

Summary Paragraph
Engineering and characterizing proteins can be time-consuming and cumbersome, motivating the development of generalist CRISPR-Cas enzymes1–4 to enable diverse genome editing applications. However, such enzymes have caveats such as an increased risk of off-target editing3,5,6. To enable scalable reprogramming of Cas9 enzymes, here we combined high-throughput protein engineering with machine learning (ML) to derive bespoke editors more uniquely suited to specific targets. Via structure/function-informed saturation mutagenesis and bacterial selections, we obtained nearly 1,000 engineered SpCas9 enzymes and characterized their protospacer-adjacent motif7 (PAM) requirements to train a neural network that relates amino acid sequence to PAM specificity. By utilizing the resulting PAM ML algorithm (PAMmla) to predict the PAMs of 64 million SpCas9 enzymes, we identified efficacious and specific enzymes that outperform evolution-based and engineered SpCas9 enzymes as nucleases and base editors in human cells while reducing off-targets. An in silico directed evolution method enables user-directed Cas9 enzyme design, including for allele-selective targeting of the RHO P23H allele in human cells and mice. Together, PAMmla integrates ML and protein engineering to curate a catalog of SpCas9 enzymes with distinct PAM requirements, and motivates the use of efficient and safe bespoke Cas9 enzymes instead of generalist enzymes for various applications.
Introduction
The properties of CRISPR-Cas enzymes have been extensively engineered for genome editing applications8,9. DNA-targeting Cas nucleases recognize genomic targets through the readout of protospacer adjacent motifs (PAMs) that are typically short in sequence (~2-4 nucleotides)7, which then initiates guide RNA (gRNA) pairing with the target site10–12 (Fig. 1a). Streptococcus pyogenes Cas9 (SpCas9) requires a 3’ NGG PAM12,13, restricting use of this enzyme to PAM-encoding genomic sequences. For many genome editing applications, precise positioning of the Cas enzyme is critical (e.g. for allele-specific editing, base editing, modifying regulatory elements, etc.; Extended Data Fig. 1a and Supplementary Note 1). SpCas9 variant enzymes 1–4,14,15 and other Cas orthologs16–20 capable of recognizing non-canonical PAMs have therefore been engineered, and can be categorized into two main classes: altered PAM enzymes that shift the PAM preference away from NGG, and relaxed PAM enzymes that expand editing to new PAMs while retaining activity against NGG (Extended Data Figs. 1b–d and Supplementary Note 2). Relaxation of the PAM is the most common engineering trajectory, leading to the creation of generalist enzymes1–4. PAM-relaxed enzymes are convenient because a single enzyme can be used across many applications, however, expanded access to the genome results in poorer specificity compared to wild-type (WT) SpCas93,5,6,21 and extended genome searching can result in slower cleavage kinetics22. Instead, PAM-selective enzymes should enable efficient on-target editing while minimizing off-targets, yet few have been developed. A large collection of selective enzymes that target different PAMs and collectively enable broad access to the genome would be an optimal solution for efficient, safe, and versatile genome editing (Fig. 1b).
Figure 1. Scalable characterization of hundreds of SpCas9 PAM variant enzymes.

(a) Schematic of target site recognition by an SpCas9-sgRNA complex. (b) Representation of the balance between targeting range and genome-wide specificity for engineered SpCas9 enzymes. (c) Schematic of the workflow to engineer SpCas9 enzymes via directed evolution. SpCas9 enzymes were obtained from a saturation mutagenesis library (harboring 6 amino acids with NNS codons; SpCas9(6AA)) either via bacterial positive selection (against 16 different substrates encoding NGNN PAMs) or by randomly picking unselected library members. SpCas9 enzymes were cloned into a mammalian expression plasmid, sequenced by a whole-ORF sequencing workflow, and subjected to the HT-PAMDA assay47 for comprehensive PAM characterization. (d) Heatmap representations of the PAM profiles of 634 SpCas9 enzymes obtained through the 16 selection experiments on NGNN PAMs, determined using HT-PAMDA (where the rate constant (k) on a PAM is a measure of targeting efficiency). PAM profiles were hierarchically clustered, with the 8 largest clusters highlighted and analyzed using sequence logos to display the amino acid composition of the cluster (right panel). PAM profiles for representative enzymes from each cluster are shown (left panel). HT-PAMDA datasets are the mean of n = 2 biological replicates using different target sites. (e) Fraction of PAM variant enzymes maximally active against the specific NGNN PAM that they were selected/designed against (rank = 1st) or where the PAM selected against was within the top 4 most active PAMs (rank = 2nd−4th), as determined by HT-PAMDA. Enzymes obtained from bacterial selections (left) and enzymes rationally designed based on most enriched amino acids from selections (right). (f) SpCas9 enzymes categorized by general PAM preference based on HT-PAMDA data (clustered as in panel d). Enzymes were labeled as inactive when no PAM had a k > 10−4.
Generating such a catalog of altered PAM enzymes would require overcoming certain challenges associated with strategies typically used to engineer Cas enzymes1–4,14,17–23. Predicting the functional impact of multiple simultaneous mutations remains challenging in the context of rational protein engineering, and experimental selection strategies that enable directed evolution can be laborious and time consuming, often yielding few engineered enzymes per evolution campaign (Supplementary Note 2). We therefore turned to machine learning (ML) to augment our experimental engineering strategies. Computational predictions can be used to screen larger numbers of enzymes bearing more diverse combinations of amino acid substitutions compared to experimental methods alone, increasing the probability of identifying optimal enzymes across a deeper mutational space23–31. ML has been applied to engineer various proteins including antibodies32–35, zinc fingers36–38, adeno-associated virus capsid tropism39–41, and to prioritize activity altering mutations for SaCas926 or SpCas942. We hypothesized that with sufficient training data, the relationship between SpCas9 amino acid sequence and PAM recognition should also be learnable. The ability to accurately predict the PAM of a Cas enzyme based on amino acid sequence would enable systematic exploration of a larger sequence space accessible only by simultaneously mutating multiple residues involved in PAM interaction.
Here we undertook a scalable protein engineering campaign to deeply profile the contribution of SpCas9 PI-domain amino acids to the PAM requirement. By generating experimental PAM profiles for hundreds of engineered SpCas9 enzymes, we produced training data for a PAM machine learning algorithm (PAMmla) to relate amino acid sequence to enzyme function. PAMmla can predict PAM variant enzymes with tunable activities and specificities, which we test in proof-of-concept experiments in human cells and in mice. Together, integration of ML with protein engineering enabled the development of a collection of bespoke PAM-selective enzymes, towards the optimization of highly safe and effective CRISPR genome editing technologies.
Results
Scalable characterization of SpCas9 PAM variants
To generate datasets necessary to train an ML model to link protein sequence to function (PAM specificity), we sought to engineer and characterize a sufficiently large collection of novel SpCas9 enzymes with altered PAM preferences. We generated a saturation mutagenesis library of SpCas9 variant enzymes using a structure and function-informed approach based on SpCas9 structures7 and previous engineering efforts3,14,43,44, simultaneously mutating 6 amino acid residues within the SpCas9 PAM interacting (PI) domain (the SpCas9(6AA) library) resulting in a library with theoretical complexity of up to 64 million enzymes (Fig. 1c, Extended Data Fig. 2a, and Supplementary Note 3). We selected residues D1135, S1136, G1218, E1219, R1335, and T1337 for saturation mutagenesis leading to an SpCas9(6AA) library of plasmids, and individual enzymes were named based on amino acid identities at these six positions. These six amino acids modulate specificity for the 3rd and 4th PAM bases in the context of the engineered enzymes SpCas9-VRER, SpCas9-VRQR and SpG3,14,43 (Supplementary Note 3). Expansion of our library to also mutate R1333, the residue which contacts the second guanine of the SpCas9 PAM10, largely abrogated enzyme activity (Supplementary Note 3). We therefore focused our efforts on altering the 3rd and 4th positions of the PAM.
To identify SpCas9 PAM variant enzymes with activity on non-canonical PAMs, we performed an extensive set of bacterial-based positive selection assays14,45 to select for enzymes capable of cleaving target sites bearing each of the 16 possible NGNN PAMs with a fixed G nucleotide at the second position (Extended Data Figs. 2b,c and Supplementary Note 4). Across the 16 experiments using the SpCas9(6AA) library, we obtained 634 unique SpCas9 enzymes that survived the selections (Supplementary Table 1). We observed PAM-dependent enrichment of amino acids across the enzymes depending on the 3rd or 4th nucleotide of the PAM used for the selection (Extended Data Fig. 2c).
