from the authors by request). Only bins included in the output from all methods were used for accuracy calculations.
Effect of purity and subclone size on accuracy
To estimate ASCENTs accuracy at different levels of tumor purity, we compared tumor cells from ALL40 mixed with normal cells at different ratios, with ASCENTs result from the full ALL40 data set as the ground truth. To estimate ASCENT’s accuracy with different subclone size, we downsampled the clones to 3, 5, and 8 cells, and calculated accuracy with the full ALL40 data set as the ground truth.
External samples
Two external samples were processed, a primary ductal carcinoma in situ (DCIS) and an untreated invasive ductal carcinoma (IDC).
FASTQ files from a DCIS; P4P, processed by ARC-well [5], were downloaded from SRA (Biosample SAMN30027787). Sample was run through the ASCENT pipeline with slight modifications. As very few cells had enough reads to calculate log-odds ratios to adjust copy numbers, cells were limited to a scale factor between 2 and 4.5, as the ploidy of this sample is estimated to be around 2.8 in the original publication. Duplicate rate threshold was increased to 75%, and no normal cell normalization was performed during any step, as we didn’t have method/sequencer-matched normal cells. Clone refinement was performed with a gamma = 5, mask_high_residuals was run with max_residuals 0.35, and split_mixed_clones was not used. The max_diff_bins parameter was increased to 50 000 bins.
FASTQ files from an IDC; TN2, processed by ACT [4], were downloaded from SRA (Biosample SAMN18202193). Sample was run through the ASCENT pipeline with slight modifications to allow for single-end reads. No normal cell normalization was performed, as with P4P, and clone refinement was performed with gamma = 1; other settings were identical as for P4P.
Reagents
| Reagent | Company | Category number |
|---|---|---|
| RPMI medium 1640 (1X) | Gibco | 21875-034 |
| Fetal bovine serum (FBS) | Gibco | A5256801 |
| Penicillin/streptomycin | Gibco | 15140122 |
| Hoechst | Thermo Fisher | H21486 |
| 1X PBS | Gibco | 14190-94 |
| 40 μM cell strainer | Corning | CLS352350 |
| CD34 | Sony Biotechnology | 2318045 |
| CD10 | Sony Biotechnology | 2161035 |
| CD20 | Sony Biotechnology | 2111525 |
| CD45 | Sony Biotechnology | 2442565 |
| CD38 | Sony Biotechnology | 2117545 |
| CD19 | Sony Biotechnology | 2111075 |
| Lymphoprep | Stemcell | 18061 |
| Dead cell removal | Miltenyi | 130-090-101 |
| Proteinase K | Thermo Fisher | EO0491 |
| Tween-20 | Merck | p-7949 |
| psfTn5 | Addgene | 79107 |
| 10% SDS | Merck | 71736-100ML |
| MaximaH reverse transcriptase | Thermo Fisher | EP0753 |
| Magnesium chloride solution for molecular biology (1.00 M) | Merck | M1028 |
| Triton X-100 | Merck | 93426 |
| dNTP mix | Thermo Fisher | R0192 |
| Betaine 5 M | Merck | B0300 |
| ERCC RNA Spike-In Mix | Thermo Fisher | 4456740 |
| USB Dithiothreitol (DTT, 0.1 M) | Thermo Fisher | 707265ML |
| RNase inhibitor | Takara | 2313 A |
| Armadillo 384 well plates | Thermo Fisher | AB2384 |
| Speedbead Mag Carboxyl Modified particle | Merck | GE65152105050250 |
| PEG 8000, 250 g | Merck | 89510-250G-F |
| UltraPure DNase/RNase-Free Distilled Water | Thermo Fisher | 10977035 |
| Sodium chloride solution, 5M | Invitrogen | 71386-1L |
| Qubit DNA HS | Thermo Fisher | Q33231 |
| Lambda exonuclease | BioNordika | M0262L |
| UltraPure Tris–HCl, 1 M, pH 8 | Invitrogen | 15568-025 |
| 0.5M EDTA, pH 8 | Sigma–Aldrich | 15575-038 |
| TAPS 0.2M buffer solution, pH 8.5 | Thermo Fisher | J63268.AE |
| KAPA HiFi HotStart ReadyMix | Roche | 7958935001 |
| Lambda exonuclease | BioNordika | M0262L |
| KAPA HiFi PCR Kit with dNTPs | Roche | 7958846001 |
| Buffer EB | Qiagen | 19086 |
Biological resources
HCT116 cell line was obtained from ATCC, category number CCL-247.
Database
Encode unified exclusion list for Grch38 was obtained from https://www.encodeproject.org/files/ENCFF356LFX/
Genome references for Grch38, d1, and vd1 were obtained from https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files
Genome references for T2T-CHM13v2.0 were obtained from
https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_009914755.1/
Software versions
Software is version controlled within conda environments of ASCENT, and available in git repository.
Trim Galore 0.6.10
BWA 0.7.17
Samtools 1.21
Picard 2.22.0
Bedtools 2.31.1
Cellsnp-lite 1.2.3
htslib 1.21
Eagle v2.4.1
Medicc2 1.0.2
Bcftools 1.21
R 4.4.2
Python 3.12.9
Dbscan 1.2.2
Uwot 0.2.3
Copynumber 1.38.0
Scater 1.34.1
GenomicRanges 1.58
Results
Development of an end-to-end method for tagmentation-based single-cell WGS
We set out to establish a pipeline that would result in high resolution CNV detection, allele specific, absolute copy number calling, and accurate clone assignment. Ultra-low coverage scWGS (0.01×–0.1×) enables CNV detection in single cells, and is critically dependent on accurate initial detection of breakpoints using a segmentation algorithm. We benchmarked two segmentation algorithms, joint and hypothesis-free (Fig. 1A). During joint segmentation (implemented in multipcf (mpcf) from the copynumber package [15]), information is shared across cells, greatly enhancing performance for data sets where many cells share the same breakpoints, which is the case for most cancers. Some unconventional patient samples, and samples originating from experiments where double-stranded breaks have been induced at random will not have common breakpoints. Here, hypothesis-free segmentation, an algorithm that considers each cell independently (see “Materials and methods” section), might be a better alternative to identify segments, and we provide a fast C++/R implementation of this approach.