Next, we linked amino acid sequence to PAM specificity for the 634 recovered SpCas9 enzymes. To identify the amino acid substitutions in the SpCas9 enzymes, we performed an arrayed plasmid sequencing method for rapid and inexpensive whole open reading frame (ORF) identification based on multiplex PCR46 (Supplementary Fig. 1). The PAM specificities of each of the 634 SpCas9 enzymes were then characterized using a high throughput PAM determination assay47 (HT-PAMDA) (Figs. 1c,d, Supplementary Fig. 2, and Supplementary Table 1). HT-PAMDA comprehensively measures rate constants (k) of nuclease cleavage for an enzyme across a library of substrates encoding all possible PAMs, providing kinetic data to quantify the global PAM profile of an enzyme.
HT-PAMDA experiments revealed that the bacterial selections performed using the SpCas9(6AA) library identified enzymes capable of recognizing each of the 16 NGNN PAMs (Fig. 1e and Supplementary Fig. 2). However, few enzymes were most active on the specific PAM that they were selected on (Supplementary Figs. 2 and 3), highlighting a limitation of the bacterial-based selection approach. Clustering the enzymes by their HT-PAMDA-determined PAM profiles revealed 8 main classes of PAM variant enzymes that were each associated with specific amino acid substitutions (Fig. 1d).
The two most common classes of SpCas9 enzymes represented in our dataset from the SpCas9(6AA) library selections were those with relaxed NGN PAM (similar to SpG or SpCas9-NG) or NGG PAM (similar to wild type SpCas9) but with up to four amino acid substitutions compared to previously described enzymes (Supplementary Figs. 4, 5). Most interestingly, our bacterial-based selections also revealed a variety of enzymes with preferences for non-canonical nucleotides in the 3rd and 4th positions of the PAM. In many cases we observed a nucleotide preference at the 4th nucleotide of the PAM, extending the PAM (specifying 3 bases instead of 2). A preference for an extended PAM may hold advantages for minimizing off-targets, as has been demonstrated for other previously described enzymes14. Together, these observations highlight the previously uncharacterized plasticity of the SpCas9 PI domain.
While our bacterial selection experiments led to the evolution of SpCas9 enzymes with novel PAM requirements, in general, an enzyme’s most efficiently targeted PAM did not always correlate with the PAM on which that enzyme was selected (Fig. 1e, Supplementary Figs. 2 and 3). We therefore sought to rationally design more optimal PAM-selective enzymes by testing “consensus” enzymes for each of the 16 NGNN PAMs, assessed by combining the most enriched amino acids at each of the six positions recovered from each selection (Extended Data Fig. 2c). When assessing the PAM requirements of the consensus enzymes using HT-PAMDA (Supplementary Fig. 6a), most enzymes did not efficiently target the PAM for which they were designed, with only 4 of 31 consensus enzymes exhibiting maximal efficiency against the PAM on which they were selected (Fig. 1e and Supplementary Fig. 6b). The rationally designed consensus enzymes generally had weaker efficiencies than the enzymes derived from bacterial selections (Fig. 1e and Supplementary Fig. 6a). These results indicate that mutations at these 6 positions do not contribute to PAM preference independently, but rather interact with one another in an epistatic manner; selections and rational design alone are thus insufficient to systematically obtain enzymes with desired PAMs.
To approximate the fraction of the 64 million enzymes encoded with our SpCas9(6AA) library that retained editing activity on any PAM, we performed HT-PAMDA on 135 randomly chosen library members. Unexpectedly, we found that ~18% of the enzymes in the SpCas9(6AA) library were capable of editing on at least one PAM, many of them with non-canonical PAM preferences (Fig. 1f). This observation suggested that amongst the millions of possible combinations of residues in our 6-position library, more than 10 million enzymes may be functional, the vast majority of which remain uncharacterized.
Learning PAM preference from AA sequence
Since we could experimentally characterize only a small number of enzymes from the SpCas9(6AA) library (~0.001%), and our data suggests that a substantial fraction of the enzymes may be active on at least one PAM (>15%; Fig. 1f), we envisioned that many additional enzymes with useful PAM requirements remained to be characterized. This, together with the observation that enriched amino acid substitutions were often neither necessary nor sufficient to predict the PAM preference of most enzymes, led us to seek a more systematic method to investigate the relationship between amino acid sequence and PAM specificity. We therefore sought to explore the entire fitness landscape of our SpCas9(6AA) library via ML.
We utilized our two HT-PAMDA datasets as training data for an ML model, including functional enzymes derived from our bacterial selections on the 16 NGNN PAMs, and random (non-selected) enzymes from the SpCas9(6AA) library. We reasoned that including some inactive enzymes in our training set would provide information about combinations of amino acids that lead to non-functional enzymes. To account for the imbalanced nature of our training data towards NGG targeting enzymes and relaxed variants that are most active on NGTG PAMs (Fig. 1f), we assigned a label to each training example based on its most active PAM and randomly over-sampled to balance across PAM classes.
We trained ML models to predict the k on each of the 64 PAMs (positions 2-4) when provided with a 6AA sequence as input (Fig. 2a), and compared the ability of different ML models to extrapolate to amino acid combinations not encountered in the training set. Linear regression, random forest, and neural network models were tested in combination with different feature encodings as inputs to the model (one-hot encoding of each amino acid substitution; one-hot encoding of all single plus pairwise amino acid combinations; and Georgiev48, a physiochemical descriptor that can improve performance of some ML models for protein engineering49) (Extended Data Fig. 3a). Model architectures and amino acid encoding were compared using an internal 5-fold cross-validation (Extended Data Fig. 3a). The use of pairwise amino acid features was necessary for the linear and random forest, but not neural network, models to achieve generalizability to enzymes dissimilar from the training set (hamming distance from training set = 4). The neural network model with one-hot encoding was the most generalizable while requiring the simplest input feature set; thus this model was named the PAM machine learning algorithm (PAMmla) and was used for subsequent analyses.
Figure 2. Development of a machine learning model to predict SpCas9 PAM preference from amino acid sequence.
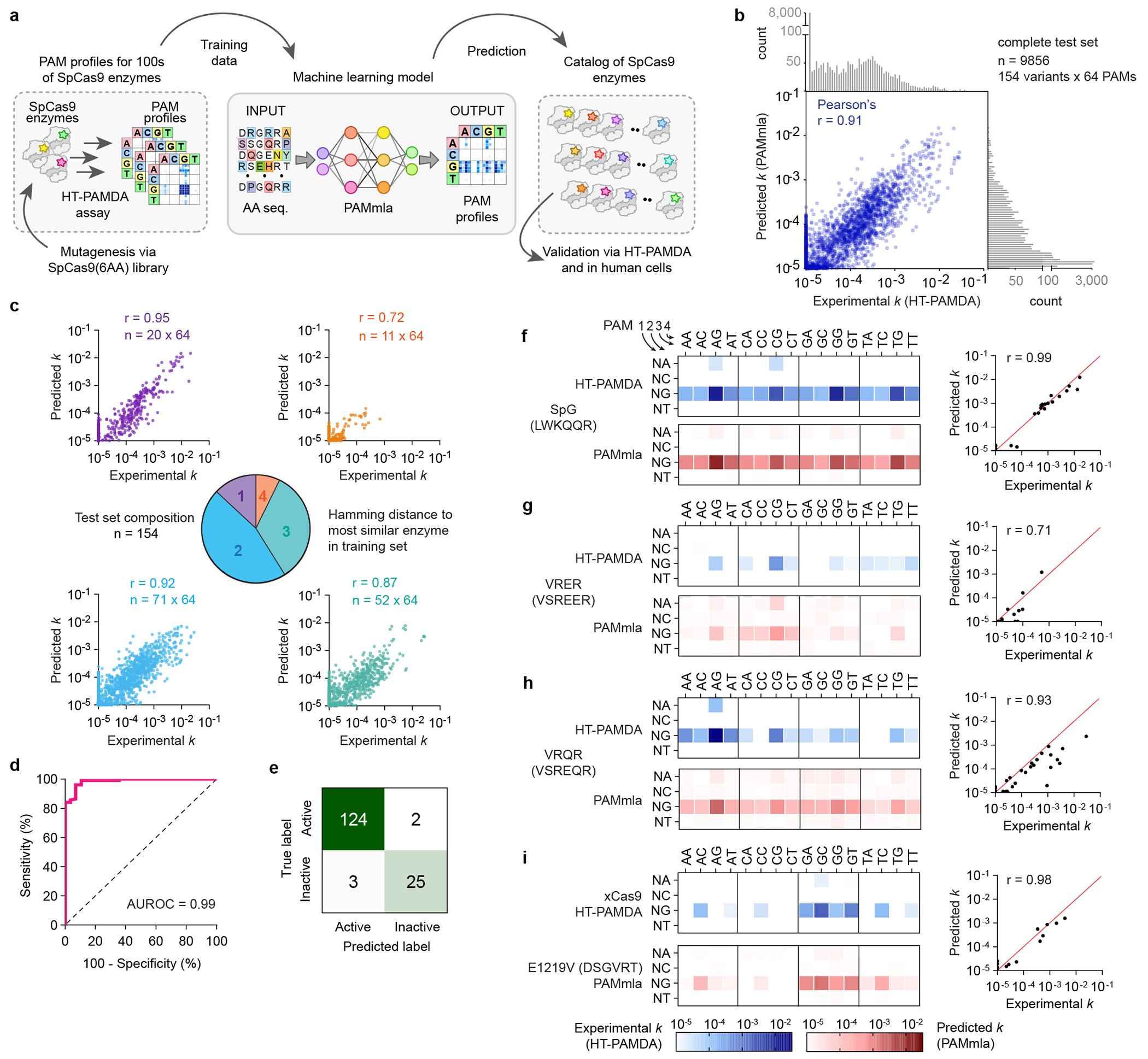
(a) Schematic a machine learning model that uses HT-PAMDA data from SpCas9 enzymes as the training data to then predict the PAM requirements for novel enzymes bearing combinations of amino acid at SpCas9(6AA) positions in the PI domain. (b) Correlation between the PAM machine learning algorithm (PAMmla) model predictions and experimentally determined rate constants (k) by HT-PAMDA, on a test set comprising 20% of the HT-PAMDA dataset held out from training. (c) Model performance via prediction of ks using PAMmla compared to HT-PAMDA determined ks, amongst different test sets by enzyme similarity to most similar sequence during training. (d) Receiver operating characteristic curve for binary classification of test set enzymes as active or inactive; enzymes are defined as inactive if the maximum HT-PAMDA k on any PAM is < 10−4.3. (e) Classification results on the test set when the threshold for identifying inactive enzymes is a maximum PAMmla predicted k < 10−4.3. (f–i) Comparison of experimentally determined PAM profiles (via HT-PAMDA; top panels in blue) to predicted PAM profiles (via PAMmla; bottom panels in red) with correlation between experimental and predicted ks (right panels), for previously published enzymes, including SpG3 (panel f), VRER14 (panel g), VRQR14,43 (panel h), and xCas92 (panel i). HT-PAMDA datasets are the mean of n = 2 biological replicates using different target sites.
We evaluated PAMmla on a test set comprising a random 20% of enzymes from our HT-PAMDA dataset that were held out from training, revealing accurate k predictions for unseen SpCas9 enzymes (Pearson’s r = 0.91) (Fig. 2b). We also evaluated PAMmla using two additional random train test splits that showed similar correlations (both r = 0.92; Extended Data Fig. 3b), demonstrating that these results are consistent regardless of the allocation of enzymes into training and testing sets. The test set was further sub-divided into progressively more challenging subsets containing enzymes with increasing numbers of mutations relative to the most similar enzyme in the training set, demonstrating generalizability to enzymes that are dissimilar to those on which the model was trained (Fig. 2c). Across different PAM classes, the model performed relatively consistently indicating the ability to generate accurate predictions, including for PAM classes which had relatively few training examples (Extended Data Figs. 3c–f). PAMmla models trained with over-sampling to balance PAM classes within the training set improved performance on under-represented PAM classes in the test set (Extended Data Fig. 3g).
In addition to accurately predicting PAM preferences of active Cas9 enzymes, PAMmla was also capable of distinguishing active from inactive enzymes with high accuracy (AUROC = 0.99; Fig. 2d). With “inactive” enzymes defined using a cutoff of maximum rate constant < 10−4.3, PAMmla correctly classified enzymes as active vs inactive for 149 of 154 enzymes (97%) in the test set (Fig. 2e). We selected a k = 10−4.3 to define a non-targetable PAM since the HT-PAMDA k values for bona fide inactive enzymes are noisy between ks of 10−5 to 10−4.3 (Extended Data Figs. 3h,i and Supplementary Note 5).
We tested PAMmla’s ability to recapitulate HT-PAMDA-determined PAM profiles of preexisting rationally designed or evolved SpCas9 PAM variant enzymes not included in the training or test sets. We were able to generate accurate PAM profiles for SpG, VRER, and VRQR (Pearson’s r = 0.99, 0.71, and 0.93, respectively), each of which contain two mutations relative to the closest member of the training set (Figs. 2f–h). We also predicted the PAM profile of xCas9, an enzyme that contains only a single PI domain mutation, E1219V, but additional mutations outside of the PI domain2. When predicting impact of the single E1219V mutation, PAMmla produced a predicted PAM profile remarkably similar to the experimental data for xCas9 (Fig. 2i), supporting previous evidence that the single E1219V mutation was the major contributor to xCas9’s altered PAM3.
ML-assisted prediction of PAM variant enzymes
We then utilized PAMmla to discover novel PAM variant enzymes by predicting the PAM profiles for each of the 64 million possible enzymes in the SpCas9(6AA) library (Supplementary Fig. 7). The resulting enzymes were sorted by their predicted PAM profiles on activity alone (kPAM) or selectivity (kPAM/sum(kall_PAMs)) for each of the 16 NGNN PAMs (Fig. 3a). Amongst the top predicted enzymes for each criterion and PAM, we chose between three and ten enzymes to experimentally validate PAMmla predictions using HT-PAMDA (for 281 enzymes; Fig. 3b, Supplementary Table 2 and Supplementary Figs. 8a–c). Amongst this group of enzymes, the PAMmla predictions for each PAM correlated with experimentally obtained ks (Pearson’s r = 0.90) (Fig. 3c, Supplementary Figs. 8a–c and 9). Most of the top predicted enzymes encoded 2 or 3 mutations (and some up to 4) relative to the most similar previously characterized variant (Fig. 3d), indicating that PAMmla can accurately predict the PAM requirements of enzymes dissimilar from the training set.
Figure 3. Characterization of the PAM requirements of PAMmla-predicted enzymes.

(a) Schematic of predicting and validating PAMmla enzymes. (b) Experimentally determined PAM profiles for 253 active PAMmla predicted enzymes using HT-PAMDA (enzymes with no k > 10−4 not shown). HT-PAMDA profiles were clustered hierarchically and amino acid enrichment motifs for the 10 largest clusters are shown (sequence logos; right panels). Expanded HT-PAMDA profiles for representative enzymes from each cluster are shown and PAMmla predicted rate constants (ks) are compared to experimentally determined ks (left panels). HT-PAMDA datasets are the mean of n = 2 biological replicates using different target sites. PAMmla datasets are the mean of n = 3 predictions from separate training instances of the model. (c) Correlation between predicted and experimentally determined ks (via PAMmla and HT-PAMDA, respectively) for 281 PAMmla predicted enzymes from panel b. Each data point represents the k of an enzyme on one of 64 NNNN PAMs. (d) Distribution of amino acid hamming distances from the training set for enzymes from panel b. (e) Categorization of enzyme clusters from panel b; inactive enzymes had no k > 10−4 as determined by HT-PAMDA. (f) Distribution of SpCas9 enzymes based on their experimental ks, with enzymes from 3 categories: random from the SpCas9(6AA) library, a bacterial selection, or PAMmla to maximize activity on an NGNN PAM. The plotted k is the rate constant of the PAM used in bacterial selections or the query for maximized PAMmla predictions. (g) Fraction of PAM variant enzymes maximally active against the specific NGNN PAM that they were selected/predicted against (rank = 1st) or where the PAM selected against was within the top 4 most active PAMs, as determined by HT-PAMDA. The three categories of enzymes analyzed are PAMmla enzymes by maximizing activity on the 16 NGNN PAMs, PAMmla enzymes by sorting for selectivity for each of the 16 NGNN PAMs, and enzymes from bacterial selections on each of the 16 NGNN PAMs.
Clustering the experimentally determined PAM profiles from the PAMmla predicted enzymes revealed 10 main clusters (Figs. 3b,e). Clusters comprised new enzymes with novel PAM requirements compared to those previously seen (e.g., NGTC, NGCT, NGCM (M = A or C), and relaxed with NGG anti-preference), along with sequence-diversified examples of enzymes with PAM profiles similar to those obtained from bacterial selections (e.g. NGG, NGN, NGC). These classes of enzymes provide new insight into combinations of amino acids within the SpCas9(6AA) library that enable useful PAM profiles.