Figure 1.

End-to-end method for tagmentation-based single-cell WGS. (A) Heatmap that illustrates the two segmentation algorithms. Joint segmentation, which is superior on samples with recurrent breakpoints; alternatively, hypothesis-free segmentation for samples that do not share breakpoints. (B) CNV plots (on top) show initial (left y-axis) and adjusted (right y-axis) copy number profiles of cells 1 and 2. Beeswarm plots of log-odds ratios highlight the values for each cell, showing cell 1 is above the cutoff, and the copy number profile will therefore be adjusted. (C) Clustering of copy number profiles results in clones, which can be used to refine the CNVs. Allele-specific copy numbers can be called per segment per clone, and clonal evolution can be reconstructed.
Regardless of which approach is used to identify segments, the next step of processing scWGS is to transform the per-bin read counts into per-segment integer copy numbers. In ASCENT, this is achieved by employing an ML approach to identify the per-cell scale factor that achieves the best internal consistency between bin counts and copy states. However, certain errors are not observable using this process. In particular, doubling or halving all copy states will result in indistinguishable profiles. Therefore, we introduce a correction step where a statistic based on standardized overlap is used to obtain absolute copy numbers (Fig. 1B).
If the sample has subclonal structure, the pipeline concludes with phylogenetic reconstruction. Several methods have been developed to enable lineage tracing from scWGS data [36, 37]. However, accurate CNV-based reconstruction requires knowledge of allele-resolved segments to resolve common driver events in tumor evolution, such as whole genome doubling (WGD) or copy-neutral LOH [38, 39]. ASCENT uses a new approach to haplotype segments on a per-clone basis, which increases sensitivity and minimizes errors at ultra-low coverage while avoiding the previous requirement to have matched normal reference data. Phylogenetic reconstruction based on the allele-resolved profile is done using Medicc2 [34], the only WGD-aware phylogenetic tool designed for scWGS data. (Fig. 1C). A detailed step-by-step overview of ASCENT is shown in Supplementary Fig. S1.
Ultra-low coverage scWGS breakpoint detection at high resolution
To benchmark our pipeline, we performed DNTR-seq on a bone marrow sample taken at diagnosis of a pediatric acute lymphoblastic leukemia (ALL) patient, ALL40. Out of 384 cells, 324 passed initial QC (minimum 60 000 bam read pairs and <35% duplicates (Supplementary file S1). To evaluate the segmentation algorithms, we performed downsampling and tested different bin sizes. For this analysis, we used all cells with at least 400 000 counts in high-quality bins that were confirmed to be in G1 based on joint analysis of cell cycle marker genes. Fixed bin sizes ranging from 10 to 500 kb, and counts between 20 000 to 400 000 were tested. To evaluate the different conditions, we prepared a reference copy number profile, based on all high-quality cells sequenced and refined on a per-clone basis (see “Materials and methods” section). Downsampled aneuploid cells were compared to the reference (Fig. 2A) (see diploid cells in Supplementary Fig. S4B). As expected in clonal disease, the joint segmentation algorithm (mpcf) performed better than the hypothesis-free (LLR) in all conditions with read depth over 30 000. Sensitivity parameters were scaled between bin resolutions to match the difference in per-bin depth of sequencing by simple linear interpolation (see “Materials and methods” section for details). Using this approach, all different resolutions tested were stable even at very low read counts, with no loss of sensitivity down to 100 000 reads, and all resolutions except for 10 kb were stable even down to 50 000 reads per cell. A common assumption is that larger bins remove noise associated with sparse single cell data, but our analysis indicates that, given reasonable parameters, both joint and hypothesis-free segmentation result in more accurate results when using higher resolution bins than low resolution. We conclude that even at low read counts, it is not beneficial to perform joint segmentation at low resolution, and that low read counts are sufficient for segment detection.
Figure 2.

High-resolution segmentation. (A) Downsampled bin counts at different resolutions, segmented with either hypothesis free segmentation (LLR) or joint segmentation (mpcf). Y-axis is the number of bp different from reference. Each point represents a single leukemic cell from ALL40, n = 47. (B) Comparison of CNV calls in bulk WGS, LLR at 80 kb resolution and 400 000 subsampled reads, mpcf at 500 kb resolution and 400 000 subsampled read, and the mpcf post refinement (pr) (10 kb resolution, refined breakpoints on clone level). Each panel shows GC-corrected 10 kb bin counts from clone 4 from ALL40 (n = 169 cells). A solid line and the color of dots indicates which copy number was called in each condition. Vertical lines indicate breakpoints determined by bulk WGS. Sizes of segments are indicated underneath. Region plotted is Chr12:0.2–160 Mb. (C) Same as panel (B), but for Chr22:17–28 Mb. (D) Single-cell copy number heatmap of ALL40, prepared with DNTR-seq on non-fixed cells, showing mpcf segments post refinement. Cells are organized into clones, indicated by color, and copy number legend indicates copy state. Ploidy indicates which cell’s copy number had to be adjusted to achieve the correct absolute copy number. Cells at the bottom of the plot passed sequencing QC but were not assigned to a clone. (E) Detail of a subset of CNV features that distinguish the minority clone (2). Vertical lines show segments detected with mpcf post refinement. Dots show GC-corrected 10 kb bin counts. Color of dots and horizontal line indicate which copy number was called. (F) Benchmarking of ASCENT, scAbsolute, Alleloscope, and CHISEL based on ALL40. Comparison is done between cells in clone 4 (major clone) and ground truth (WGS). Total copy number and allele-specific copy number are compared for methods, if applicable. Y-axis denotes F1 score (total bins—correctly called bins)/total bins. (G) Single-cell copy number heatmap of Tn2, prepared with ACT on non-fixed cells, showing mpcf segments post refinement, as in panel (D). (H) Detail of a subset of CNV features from two different clones, showing segments post refinement, as in panel (E).
Segmentation errors can occur because of insensitive edge detection or because of false positives or negatives. At low resolution, insensitive edge detection is impossible to avoid, as breakpoints are often within the bins. Nevertheless, mpcf at 500 kb resolution still detected every segment on Chr12 identified using bulk WGS of ALL40 and the post-refinement (PR) reference from Fig. 2A. The hypothesis-free segmentation performed worse and showed both insensitive edge detection as well as failure to find a 3 Mb segment (Fig. 2B). While joint segmentation generally achieves high sensitivity, it has difficulties finding segments below 500 kb in single cells. ASCENT uses a subsequent refinement step, where cells are classified into clones and re-analyzed as pseudobulk. In this analysis, smaller segments are readily identified; e.g. we could readily identify a 140 kb segment on Chr22, verified by bulk WGS (Fig. 2C and Supplementary Table S1). Of the segments identified by bulk WGS that exceeded our detection threshold (100 kb), we successfully detected 11 out of 13, only failing to detect one pericentromeric event, where we do not have good coverage, and one event with borderline significance in the bulk WGS analysis, which is in a TCR region. Additionally, we identified four segments that were not observed in the bulk WGS data (Supplementary Table S1). Leveraging joint segmentation followed by density-based clustering, we found four genetic subclones, in addition to diploid cells (Fig. 2D). The smallest clone was only eight cells, which was identified by a deletion on chromosome 11, q arm, and an amplification on chromosome 16, p arm. When analyzed on the pseudobulk level, we additionally identify small deletions on chromosomes 1 and 4 (Figs 2E). None of the breakpoints found exclusively in this minority clone could be found with bulk WGS, which is expected, as it consists of 3% of the sampled tumor cells. Recently, a reference based on long-read sequencing of a haploid human genome was assembled (the telomere-to-telomere [T2T] reference) [27], which includes corrected reference sequences of previously poorly mapped regions such as fragile areas around centromeres. To test whether this reference could be used to enhance CNV detection, we aligned ALL40 against the T2T genome. While this did improve coverage in, e.g. the p arm of Chr22 (Supplementary Fig. S6A and B), the data were too sparse to derive additional results and introduced new artifacts. This is likely due to short-read sequences failing to map uniquely in the improved regions.
Benchmarking ASCENT against scWGS analysis pipelines
In order to evaluate the performance of ASCENT against other methods specifically developed for scWGS, we analyzed ALL40 with ASCENT, scAbsolute [19], Alleloscope [18], and CHISEL [17]. To determine accuracy, we compared all single cells from the major clone against bulk WGS. Accuracy of allele-specific copy numbers was calculated for methods that support it (Fig. 2F). Importantly, CHISEL depends on SNP analysis for segmentation, and ultra-low coverage sequencing at this cell number does not provide enough heterozygous SNPs for CHISEL to run well, resulting in almost universally erroneous copy numbers, and a median accuracy below 0.01. Alleloscope segments on pseudobulk level with HMM-copy [13], based on counts only, and requires fewer SNPs than CHISEL for haplotyping. However, the authors recommend at least 1 million SNPs, which is five times more than this sample. This results in a median accuracy of 0.85 for allele-specific CN, and 0.94 for total CN. scAbsolute uses HMM-copy on single cell level for segmentation, which results in largely accurate segmentation with a median F1 score of 0.996, compared to ASCENTs 0.999. scAbsolute does not perform any haplotyping.
Accuracy of ASCENT in low tumor purity and in small cell numbers
In some cases, it is not possible to enrich tumor cells, and tumor purity of certain samples can therefore be very low. To determine the performance of ASCENT in this scenario, we simulated low tumor purity samples by analyzing mixes of known tumor purity ranging between 10% and 50%. Accuracy remained high at all tested purities (Supplementary Fig. S7A). Minority clones, such as clone two in ALL40, can be biologically meaningful, and we therefore tested the number of cells necessary for high accuracy of clonal detection and segmentation. We downsampled the clones of ALL40 and saw that while the accuracy is still high with only three cells, there is a considerable drop from five to three, and we therefore use five cells as a minimum clone size (Supplementary Fig. S7B).