Sorting PAMmla predicted enzymes for maximized efficiency on each of the 16 NGNN PAMs resulted in enzymes with higher ks on the PAM of interest than those obtained from bacterial selections (Fig. 3f). While highly efficient enzymes with activity on a PAM of interest with k > 10−2.5 (similar to the maximum k of SpG) were rarely recovered from bacterial selections (less than 9% of enzymes), the majority of PAMmla derived enzymes (56%) designed to maximize activity had k > 10−2.5 on the PAM for which they were sorted and had maximal efficiency on that PAM (Fig. 3f and Supplementary Fig. 9). Furthermore, when we sorted PAMmla predictions for selectivity on a PAM of interest, the resulting enzymes were more specific for the target PAM than enzymes derived from bacterial selections (Fig. 3g). Approximately 60% of PAMmla enzymes designed to maximize selectivity had a maximal experimentally determined k on the specific 4-nt PAM for which sorting was performed (with 91% of PAMmla enzymes having the intended PAM in the top four; Fig. 3g and Supplementary Fig. 9). In contrast, only 26% of enzymes from bacterial selections exhibited maximal activity on the PAM from which they were selected (Fig. 3g and Supplementary Fig. 3). When ranked amongst PAMmla predictions of 64M enzymes, bacterial selection-derived enzymes ranked on average in the 95th percentile in terms of activity (outranked by 3.2M enzymes) and the 70th percentile in terms of selectivity (outranked by 18.9M enzymes) (Supplementary Figs. 10a,b), highlighting the utility of PAMmla to predict superior enzymes versus those derived from experimental selections. Thus, PAMmla predictions can be used to obtain enzymes tailored to the PAM of interest rather than generalist enzymes with relaxed PAMs, which tend to be the most common outcome of experimental engineering methods.
We investigated potential functional roles of the amino acid substitutions in PAMmla predicted enzymes (Supplementary Note 6). Using SHAP50 analysis of ML feature importance (Extended Data Fig. 4) and structural modeling (Extended Data Fig. 5), we observed that mutations in PAMmla enzymes can be divided into 3 categories that either: (1) alter 3rd PAM position preference (Extended Data Figs. 5a–c); (2) alter 4th PAM position preference (Extended Data Figs. 5d–f); or (3) modify PAM recognition via base-independent interactions (Extended Data Figs. 5g–I). Generally, more selective PAMmla enzymes harbored mutations that form base-specific contacts with the DNA bases of the PAM, whereas PAMmla enzymes with broader PAM specificity shared non-specific activity-potentiating mutations with previously described PAM-relaxed enzymes that form nonspecific contacts with the DNA backbone3,6. Many PAMmla enzymes shared combinations of mutations from both categories, potentially achieving a balance between specifying novel PAMs and retaining high activity.
Testing PAMmla-predicted enzymes in human cells
We tested the editing efficiencies of 22 PAMmla generated enzymes in HEK 293T cells, prioritizing those with preferences for PAMs which have not been previously described in the literature: NGAT, NGCM (M = A or C), NGCN, NGTN, NGTC, NGDC (D = A, G, or T), and NGTG (Extended Data Fig. 6a and Supplementary Figs. 8a–c). We also assessed 14 enzymes from the training set that were identified from our bacterial selection experiments. For both enzyme classes, predicted PAM requirements correlated well with their HT-PAMDA determined profiles (Supplementary Fig. 11). Nuclease forms of these 36 enzymes were assessed for generating insertion or deletion mutations (indels) across 32 endogenous target sites in HEK 293T cells compared to SpG, which is an engineered SpCas9 nuclease enzyme variant capable of targeting sites with NGN PAMs3. In general, PAMmla enzymes exhibited similar or improved average editing efficiencies compared to SpG (Fig. 4a and Extended Data Fig. 6b). Although we only highlight a single enzyme from each PAM class (Fig. 4a), for each target site that we assessed several PAMmla enzymes often achieved similar or greater editing efficiencies to SpG while maintaining a more selective PAM preference (Extended Data Fig. 6b and Supplementary Figs. 12a–g). In some cases, PAMmla enzymes were substantially more efficient at creating indels compared to the most similar variants from the training set. For instance, the PAMmla enzyme LWKYQS was capable of ~1.5-fold higher average editing efficiencies on sites with NGCM PAMs compared to its nearest neighbor from the training set, LWKYSS, which contains only one amino acid difference (Extended Data Fig. 6b, NGCM variants). Editing efficiencies varied by target site (Supplementary Figs. 12a–g), suggesting spacer-specific differences and thus the potential utility of testing multiple PAMmla enzymes for each target of interest. We also tested all PAMmla enzymes on a control site bearing an NGG PAM to assess remaining activity against the ancestral PAM (Fig. 4b and Supplementary Fig. 12h). PAMmla enzymes predicted to poorly target NGG exhibited greatly diminished activity on the control NGG PAM target site (Supplementary Fig. 12i), indicating that PAMmla can generate enzymes that minimize editing on the canonical PAM. Together, these observations demonstrate that PAMmla-predicted enzymes improve human cell-based editing efficiencies compared to SpG or those derived from bacterial selections, and that PAMmla can be used to generate enzymes with altered rather than relaxed PAM specificities.
Figure 4. Genome editing and off-target analysis in human cells with PAMmla-predicted enzymes.

(a,b) Nuclease-mediated editing at endogenous sites in HEK 293T cells for each PAMmla derived enzyme (colored bars) compared to SpG and WT SpCas9, across sites harboring preferred PAMs (panel a) or NGG PAMs (panel b). Data points represent 3 independent biological replicates on 3-to-11 sites per PAM (Supplementary Fig. 12). (c,d) Summary of ABE8e and TadCBEd base editing efficiencies (panels c and d respectively) for all PAMmla enzymes from panel a on their preferred PAMs compared to SpG and SpRY (Supplementary Figs. 13, 14). Data points represent 3 independent biological replicates on 3 genomic sites per PAM. Bars = data mean. (e) Modification of endogenous sites in HEK 293T cells from GUIDE-seq-2 transfections containing the dsODN tag. Percent modification assessed by targeted sequencing; n = 3 technical replicates. (f) Number of GUIDE-seq-2 detected off-target sites for PAMmla enzymes, SpG, or SpRY, normalized to the number of off-target sites for SpRY. (g, h) Fraction of on- and off-target GUIDE-seq-2 reads with sgRNAs targeted to sites with few or many off-target sites (panels g and h, respectively). (i) Schematic of CYBB T326I mutation with the A8 sgRNA target site encoding an NGAT PAM shown, with intended edit position and bystander edit labeled in blue or red numbering, respectively. (j). Base editing efficiencies to correct the CYBB T326I mutation in a patient-derived B cell line. Base editing assessed by targeted sequencing; mean, SD, and individual data points shown for n = 3 independent biological replicates; all bases edited at >1% efficiency are shown. (k) Fraction of GUIDE-seq-2 reads attributed to on- and off-target sites for KWRQLC and SpG variants in GUIDE-seq-2 experiments using the CYBB T326I A8 sgRNA in HEK 293T cells (see Extended Data Figs. 7d,e).
To maximize the efficiency of base editing, precise positioning of Cas9 enzymes over the target base is required51. For each of the seven PAM categories tested above, we therefore selected a single PAMmla-derived enzyme to assess whether they could be beneficial for A-to-G and C-to-T editing efficiencies in the contexts of ABE8e52 and TadCBEd53 architectures, respectively. For ABE8e and TadCBEd enzymes, we observed efficient base editing in HEK 293T cells at 3 genomic loci harboring the preferred PAM of each of the corresponding Cas9 variants (Figs. 4c,d, Extended Data Figs. 6c,d, and Supplementary Figs. 13 and 14). The base editing efficiencies of PAMmla-generated BEs were superior to the PAM-relaxed SpRY in both ABE8e and TadCBEd contexts, while PAMmla enzymes were more active versus SpG as ABE8e constructs and similarly efficient as TadCBEd constructs depending on the site (Figs. 4c and 4d, respectively). We additionally tested PAMmla enzymes on a therapeutic base edit for sickle cell disease, achieving comparable on-target editing and fewer bystander edits compared to a previously reported approach54 (Supplementary Figs. 15a–h, Supplementary Note 7). Together, these observations demonstrate that compared to PAM-relaxed enzymes, PAMmla derived editors are generally more efficient nucleases and BEs.
Specificity of PAMmla-predicted enzymes
SpCas9 enzymes with relaxed PAM requirements are more prone to off-target editing since they search a larger fraction of the genome and thus encounter a larger number of potential off-target sites3,6,21. In contrast, PAMmla-predicted enzymes with altered PAM requirements should encounter fewer potential off-targets due to their more limited targeting ranges. To test this hypothesis, we utilized a cell-based unbiased genome-wide off-target assay, GUIDE-seq-2 (ref. 55), to identify nuclease-cleaved off-target sites. We observed comparable or greater on-target editing using PAMmla enzymes compared to SpG and SpRY at the sites chosen for GUIDE-seq-2 (Fig. 4e and Extended Data Fig. 7a), and yet the total number of off-target sites were reduced with PAMmla enzymes versus SpG and SpRY (ranging from 26%-93% reduction compared to SpG, and 49%-96% reduction compared to SpRY; Fig. 4f). PAMmla enzymes resulted in a higher proportion of on-to-off-target GUIDE-seq-2 reads relative to SpG and SpRY for all sites, including those with modest and high numbers of off-target sites (Figs. 4g and 4h, respectively). Off-target sites detected for each PAMmla-predicted enzyme were largely also detected in the SpG or SpRY sample with the same sgRNA, though some off-targets were specific to the altered PAM variant enzymes (Extended Data Fig. 7b and Supplementary Note 8). The PAMs observed at off-target sites for each PAMmla-derived enzyme reflected their cognate HT-PAMDA-determined PAMs, while the aggregate PAMs observed for SpG and SpRY were more relaxed (Extended Data Fig. 7c). These results demonstrate that PAMmla enzymes with altered PAMs can reduce genome-wide off-targets compared to PAM-relaxed enzymes.
To verify the on- and off-target benefits of PAMmla versus generalist enzymes in human patient-derived cells, we sought to correct a CYBB mutation in cells from an individual with X-linked Chronic Granulomatous Disease (X-CGD). X-CGD patients are highly susceptible to invasive infections and hyperinflammation that results in significant morbidity and early mortality. CYBB mutations may be correctable through ex vivo editing of hematopoietic stem cells56,57. Using patient-derived Epstein Barr virus-transformed B cells harboring the CYBB T362I mutation, we identified a target site with an NGAT PAM that positions the causative G-to-A mutation at position 8 within the protospacer (Fig. 4i) and electroporated mRNA encoding either ABE8e-SpG or the PAMmla-derived ABE8e-KWRQLC enzyme along with the A8 sgRNA. Both ABEs resulted in highly efficient levels of T362I correction (up to >90%) with minimal bystander editing (Fig. 4j). GUIDE-seq-2 assessment of off-targets using the CYBB T362I-targeted sgRNA in HEK 293T cells revealed only a single low-likelihood off-target site detected when using the PAMmla predicted KWRQLC enzyme compared to 5 off-target sites with SpG (Extended Data Fig. 7e), resulting in a superior on-to-off-target ratio for the PAMmla enzyme compared to the PAM relaxed SpG (Fig. 4k).
PAMmla-enabled allele-specific editing in vivo
One potential application of PAMmla is to predict SpCas9 enzymes for allele-specific editing58 by maximizing activity on a PAM of interest while minimizing targeting on another. Sorting the PAMmla predictions for all 64 million SpCas9(6AA) enzymes is cumbersome, so we developed an in silico directed evolution (ISDE) approach that utilizes PAMmla predictions to computationally engineer SpCas9 variant enzymes in a stepwise manner (Extended Data Fig. 8a). ISDE enables prediction of optimal enzymes with greater computational efficiency, enabling rapid sorting by customizable fitness metrics (Extended Data Figs. 8b–d and Supplementary Note 9). As a proof-of-concept, we sought to utilize PAMmla-ISDE to evolve a customized editor to target the human Rhodopsin (RHO) P23H mutation causative of retinitis pigmentosa59–61. As a dominant negative mutation, knock-out of the mutant allele while leaving the WT allele intact can rescue function in heterozygous genotypes62,63. Single nucleotide mutations can be difficult to target in an allele-specific manner via the sgRNA due to the propensity of SpCas9 to tolerate single base pair mismatches between the sgRNA and target DNA58,64. However, if a disease-causing point mutation generates a novel PAM, a high specificity PAM proof-reading step may be exploited for allele-specific editing58,65,66.
For allele selective editing of RHO P23H, we utilized PAMmla-ISDE to design enzymes that target an NGTG PAM on the mutant allele and that minimize editing of NGGG on the WT allele (Fig. 5a). Neither WT SpCas9 nor SpG are useful, since SpCas9 should efficiently target the WT P23 allele and SpG should target both P23 and P23H alleles equally. We initiated PAMmla-ISDE starting with the WT SpCas9 sequence and evolved enzymes by sorting for maximized activity on NGTG PAMs while requiring a k < 10−3.7 on NGGG. The PAMmla-ISDE fitness plateaued after four cycles of mutagenesis and prediction (Figs. 6b,d and Supplementary Fig. 16a). The most optimal enzyme, MRRWMR, was predicted by PAMmla to strongly target NGTG and weakly target NGGG (ks of = 10−2.1 and 10−3.7, respectively; Fig. 5b). Interestingly, MRRWMR was also the most optimal enzyme via ISDE using SpG as a starting sequence and different parameters (Supplementary Fig. 16b). We also performed a stricter evolution via IDSE to further minimize targeting of NGGG, requiring a k < 10−4 on NGGG, which resulted in several enzymes including KRHWMR after 4 rounds of evolution (Figs. 6c,e and Supplementary Fig. 16c).
Figure 5. In silico directed evolution of an allele-specific editor for the RHO P23H allele.

(a) Schematic of allele-specific editing of heterozygous RHO P23H alleles. (b,c) Predicted PAM profiles of enzymes resulting from PAMmla-enabled ISDE, using WT SpCas9 as a starting sequence and seeking to maximize activity on NGTG while minimizing on NGGG (k < 10−3.7 and k < 10−4 in panels b and c, respectively). Only the evolutionary trajectories leading to MRRWMR and KRHWMR are shown (Supplementary Fig. 16). (d,e) Fitness functions used to perform the in silico directed evolution experiments in panels b and c respectively; the top 10 enzymes from each round are in gray and the trajectory leading to MRRWMR or KRHWMR are in red. (f) Modification of the WT RHO and P23H alleles in a HEK 293T cell line harboring a 2:1 P23H:P23 allele ratio (see also Extended Data Fig. 9b). Editing assessed by targeted sequencing and CRISPResso2; mean and s.d. shown for n = 3 biological replicates; for reads containing indels that span the P23H mutation, edited counts were distributed using the ratio of WT to mutant as observed for the identifiable edited reads (Extended Data Fig. 9d). (g) Fraction of on- and off-target GUIDE-seq-2 reads for PAMmla predicted enzymes, SpG, and SpRY when paired with the RHO P23H sgRNA in homozygous P23H HEK 293T cells (Extended Data Figs. 10d,e). (h,i) Mutations shared between MRRWMR and KRHWMR modelled on the structure of VRER (PDB: 5FW3) interacting with NGTG or NGGG PAMs (panels h and i, respectively). Protein surface is colored by lipophilicity potential. Hydrogen bonds are represented by dashed lines and Van der Waals interactions are represented by green squiggles. (j, k) Force plots depicting SHAP values50 for MRRWMR activity on NGTG or NGGG PAMs (panels j and k, respectively). (l) In vivo modification of the RHO P23H or WT alleles in heterozygous humanized P0-P2 mouse pups via subretinal plasmid injection and electroporation. Editing assessed by targeted sequencing of BFP+ sorted retinal cells. Mean and s.d. shown for n = 7, 10, and 4 mice injected with KRHWMR, MRRWMR, or SpG respectively; unidentifiable reads containing indels that span the P23H mutation were discarded ( Extended Data Fig. 10g).