ASCENTs performance on highly complex samples of different origin
Sample quality and subclonal complexity can severely affect the performance of scWGS analysis methods. To test the generalizability of ASCENT, we analyzed two breast cancer samples: “Tn2” prepared with ACT [4] sequenced with single-end reads (Fig. 2G) and “P4P,” originating from Formalin-Fixed Paraffin-Embedded (FFPE) tissue, prepared with ARC-well [5] (Supplementary Fig. S7C). The processing of Tn2 results in visually similar clones as found in the original publication [4], and by segmenting pseudobulk at 10 kb resolution, we identify multiple small subclonal CNVs (Fig. 2G and H), underlining the benefits of processing data at high resolution. Analysis of P4P reveals the same CNV patterns as the original publication [5], e.g. a subclonal deletion on Chr 13 and an amplification of Chr 19 q arm, but our fine grained analysis identifies more clones (21 versus 9) (Supplementary Fig. S7C). Together, these results highlight ASCENT’s ability to run on single-end and paired-end data, from highly complex tumors, different wet-lab protocols, and on lower quality DNA originating from FFPE samples.
Absolute copy number calling of segments in single cells
In principle, the copy number of a segment can be estimated from data either based on the relative density of reads in the segment compared to others, or by directly using allelic frequencies of SNVs measured at high depth of coverage. Using SNVs is impractical in single-cell studies because of the high coverage required, but has been used for small datasets sequenced deeply using a random priming method [40]. The direct tagmentation approach is ideally suited for read density estimation of copy number calling due to its even coverage. We solve the actual (integer) copy numbers of all segments in a cell jointly using a ML estimation, by determining the scale factor that minimizes residuals between real-valued and integer copy numbers (see “Materials and methods” section and Supplementary Fig. S2B). However, there are common cases where the ML approach on its own will fail. First, the solution represents the minimum scale factor consistent with the data—perfect multiples would be as likely in principle but will usually not be selected due to scaling of residuals. This error is common in highly proliferative cells, where S/G2/M cells are abundant. Second, due to noise and imperfections in the data, the scale factor might be called at twice or half the correct ploidy even when it is not a perfect multiple. For example WGD in cancer is often quickly followed by loss of small segments of the genome. Given a cell with less than perfect data, the signal from a small region might not be sufficient, and the ML estimation might result in half the correct scale factor. To ensure the accuracy of our integer copy number calls, we therefore need to use an orthogonal property of the data.
In ASCENT, we use the fact that, in contrast to random priming, the direct tagmentation approach is destructive to the original DNA template, precluding overlapping copies from the same template. In a haploid genomic region, any observed overlap is artifactual, while they are possible in a diploid region and get relatively more common at higher ploidy levels in a predictable manner (see “Materials and methods” section for details). The degree of overlap between fragments can therefore be used as a measure of the number of DNA templates (Fig. 3A). By analyzing segments from HCT116 cells in G1 of known ploidy we could compare measured, simulated and calculated standardized overlaps (Fig. 3B). As expected, the three copy states of HCT116 cells had separate distributions. HCT116 cells have a total length of segments of around 140 and 150 million bp for copy state 1 and 3 respectively, and 2.2 billion bp copy state 2. Simulations in Fig. 3B were performed with the corresponding genome sizes to correctly simulate the spread of values. In the calculated and simulated standardized overlap scores, noise is included to represent the effect of amplification and sequencing artifacts. The noise level can be estimated from data if the base ploidy is known; however, we found that results were robust across a wide range of noise values, and a 10% noise level was used for all calculations. To show the robustness of our calculation, we simulated the overlaps at different genome sizes, with different noise levels (Supplementary Fig. S5). Based on these simulations, we concluded that an effective genome size of 10 million is enough for an accurate estimation of standardized overlap statistic, and in our calculations we therefore required each ploidy state used for the calculation to exist in at least one hundred 10 kb bins to be used in a cell. As expected, the standardized overlap did not correlate with the sequencing depth, duplication rate, or insert size (Fig. 3C). However, sequencing chemistry had an effect, since we observed a statistically significant increase in overlap at copy state one, representing purely artifactual overlaps, in a library sequenced using the newer Illumina chemistry using patterned flow cells. This finding is consistent with reports that duplicate errors arise due to in-flow cell mispriming (index hopping) on sequencers using this chemistry [41] (Fig. 3D).
Figure 3.

Absolute copy number calling. (A) Scaled integer copy numbers for a single HCT116 cell. Templates with possible overlaps are shown for each copy state. (B) Standardized overlaps (SOs) for three different copy states. The calculated theoretical overlap and simulated data has 10% noise level; and measured values are from HCT116 cells in G1. Simulation is performed from as many base pairs as each respective ploidy of an HCT116 cell contained. (C) SO at 2× coverage for copy states of G1 HCT116 cells plotted against QC measures: number of bam read pairs, duplicate fraction per bam file, and the average insert size. Pearson correlation is shown as a line with an R2 value in each plot. (D) G1 HCT116 cell library was sequenced on two different types of sequencers, one with a patterned flow cell (NovaSeq 6000) and another with a non-patterned flow cell (NextSeq 550). P-value was calculated with a paired t-test. (E) Flow cytometry plot, indicating cell phase classification by DNA content (Hoechst staining intensity) on cycling HCT116 cells. (F) Log-odds ratios of doubled/initial or halved/initial copy number of HCT116 cells sorted by cell phase, and cells from ALL40. Red horizontal line indicates P < .01, which was used as a cutoff for adjusting copy number. Shaded areas indicate cells whose copy numbers will be adjusted.
To test the accuracy of our calculations on real genomic data, we analyzed HCT116 cells in different cell-cycle phases (Fig. 3E). By using our closed-form solution of expected overlap at correct copy states, we calculated log-odds ratios to select the correct ploidy. With a threshold of P < 1% (log-odds ratio of 4.6), we correctly detected copy numbers of cells that were sorted as S phase, G2M or as multiple cells and adjusted their ploidies (Fig. 3F and Supplementary Fig. S8). These calculations also enabled us to detect cycling cells from ALL40. At the same P-value cutoff of 1%, six cells were corrected. For all cells where messenger RNA (mRNA) libraries were available (5 out of 6), the cell cycle markers confirmed that the cell was in G2M phase. We compared our absolute copy calling to a previously published method, scAbsolute [19] by applying it to our cell-cycle annotated HCT116 data set, but found that the method failed to identify the tetraploid cells (89% false-negative rate for G2M cells, Supplementary Fig. S9).