Initial experiments with MRRWMR on three unrelated genomic sites harboring NGTG PAMs achieved similar editing efficiencies compared to SpG, with minimal editing on a site bearing an NGGG PAM (Extended Data Fig. 9a). We then generated a heterozygous RHO P23H HEK 293T cell line to assess allele-selective editing with the PAMmla predicted enzymes on the target of interest (Supplementary Figs. 17a–f). We tested a total of four ISDE-derived enzymes from either campaign in the RHO P23H cell line and observed a preference for editing the mutant over the WT allele compared to SpG for all ISDE enzymes (Extended Data Fig. 9b). The MRRWMR enzyme led to the most efficient mutant allele disruption and KRHWMR resulted in superior allele-specific discrimination with nearly undetectable editing of the WT allele in heterozygous and WT cells (Fig. 5f and Extended Data Figs. 9b,c). The ISDE-derived MRRWMR and KRHWMR enzymes exhibited ~2.5- and ~40-fold preferences for mutant P23H over WT RHO alleles respectively (Extended Data Fig. 9e). Use of other recently described Cas9 ortholog nucleases PrCas967, CoCas968, and GeCas968 harboring PAM preferences that should target the P23H site failed to elicit detectable editing at the RHO locus (Supplementary Figs. 18a–c and Supplementary Note 10).
Next, we performed off-target analyses via GUIDE-seq-2 to investigate specificity improvements with the allele-selective PAMmla enzymes. On two unrelated genomic loci we observed enhanced specificity with MRRWMR over SpG and SpRY (Extended Data Figs. 10a–c). When using the RHO P23H sgRNAs in a homozygous P23H cell line, both MRRWMR and KRHWMR also improved on-target specificity by minimizing off-target reads and the number of detected off-target sites compared to SpG and SpRY (Fig. 5g and Extended Data Figs. 10d–f).
To examine potential structural determinants of the PAM-selective preferences of MRRWMR and KRHWMR, we modeled their mutations and investigated their contributions to PAMmla predicted rates on NGTG and NGGG PAMs using SHAP (Figs. 6h-k, Extended Data Figs. 11a–e). For both enzymes, PAM selectivity for a 3rd PAM position T over G largely resulted from the E1219W and R1335M side chains forming a hydrophobic pocket, enabling discrimination via favorable hydrophobic interactions with the T3 methyl group of an NGTG PAM and unfavorable interaction with the polar G3 side chain of NGGG (Figs. 6h,i). SHAP analysis supports that these mutations positively impact recognition of NGTG and negatively impact activity on NGGG (Figs. 6j,k and Extended Data Figs. 11b–e). Additional common mutations between the two PAMmla enzymes S1136R and T1337R contribute to PAM selectivity and/or potentiation of activity (Extended Data Fig. 11 and Supplementary Note 11).
We assessed the in vivo editing efficiency and allele-specificity of MRRWMR and KRHWMR in mouse retinas. Subretinal plasmid injections in humanized heterozygous WT-hRHO-GFP/P23H-hRHO-RFP mice at P0–P2 were performed prior to in vivo electroporation. Analysis of on-target editing in transfected cells with MRRWMR revealed mean on-target efficiency of 37% on the P23H allele (up to 59%) and 7.6% on the WT allele, leading to 4.8-fold selectivity for editing the mutant allele (Fig. 5l and Extended Data Fig. 10h). Consistent with results in cells, KRHWMR resulted in lower in vivo editing (~20%) but exhibited increased specificity for the P23H over wild type allele (9.5-fold) (Fig. 5l and Extended Data Fig. 10h). Together, these findings demonstrate how PAMmla-ISDE can predict novel enzymes for therapeutically relevant edits with no intervening engineering or evolution. PAMmla-nominated enzymes enable cell-based and in vivo allele-selective editing not possible with previously available SpCas9 enzyme variants or Cas9 orthologs.
Discussion
Here, we optimized an experimental workflow to develop an ML model, PAMmla, capable of predicting the PAM requirements of millions of SpCas9 enzymes from amino acid sequence alone. PAMmla enables the discovery of novel Cas9 enzymes that are effective in human cells and in mice, with advantages over generalist PAM-relaxed enzymes including higher levels of on-target editing and reduced genome-wide off-targets (likely due to a narrower genome search space; Supplementary Note 12). To expedite the customization of SpCas9 enzymes for research or translational uses, we developed a webtool to generate predictions based on the PAMmla model, and prioritize enzymes with user-defined properties via ISDE (https://pammla.streamlit.app/).
ML combined with scalable experimental assays enables the exploration of a large and diverse protein sequence space to identify useful enzymes23–40,69,70. PAMmla offers new insight into the plasticity of how Cas9 specifies a nucleic acid target. Prior to PAMmla, there were few examples of PAM-altered enzymes14 relative to the more common PAM-relaxed enzymes1–4. This discrepancy suggests that relaxing the PAM may be the simplest evolutionary trajectory to permit new targeting capabilities for a Cas enzyme, perhaps because PAM-altered enzymes may require several specific simultaneous mutations that function epistatically to specify new PAMs. PAM-altered enzymes are therefore less likely to be discovered during experimental engineering approaches (e.g. directed evolution) that do not incorporate a counter-selection step to preserve PAM selectivity.
Recent models trained on evolutionary sequence data have predicted variant effects with comparable performance to experimental assays71,72. However, we found that several evolutionary sequence-based models71–73 were not predictive of activity of our enzyme set on non-canonical PAMs (Supplementary Fig. 19). Previous ML-based approaches have sought to predict sequence-diversified Cas9 enzymes42,74 or improved SaCas9-KKH enzymes26 (Supplementary Note 13). While these studies support the use of ML for engineering Cas enzymes, modeling is largely limited to activity on the canonical PAM. These data suggest that natural sequence information alone is likely insufficient to train models capable of accurately predicting enzymes with PAM requirements divergent from those seen in nature.
Together, PAMmla enables user-specifiable design of bespoke SpCas9 PAM variant enzymes and motivates a transition from generalist PAM-relaxed enzymes to improve genome editing efficiency and safety for various applications. More broadly, the development of PAMmla highlights the synergies between experimental engineering and ML for the rapid and scalable interrogation and optimization of proteins.
Methods
Plasmids, oligonucleotides strains, and cloning
Oligonucleotide sequences are available in Supplementary Table 3. Descriptions of plasmids used in this study are available in Supplementary Table 4; new plasmids generated during this study have been deposited with Addgene (https://www.addgene.org/Benjamin_Kleinstiver/), and were validated using Sanger sequencing, whole plasmid sequencing (Primordium Labs), or a custom whole-ORF sequencing method (described below). Target site sequences for sgRNAs and epegRNAs are available in Supplementary Table 5.
A saturation mutagenesis plasmid library was generated by randomizing six amino acid positions in the SpCas9 coding sequence (D1135, S1136, G1218, E1219, R1335, and T1337)44. A parental SpCas9 bacterial expression plasmid pACYC-T7-SpCas9-T7-EGFPgRNA1 (BPK848; Addgene plasmid ID 181745) was used to generate the library by cloning type IIS restriction enzyme cassettes into the sequence near amino acid positions D1135/S1136 (BspMI enzyme cassette), G1218/E1219 (SapI enzyme cassette), and R1335/T1337 (BsaI enzyme cassette) to create the library entry plasmid pACYC-T7-SpCas9(BspMI/SapI/BsaI_cassettes)-T7-EGFPgRNA1 (BPK1807). The entry plasmid BPK1807 was then subjected to sequential cloning steps (restriction digests followed by ligations with library oligos encoding NNS codons (where ‘N’ is any nucleotide and ‘S’ is C or G) that were pre-annealed with adapter oligos to reform restriction site overhangs; Supplementary Table 3). Plasmid BPK1807 was digested with BspMI and subjected to a ligation using annealed oligonucleotides oBK1107 (NNS at D1135 and S1136), oBK1101, and oBK1102, resulting in plasmid library BPK1993 (of approximate complexity of 2.61e6 library members); plasmid BPK1993 was digested with SapI and subjected to a ligation using annealed oligonucleotides oBK1701 (NNS at G1218 and E1219), oBK1103, and oBK1104), resulting in plasmid library BPK2057 (of approximate complexity of 1.05e6 library members); plasmid BPK2057 was digested with BsaI and subjected to a ligation using annealed oligonucleotides oBK1110 (NNS at R1335 and T1337), oBK1105, and oBK1106, resulting in the final saturation mutagenesis library plasmids pACYC-T7-SpCas9(6AA_NNS)-T7-EGFPgRNA1 BPK2097 and MNW94 (of approximate complexity of >3.51e6 library members with NNS at codons D1135, S1136, G1218, E1219, R1335, and T1337). The SpCas9(6AA) library was verified by Sanger sequencing and targeted sequencing.
Target plasmids for bacterial-based positive selection assays containing an arabinose-inducible ccdB toxin gene were generated by cloning duplexed oligonucleotides into XbaI and SphI-digested p11-lacY-wtx1 (Addgene ID 69056) as previously described14. The derivative ccdB-expressing plasmids contain an EGFP-derived protospacer sequence (GGGCACGGGCAGCTTGCCGG) adjacent to each of the 16 possible NGNN PAMs varying at positions 3 and 4 (Supplementary Table 4).
Bacterial strains for positive selection assays were generated by separately transforming chemically competent BW25141(λDE3)75. E. coli with each of the 16 p11-lacY-wtx1 plasmid derivatives harboring each of the NGNN PAMs. To make electrocompetent cells harboring each toxic plasmid, single colonies from each transformation were grown overnight in 5 mL LB supplemented with 100 μg/mL carbenicillin and 10 mM dextrose. Overnight cultures were diluted in 500 mL LB + carbenicillin + dextrose and grown to an OD600 of 0.5-0.8. After chilling on ice for 30 minutes, cultures were pelleted at 4 °C at 4000g for 15 minutes and resuspended in 500 mL ice cold H2O. Pelleting and resuspension were repeated three more times using 250 mL ice cold H2O, 10 mL ice cold 10% glycerol, and 1 mL 10% glycerol. Aliquots were frozen in liquid nitrogen and stored at −80 °C.
SpCas9 sgRNA expression plasmids for human cell experiments were cloned by digesting pUC19-U6-BsmBI_cassette-SpCas9gRNA (BPK1520; Addgene ID 65777)14 with BsmBI at 55 °C overnight and performing ligations with annealed oligos encoding the sgRNA spacer (Supplementary Table 5). SpCas9 nuclease expression plasmids (Supplementary Table 4) for human cell experiments were generated by digesting pCMV-T7-SpCas9-P2A-EGFP (RTW3027; Addgene plasmid ID 139987)3 with PmlI and XhoI, and inserting the modified SpCas9 PAM interacting domain via four PCR products with mutations contained in the primer overlaps followed by isothermal assembly76. A-to-G base editor (ABE) expression plasmids utilizing the ABE8e architecture52 were generated by digesting pCMV-T7-ABE8e-nSpCas9-P2A-EGFP (KAC978; Addgene plasmid ID 185910)77 with XcmI and PmlI, and inserting the modified SpCas9 PAM interacting domain via PCR and isothermal assembly. C-to-T base editor (CBE) expression plasmids utilizing the TadCBEd architecture53 were generated by digesting pCMV-T7-TadA-CDd-nSpCas9-P2A-EGFP (BKS327; Addgene plasmid ID 223123) with XcmI and PmlI, and inserting the modified SpCas9 PAM interacting domain via PCR and isothermal assembly76. SpCas9-based prime editor epegRNA78 expression plasmids were generated via cloning into pUC19-U6-[BsmBI]-tevopreQ1-term (LM1138) (Supplementary Table 5), and second strand nicking sgRNAs were generated via cloning into BPK1520 (Supplementary Table 5). A modified prime editor plasmid76 harboring a co-translationally expressed EGFP protein (pCMV-T7-PEmax-P2A-EGFP; LM1589) was generated via SpRYgest79 followed by isothermal assembly76.
Animal care and models
Our animal study followed the tenets of the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the guidelines of the Massachusetts Eye and Ear for Animal Care and Use (under IACUC protocol number 2021N000059). The humanized WT-hRHO-GFP and P23H-hRHO-RFP mice were gifted from Dr. Theodore G. Wensel80,81. The two strains were crossbred to generate heterozygous hRHO-WT-GFP/hRHO-P23H-RFP mouse line, in which the humanized WT-hRHO allele is fused to GFP and the humanized P23H-hRHO mutant allele is fused to the fluorescent protein TagRFPt. All experiments were performed on P0-P2 heterozygous pups. Equal numbers of male and female mice were used. Mice were housed under a 12-hour light/dark cycle at an ambient temperature of 20-22°C and relative humidity of 40-60%.
Bacterial-based positive selection experiments
To perform positive selections, 100 μL of electrocompetent BW25141(λDE3) E. coli harboring p11-lacY-wtx1 plasmid derivatives harboring each of the NGNN PAMs were each electrotransformed with 100 ng of the SpCas9(6AA_NNS) plasmid library (MMW94), which also expresses an sgRNA targeting the protospacer sequence GGGCACGGGCAGCTTGCCGG and chloramphenicol resistance marker. Following a 60-minute recovery in 3 mL Super Optimal broth with Catabolite repression (SOC) media, transformations were spread on LB plates containing either 25 μg/mL chloramphenicol and 10 mM dextrose (non-selective) or 25 μg/mL chloramphenicol + 10 mM arabinose (selective). Transformation efficiency was estimated based on colony count from non-selective plates.
Colonies from selective plates were picked and used as template for colony PCR to be transferred to mammalian expression plasmids for the HT-PAMDA assay. Colony PCR was performed on the PAM interacting domain sequence of ~24-48 colonies that each of the 16 bacterial selections using primers oRAS122 and oBK591 (Supplementary Table 3). PCR products were purified by paramagnetic beads (generated as previously described82,83 and cloned into a gel-purified PvuII- and XhoI-digested mammalian SpCas9 expression vector pCAG-hSpCas9-P2A-EGFP (MSP2582; Addgene plasmid ID 223067) by isothermal assembly. We also cloned randomly chosen variants from the saturation mutagenesis SpCas9 (6AA_NNS) plasmid library (MMW94) without being subject to the bacterial selection strategy. The library was cloned en masse from MMW94 into PvuII- and XhoI-digested MS2582 by isothermal assembly. The cloning reactions for PAM variant enzyme plasmids derived from bacterial selections or that were randomly chosen were transformed into electrocompetent XL1-Blue E. coli and plated on LB + carbenicillin. Single colonies were mini prepped (Qiagen) for arrayed sequencing via multiplex PCR (described below).
Arrayed sequencing of SpCas9 variants via multiplex PCR
Two pools of staggered amplicons covering the entire expression construct (from CAG promoter, SpCas9 coding sequence, P2A linker, and EGFP ORFs) of MSP2582 were designed using PrimalScheme84 (https://primalscheme.com/) (Supplementary Fig. 1). Primers were inspected manually to ensure that none overlapped with the sites of saturation mutagenesis, SpCas9 residues D1135, S1136, G1218, E1219, R1335, T1337. 33nt flaps overlapping with Illumina P5 and P7 adapter sequences were then added to each of the forward and reverse primers respectively forming two primer pools: oPool1 (oRAS127-156) and oPool2 (oRAS157-186) (Supplementary Table 3). Two separate multiplex PCR reactions using oPool1 and oPool2 were performed for each plasmid to be sequenced using ~10 ng plasmid DNA as template. Multiplex PCR was performed as described by Quick et al.84 , with Pool1 and Pool2 PCR products then pooled together to achieve a single PCR pool per plasmid. PCR pools were bead purified and distinct i5 and i7 barcodes were added to each PCR pool in a second round of PCR as previously described12 (Supplementary Table 3). All barcoded PCR products were then combined into a single pool, bead purified, quantified by Qubit (Thermo Fisher), diluted to 0.3 ng/μL and sequenced a MiSeq sequencer using a 300-cycle v2 kit (Illumina).
Multiplex sequencing data were analyzed using custom python scripts available at https://github.com/RachelSilverstein/multiplex_seq_analysis. First, Fastq files were analyzed using “get_alignment.py” with gap_open_penalty set to 7. Briefly, reads were trimmed using TrimGalore with default settings and aligned to the reference vector map using Bowtie2. Pileup files containing aligned reads were analyzed using “identify_variants.py” with parameters set to: MAX_INSERTION_FREQ = 0.2, MAX_DELETION_FREQ = 0.2, call_variant_when_identity_below = 0.95, include_alleles_above_freq = 0.01, and MIXED_VARIANT_FRACTION = 0.05. Samples were discarded if they contained insertions or deletions, point mutations other than the intended sites of saturation mutagenesis, or mixed reads indicating more than one plasmid per colony. Unique plasmid sequences containing only mutations in the intended 6 positions were used for further analysis by HT-PAMDA (Supplementary Table 4).
Profiling the PAM requirements of SpCas9 enzymes
SpCas9 sgRNAs in vitro transcribed from roughly 1 μg of HindIII linearized sgRNA T7-transcription plasmid templates (RTW443 and RTW448; Addgene plasmid IDs 160136 and 160137, respectively47 using the T7 RiboMAX Express Large Scale RNA Production Kit (Promega). The DNA template was degraded by the addition of 1 μL RQ1 DNase at 37 °C for 15 minutes. sgRNAs were purified using beads and refolded by heating to 90 °C for 5 minutes and then cooling to room temperature for 15 minutes.