Reference-free B allele frequency in subclonal segments
There are important structural phenomena in cancer that can only be captured using SNV calls. In particular, LOH is a critical step in eliminating a non-mutated allele. However, due to the very low depth of coverage, single-cell WGS methods have often stayed away from allele-resolved copy numbers. Due to low coverage, it is not possible to directly use the B allele frequency (BAF) of single SNV to call copy numbers, but previous methods have used a properly haplotyped germ-line genome to calculate joint BAF across a segment for accurate allele specific copy calls [17].
In ASCENT, we developed an approach that uses only the single-cell data without the need for a non-tumor haplotyped reference from the same patient. To achieve allele-specific absolute copy numbers, we split all heterozygous SNPs identified from the single-cell data into 50 kb blocks and performed phasing followed by BAF calculation across segments per clone (Fig. 4A). Because of haplotype switching and the requirement for imbalances in order to call haplotypes, we implemented a set of logical rules in order to decide which allele is major and which minor per segment, and subsequently referred to the BAFs as corrected BAF (see “Materials and methods” section). To benchmark our method, we applied it to ALL40, where we used bulk WGS performed in parallel as validation. Using default parameters, ASCENT readily identified a small copy-neutral LOH on chromosome 12 (Fig. 4B), indicating an additional event separating two clones from the remaining cells; this event was confirmed by the paired bulk WGS (Supplementary Table S1). This event and the clonal LOH on Chr3 are highlighted in Fig. 4C. The same analysis on Tn2 revealed a massive LOH on several chromosomes (Supplementary Fig. S10), as had been described previously in this sample [34]. To obtain a final haplotype-aware phylogenetic tree, we used Medicc2 [34], which is ideally suited to this task because it estimates evolutionary distance based on minimal number of copy number events in an allele-aware fashion. Medicc2 is integrated into ASCENT and was used with the final allele specific copy numbers as an input (Fig. 4D).
Figure 4.

Haplotype-aware phylogeny with ASCENT. (A) Mirrored BAF of all filtered segments across clones. (B) Allele specific absolute copy number heatmap of all segments across all clones of ALL40 (prepared with DNTR-seq on non-fixed cells). Capital-lettered copy numbers indicate that allele-specific CN has been achieved, lowercase indicate segments where allele specific CN was not assigned confidently. Vertical solid lines mark chromosome boundaries, vertical dashed lines mark centromeres. Candidate driver genes in CNVs are labeled above. (C) BAF in clone 5 on Chr3, which has a deletion, and Chr12, which has an amplification, copy-neutral LOH, and a deletion. The copy numbers for clone 5 are shown below. (D) Haplotype-aware phylogeny of genetic clones of ALL40. Colors indicate clones, as in panels (A) and (B). Size of the leaf indicates number of cells in the respective clone.
Thus, by carefully designing every aspect of ASCENT to the particular strengths and weaknesses of single-cell WGS data, we can obtain accurate single-cell CNV profiles with absolute integer copy number and correct per-segment haplotypes, which we used to track the genetic history of a leukemia based on a single patient sample.
Discussion
In this work, we developed ASCENT, an absolute copy number-aware CNV caller for ultra-low depth of sequencing. ASCENT advances the state of the art in several ways: by increasing the resolution of the segmentation, by introducing true reference-free absolute copy number correction, and by implementing methods for haplotyping at ultra-low sequencing depth without matched normal cells.
Identifying correct segments is a critical first step in the analysis. We performed up-front segmentation with high-resolution bins, which we show greatly improves the accuracy of breakpoint detection. Previous methods have taken a conservative position by using large (low resolution) bins, based on the expectation that using high resolution bins would introduce more noise than low resolution bins. However, both our hypothesis-free and the joint segmentation performed better at high resolution (40k and 10k respectively) than at 200k or 500k resolutions commonly used in the field, given that sensitivity parameters were altered to match the average number of read counts per bin.
We developed a method to achieve absolute copy numbers in ultra-low coverage scWGS data, which has been obtained without preamplification. In cancer genomics, it is crucial to be able to determine absolute copy numbers, particularly for distinguishing tumor cells that have undergone WGD from those that have not. Traditional methods leveraging BAFs and read depth are effective in detecting unbalanced chromosomal alterations but may fail to identify balanced genome duplications, which lack allelic imbalance. In cases of balanced duplications, alternative approaches are necessary. Experimental techniques like Hoechst staining can estimate DNA content but may lack precision, as the canonical G1 peak additionally contains cells that are in early S phase [42]. Similarly, assessments based on nuclear size may not reliably reflect ploidy changes. However, an inherent effect of direct tagmentation is that fractional overlap can be used to infer ploidy. We and others had previously shown different methods to utilize this in ploidy estimation [1, 19], but the previously described methods were limited by the fact that they attempted to calculate an average ploidy, where all bins are considered equally, despite being used on cancer samples, which often have complicated CNVs. The scAbsolute [19] method is further limited by the fact that it requires at least 300 000 read pairs as input and that the assessment is based on visual inspection of distributions rather than statistical assessment. Furthermore, even at high depth of sequencing we were unable to obtain any clear separation of ploidy scores between 4n and 2n clones (Supplementary Fig. S9), suggesting an issue with generalizability. We have improved on previous methods by utilizing precise segmentation and initial copy number calling in order to calculate overlap fraction per copy number, and we have introduced statistical modeling based on a closed-form solution of the expected degree of overlap, which directly tests the ploidy of individual cells.
Additionally, we showed that by careful quality control and classification into clones, with subsequent breakpoint refinement on clonal level, ASCENT can accurately detect small segments close to the theoretical limit of 100 kb (based on segment size cutoff), as exemplified in our patient data.
Finally, we demonstrate that even at ultra-low coverage, ASCENT achieves allele-specific copy numbers for most segments. Previous methods require either a high-quality normal reference for pre-phasing SNPs or a much higher depth of sequencing. Consequently, there are tradeoffs to our method—given that we do not use allele-specific data as the input to segmentation, we will fail to detect copy-neutral LOH at sub-chromosome arm level if all neighboring segments are at an equal copy state. Also, since ASCENT relies on short-read sequencing, structural rearrangements such as translocations will often not be fully resolved—we will usually not detect the exact base-pair resolution breakpoint and the two sequences involved. A future improvement could include long-read sequencing to help to improve this. Performing shallow long-read sequencing in a subset of cells would also make it possible to do non-reference-based or hybrid phasing, which in turn should enhance allele-specific copy number calls. An additional limitation is that ASCENT is built specifically for direct tagmentation-based scWGS and should not be used for random-priming-based data. In particular, the theoretical underpinnings of the absolute copy number calling requires that the original template is split in non-overlapping fragments.
We benchmarked ASCENT, scAbsolute [19], Alleloscope [18], and CHISEL [17] on the ALL40 dataset, using WGS as the ground truth. Among these methods, ASCENT demonstrated the highest accuracy. CHISEL, by design, is not optimized for data with such low coverage, as it requires a minimum SNP density to perform effectively. Running CHISEL in this setting would therefore necessitate a matched bulk sample. This reliance on a high number of SNPs stems from CHISEL’s segmentation step, which incorporates both SNPs and raw counts. While this imposes certain limitations, it also represents one of CHISEL’s key strengths: enabling the detection of sub-chromosomal arm cnLOH events independently of neighboring CNVs. Alleloscope requires fewer SNPs than CHISEL; however, it still exceeds what is common practice in current high-throughput scWGS and our dataset provides. One notable limitation of Alleloscope, contributing to its suboptimal total copy number calling on the ALL40 dataset, is its reliance on pseudobulk segmentation. This approach restricts its ability to detect breakpoints associated with minority clones. However, unlike ASCENT and CHISEL, Alleloscope does not require external phasing, which can be an advantage in certain settings.The final method we evaluated, scAbsolute, estimates only the total copy number. Its recommended read depth per bin necessitates the use of large bin sizes when applied to ultra-low coverage data, which in turn reduces its segmentation resolution and overall accuracy relative to ASCENT.
ASCENT was designed to perform well on samples with different clonal complexity and library preparation protocols and qualities. Here, we demonstrate accurate analysis of a pediatric leukemia with unbalanced subclonal fractions prepared using DNTR-seq, and a clonally complex breast cancer prepared using ACT, which was sequenced using single-end sequencing. We also analyze an archival FFPE sample prepared using ARC-well, with highly damaged and degraded DNA. The formalin fixed sample produced the expected clonal structure, but due to an overwhelming number of artifactual mutation allele specific copy numbers could not be reliably called (data not shown). Development of better methods for detecting artifactual mutations might improve ASCENTs performance on such samples [43, 44].
In conclusion, ASCENT is a pipeline specifically developed for ultra-low coverage direct-tagmentation scWGS data. ASCENT runs on scWGS data alone, or combined with scRNA-seq data, as in the case of DNTR-seq experiments. ASCENT’s input is demultiplexed FASTQ files and the outputs are absolute allele specific copy numbers, refined subclones and haplotype aware phylogeny. ASCENT scales to very large data sets, but also enables complete analysis of small datasets down to a few hundred cells, enabling non specialized labs to establish subclonal structure and accurate segmentation based on scWGS using as little as a single 384-well plate.
Supplementary Material
Acknowledgements
Part of this work was facilitated by the Protein Science Facility at Karolinska Institutet, Stockholm, and BEA, the Bioinformatics and Expression Analysis core facility, which is supported by the board of research at the Karolinska Institutet. We acknowledge CSC – IT Center for Science, Finland, and UEF Bioinformatics Center, University of Eastern Finland, Finland, and Finnish Institute for Molecular Medicine Genomics core facilities for computation, Henrikki Almusa, Roger Kramer, and Sanni Moisio for running bioinformatics workflows. We want to thank the families participating, Laura Oksa for coordinating, and clinicians and research personnel participating in the pediatric leukemia sample collection.
Author contributions: Solrun Kolbeinsdottir (Conceptualization [equal], Data curation [equal], Formal analysis [equal], Investigation [equal], Methodology [equal], Software [equal], Validation [equal], Visualization [equal], Writing—original draft [equal], Writing—review & editing [equal]), Vasilios Zachariadis (Conceptualization [equal], Data curation [equal], Formal analysis [supporting], Investigation [supporting], Methodology [equal], Software [equal], Visualization [supporting], Writing—original draft [supporting], Writing—review & editing [supporting]), Christian Sommerauer (Data curation [supporting], Investigation [supporting], Writing—review & editing [supporting]), Olli Lohi (Funding acquisition [supporting], Resources [supporting]), Merja Heinäniemi (Data curation [supporting], Funding acquisition [supporting], Resources [supporting], Writing—review & editing [supporting]), and Martin Enge (Conceptualization [equal], Data curation [supporting], Formal analysis [equal], Funding acquisition [equal], Methodology [equal], Project administration [equal], Resources [lead], Software [equal], Supervision [lead], Visualization [supporting], Writing—original draft [equal], Writing—review & editing [equal]).
Contributor Information
Solrun Kolbeinsdottir, Department of Oncology-Pathology Karolinska Institutet, Stockholm 171 77, Sweden.
Vasilios Zachariadis, Department of Oncology-Pathology Karolinska Institutet, Stockholm 171 77, Sweden.
Christian Sommerauer, Department of Oncology-Pathology Karolinska Institutet, Stockholm 171 77, Sweden.
Olli Lohi, Tampere Center for Child, Adolescent, and Maternal Health Research Faculty of Medicine and Health Technology, Tampere University, and Tays Cancer Centre Tampere University Hospital, Tampere 33520, Finland.
Merja Heinäniemi, School of Medicine, University of Eastern Finland, Kuopio 70211, Finland.
Martin Enge, Department of Oncology-Pathology Karolinska Institutet, Stockholm 171 77, Sweden.
Supplementary data
Supplementary data is available at NAR online.
Conflict of interest
None declared.
Funding
This work was supported by the Swedish Research Council (2023-02912), the Swedish Cancer Society (232839 Pj), Swedish Childhood Cancer Foundation (PR2024-0108 and MT2023-0014), Research Council of Finland (341693, 321553, 341540), and the Sigrid Juselius Foundation M.E. is supported by a career grant from the Swedish Cancer Society (210391 SIA). S.K. is supported by a PhD grant from Karolinska Institutet (2018-00963). Funding to pay the Open Access publication charges for this article was provided by Swedish Research Council (2023-02912).
Data availability
Bulk WGS raw data is available under EGAC00001003137 (sample identifier ALLT-340) on the European Genome-Phenome Archive database. scWGS raw files for ALL40 are available under EGAD50000001774 (study EGAS50000001247) on the European Genome-Phenome Archive database. scWGS raw files for HCT116 cells available on SRA under PRJNA1263634.
Processed data from DNTRseq experiments (bin counts) for HCT116 and ALL40 cells are available on GitHub (https://github.com/EngeLab/ASCENT) and https://doi.org/10.5281/zenodo.15466139 as well as code to run the pipeline and other code associated with this manuscript.
References
- 1. Zachariadis V, Cheng H, Andrews N et al. A highly scalable method for joint whole-genome sequencing and gene-expression profiling of single cells. Mol Cell. 2020; 80:541–53. 10.1016/j.molcel.2020.09.025. [DOI] [PubMed] [Google Scholar]
- 2. Zahn H, Steif A, Laks E et al. Scalable whole-genome single-cell library preparation without preamplification. Nat Methods. 2017; 14:167–73. 10.1038/nmeth.4140. [DOI] [PubMed] [Google Scholar]
- 3. Laks E, McPherson A, Zahn H et al. Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencing. Cell. 2019; 179:1207–21. 10.1016/j.cell.2019.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Minussi DC, Nicholson MD, Ye H et al. Breast tumors maintain a reservoir of subclonal diversity during expansion. Nature. 2021; 592:302–8. 10.1038/s41586-021-03357-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang K, Kumar T, Wang J et al. Archival single-cell genomics reveals persistent subclones during DCIS progression. Cell. 2023; 186:3968–82. 10.1016/j.cell.2023.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu L, Wang X, Mu Q et al. scONE-seq: a single-cell multi-omics method enables simultaneous dissection of phenotype and genotype heterogeneity from frozen tumors. Sci Adv. 2023; 9:eabp8901. 10.1126/sciadv.abp8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Theunis K, Vanuytven S, Claes I et al. Single-cell genome and transcriptome sequencing without upfront whole-genome amplification reveals cell state plasticity of melanoma subclones. Nucleic Acids Res. 2025; 53:gkaf173. 10.1093/nar/gkaf173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Williams MJ, Oliphant MUJ, Au V et al. Luminal breast epithelial cells of BRCA1 or BRCA2 mutation carriers and noncarriers harbor common breast cancer copy number alterations. Nat Genet. 2024; 56:2753–62. 10.1038/s41588-024-01988-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olsen TK, Otte J, Mei S et al. Joint single-cell genetic and transcriptomic analysis reveal pre-malignant SCP-like subclones in human neuroblastoma. Mol Cancer. 2024; 23:180. 10.1186/s12943-024-02091-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao R, Davis A, McDonald TO et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet. 2016; 48:1119–30. 10.1038/ng.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leppä AM, Grimes K, Jeong H et al. Single-cell multiomics analysis reveals dynamic clonal evolution and targetable phenotypes in acute myeloid leukemia with complex karyotype. Nat Genet. 2024; 56:2790–803. 10.1038/s41588-024-01999-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ding L, Ley TJ, Larson DE et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012; 481:506–10. 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lai D, Ha G, Shah S. HMMcopy: Copy number prediction with correction for GC and mappability bias for HTS data. R Package Version 1.50.0. 2025; 10.18129/B9.bioc.HMMcopy. [DOI] [Google Scholar]
- 14. Olshen AB, Venkatraman ES, Lucito R et al. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004; 5:557–72. 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 15. Nilsen G, Liestøl K, Van Loo P et al. Copynumber: efficient algorithms for single- and multi-track copy number segmentation. BMC Genomics. 2012; 13:591. 10.1186/1471-2164-13-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McPherson A, Vázquez-García I, Myers MA et al. Ongoing genome doubling shapes evolvability and immunity in ovarian cancer. Nature. 2025; 644:1078–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zaccaria S, Raphael BJ Characterizing allele- and haplotype-specific copy numbers in single cells with CHISEL. Nat Biotechnol. 2021; 39:207–14. 10.1038/s41587-020-0661-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu CY, Lau BT, Kim HS et al. Integrative single-cell analysis of allele-specific copy number alterations and chromatin accessibility in cancer. Nat Biotechnol. 2021; 39:1259–69. 10.1038/s41587-021-00911-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schneider MP, Cullen AE, Pangonyte J et al. scAbsolute: measuring single-cell ploidy and replication status. Genome Biol. 2024; 25:62. 10.1186/s13059-024-03204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leung K, Klaus A, Lin BK et al. Robust high-performance nanoliter-volume single-cell multiple displacement amplification on planar substrates. Proc Natl Acad Sci USA. 2016; 113:8484–9. 10.1073/pnas.1520964113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen C, Xing D, Tan L et al. Single-cell whole-genome analyses by linear amplification via transposon insertion (LIANTI). Science. 2017; 356:189–94. 10.1126/science.aak9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gonzalez-Pena V, Natarajan S, Xia Y et al. Accurate genomic variant detection in single cells with primary template-directed amplification. Proc Natl Acad Sci USA. 2021; 118:e2024176118. 10.1073/pnas.2024176118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H, Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–60. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, Handsaker B, Wysoker A et al. Sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–9. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Quinlan AR, Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinforma Oxf Engl. 2010; 26:841–2. 10.1093/bioinformatics/btq033. [DOI] [Google Scholar]
- 26. Amemiya HM, Kundaje A, Boyle AP The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep. 2019; 9:9354. 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nurk S, Koren S, Rhie A et al. The complete sequence of a human genome. Science. 2022; 376:44–53. 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lawrence M, Huber W, Pagès H et al. Software for Computing and Annotating Genomic Ranges. PLoS Comput Biol. 2013; 9:e1003118. 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Campello RJGB, Moulavi D, Sander J. Pei J, Tseng VS, Cao L, Motoda H, Xu G Density-based clustering based on hierarchical density estimates. Advances in Knowledge Discovery and Data Mining. 2013; Berlin, Heidelberg: Springer; 160–72. 10.1007/978-3-642-37456-2_14. [DOI] [Google Scholar]
- 30. The 1000 Genomes Project Consortium Auton A, Brooks LD, Durbin RM et al. A global reference for human genetic variation. Nature. 2015; 526:68–74. 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang X, Huang Y Cellsnp-lite: an efficient tool for genotyping single cells. Bioinformatics. 2021; 37:4569–71. 10.1093/bioinformatics/btab358. [DOI] [PubMed] [Google Scholar]
- 32. Loh PR, Danecek P, Palamara PF et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016; 48:1443–8. 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Koenig Z, Yohannes MT, Nkambule LL et al. A harmonized public resource of deeply sequenced diverse human genomes. Genome Res. 2024; 34:796–809. 10.1101/gr.278378.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaufmann TL, Petkovic M, Watkins TBK et al. MEDICC2: whole-genome doubling aware copy-number phylogenies for cancer evolution. Genome Biol. 2022; 23:241. 10.1186/s13059-022-02794-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCarthy DJ, Campbell KR, Lun ATL et al. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017; 33:1179–86. 10.1093/bioinformatics/btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang F, Wang Q, Mohanty V et al. MEDALT: single-cell copy number lineage tracing enabling gene discovery. Genome Biol. 2021; 22:70. 10.1186/s13059-021-02291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Markowska M, Cąkała T, Miasojedow B et al. CONET: copy number event tree model of evolutionary tumor history for single-cell data. Genome Biol. 2022; 23:128. 10.1186/s13059-022-02693-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bielski CM, Zehir A, Penson AV et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet. 2018; 50:1189–95. 10.1038/s41588-018-0165-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zack TI, Schumacher SE, Carter SL et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013; 45:1134–40. 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gawad C, Koh W, Quake SR Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. Proc Natl Acad Sci USA. 2014; 111:17947–52. 10.1073/pnas.1420822111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Larsson AJM, Stanley G, Sinha R et al. Computational correction of index switching in multiplexed sequencing libraries. Nat Methods. 2018; 15:305–7. 10.1038/nmeth.4666. [DOI] [PubMed] [Google Scholar]
- 42. Zhao PA, Sasaki T, Gilbert DM High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells. Genome Biol. 2020; 21:76. 10.1186/s13059-020-01983-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Satas G, Myers MA, McPherson A et al. Inferring active mutational processes in cancer using single cell sequencing and evolutionary constraints. bioRxiv27 February 2025, preprint: not peer reviewed 10.1101/2025.02.24.639589. [DOI]
- 44. Guo Q, Lakatos E, Bakir IA et al. The mutational signatures of formalin fixation on the human genome. Nat Commun. 2022; 6:4487. 10.1038/s41467-022-32041-5. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk WGS raw data is available under EGAC00001003137 (sample identifier ALLT-340) on the European Genome-Phenome Archive database. scWGS raw files for ALL40 are available under EGAD50000001774 (study EGAS50000001247) on the European Genome-Phenome Archive database. scWGS raw files for HCT116 cells available on SRA under PRJNA1263634.
Processed data from DNTRseq experiments (bin counts) for HCT116 and ALL40 cells are available on GitHub (https://github.com/EngeLab/ASCENT) and https://doi.org/10.5281/zenodo.15466139 as well as code to run the pipeline and other code associated with this manuscript.