The high-throughput PAM determination assay (HT-PAMDA) was performed as previously described12 apart from omitting the ExonucleaseI digestion step following the first PCR. Alternative barcoding primers were used for different sequencing runs with either a four or five nucleotide unique barcode (Supplementary Table 3). Briefly, SpCas9 containing lysates were generated by transfecting 1.5x105 HEK 293T cells with approximately 700 ng Cas9 expression plasmid (containing -P2A-EGFP tag) and 1.5 μL TransIT-X2 transfection reagent (Mirus). Cells were lysed ~48 hours post-transfection and EGFP signal was measured and normalized to a standard containing 150 nM Fluorescein (Sigma). 4.375 μL of normalized cell lysates were separately complexed with 8.75 pmol of in vitro transcribed sgRNAs encoding two distinct spacers, and in vitro cleavage reactions were preformed using with the pre-formed RNPs and two distinct corresponding libraries encoding randomized PAMs (RTW554 and RTW555; Addgene plasmid IDs 160132 and 160133, respectively) 47. Cleavage reactions were terminated at time points of 1, 4, and 32 minutes. Approximately 3 ng of digested PAM library for each SpCas9 variant and reaction timepoint was PCR amplified using Q5 polymerase (New England Biolabs; NEB) and barcoded with primers containing sample-specific 4 or 5 nucleotide barcodes (Supplementary Table 3). PCR products were pooled for each time point, purified twice using paramagnetic beads, and amplified with primers containing adapters and the Illumina i5 and i7 indexes (Supplementary Table 3). Libraries were quantified via qPCR using the Universal KAPA Illumina Library qPCR Quantification Kit (KAPA Biosystems) and sequenced on a NextSeq sequencer using either 150-cycle or 75-cycle NextSeq 500/550 High Output v2.5 kits (Illumina) at the Dana-Farber Cancer Institute Molecular Biology Core Facility. Sequencing reads were analyzed as previously described3 using the HT-PAMDA data analysis pipeline available at https://github.com/kleinstiverlab/HT-PAMDA.
Additional analysis of the HT-PAMDA data was performed by clustering the PAM requirements of the characterized variants using the scipy.cluster.hierarchy.linkage() function with ‘optimal_ordering’ parameter set to True, ‘method’ set to ‘average’ and ‘metric’ set to ‘correlation’. Flat clusters were generated from the hierarchical clustering using the scipy.cluster.hierarchy.fcluster() function with maximum cluster number set to 12. Sequence logos for the amino acid composition of HT-PAMDA data clusters were generated using Logomaker85.
Data preprocessing and PAM ML models
All scripts and data in this section are available at https://github.com/RachelSilverstein/PAMmla. The HT-PAMDA-calculated rate constants (ks) for each of the 64 3-nucleotide PAMs were compiled for all SpCas9 PAM variant enzymes obtained from bacterial selections (634 enzymes) and also those chosen randomly from the SpCas9(6AA_NNS) library (135 enzymes) (Supplementary Table 4). ks < 10−5 were set to 10−5 (approximately the detection limit of HT-PAMDA). Log10 ks were normalized to center the mean at zero and standard deviation to one. Amino acid identity at the 6 randomized positions was encoded as either 1) a one-hot encoding or 2) a “Georgiev” numerical descriptor48 (Georgiev encodings were obtained from code by Ofer & Linial86). We also tested the effect of balancing different classes of PAM variants prior to training; enzymes were divided into classes based on their most preferred four-nucleotide PAM and then all classes were randomly over-sampled to match the size of the largest class. Hyperparameters, amino acid encodings, and model architecture for the final model were chosen based on maximizing R2 score in an internal the 5-fold cross validation in the training set. Briefly, the training set was randomly sub-divided into 5 subsets prior to over-sampling and each 1/5 of the data was excluded as a validation set while the remaining 4/5 was subject to over-sampling and then training. A neural network architecture was chosen for the final PAM machine learning algorithm (PAMmla), constructed using the Keras TensorFlow API87. The model consists of three Dense() hidden layers of dimension 512, 256, and 128 with ReLU activation and dropout of 0.2 between each hidden layer. Training was performed for 100 epochs using MSE loss, a batch size of 32, and Adam optimizer with learning rate of 10−4 and decay of 0.001. Final model evaluation was performed on a test set consisting of a random 20% of HT-PAMDA data which was held out from training and model optimization. Code used to preprocess data and train the PAMmla model is available in “AA_to_PAM_NN.ipynb”. Final PAMmla model weights from three different train test splits are available on the github page in the folders “220924_NN_rand_seed0_ROS”, “220924_NN_rand_seed2_ROS”, and “220924_NN_rand_seed3_ROS”. Predictions from these three models can also be generated using the PAMmla web interface https://pammla.streamlit.app/.
We also explored other potential models, including: (1) Linear regression with l2 regularization (Ridge) with alpha=10 using Scikit-learn88, or (2) RandomForestRegressor with max_depth=10, max_features=None, boostrap=True, and max_samples=0.8, also with Scikit-learn88. Hyperparameters and encodings for these models were also chosen by maximizing R2 score via 5-fold internal cross validation. In addition to the simple one-hot and Georgiev encodings used for the neural network model, we also tested additional training features including a one-hot encoding of all-pairwise mutation combinations which was necessary for optimal performance of linear and random forest models. Code used to generate linear and random forest models are available in “AA_to_PAM_linear.ipynb” and “AA_to_PAM_random_forest.ipynb” respectively.
SHapely Additive exPlanations50 (SHAP) values for the final PAMmla model were obtained using DeepSHAP89. A DeepExplainer object was fit on 200 variants sampled from the training dataset. SHAP values were visualized using the summary_plot and force_plot functions. Code used to generate SHAP plots is available in “SHAP_analysis_NN_DeepExplainer.ipynb”.
Visualization of PAMmla predictions
PAM predictions were generated using PAMmla for all 64 million possible combinations of amino acids at each of the 6 randomized library positions, D1135, S1136, G1218, E1219, R1335, and T1337. Predictions were filtered to remove variants with maximum rate constant less than 10−3; 1,890,023 sequences remained after filtering. Filtered variants were then downsampled with diversity preservation using scSampler90 with parameters: fraction = 0.01, random_split = 256. UMAP91 was then fitted to the data with parameters metric=‘correlation’, n_neighbors=10.
Human cell culture and transfections
Human HEK 293T cells (ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated FBS (HI-FBS) and 1% penicillin/streptomycin. The supernatant media from cell cultures was analyzed monthly for the presence of mycoplasma using MycoAlert PLUS (Lonza).
HEK 293T cells were seeded at a density of ~20,000 cells per well in 96-well plates ~20 hours prior to transfections. For nuclease experiments, transfections were performed using 29 ng of nuclease expression plasmid, 12.5ng of sgRNA expression plasmid and 0.3 μL of TransIT-X2 (Mirus) in a total volume of 15 μL Opti-MEM (Thermo Fisher) according to manufacturer instructions. The transfection mixtures were incubated for ~15 minutes at room temperature and distributed across the seeded HEK 293T cells. Base editor experiments with ABE8e-SpCas9 or TadCBEd-SpCas9 expression plasmids were performed using 70 ng of base editor plasmid, 30 ng of sgRNA plasmid, and 0.72 μL of TransIT-X2. Genomic DNA was collected from all transfections after ~72 hours by removing media and resuspending in 100 μL of quick lysis buffer (20 mM Hepes pH 7.5, 100 mM KCl, 5 mM MgCl2, 5% glycerol, 25 mM DTT, 0.1% Triton X-100, and 60 ng/μL Proteinase K (NEB)), heating the lysate for 6 minutes at 65 °C, heating at 98 °C for 2 minutes, as previously described3.
Generation of cell lines harboring pathogenic mutations
We generated various HEK 293T cell lines bearing therapeutically relevant mutations. First, we generated a cell line harboring the RHO P23H mutation via prime editing92. Transfections were performed as described above using HEK 293T cells with 70 ng of prime editor expression plasmid pCMV-T7-PEmax-P2A-EGFP (LM1589; Addgene plasmid ID 223136), 38 ng of pegRNA expression plasmid, 12.5 ng of nicking sgRNA expression plasmid, and 0.79 μL of TransIT-X2 (Mirus) in a total volume of 20 μL Opti-MEM. Cells were grown for approximately 72 hours prior to extracting gDNA to assess editing efficiency in bulk transfected cells (by next-generation sequencing (NGS) as described below, using PCR primers in Supplementary Table 3; see resulting data from pegRNA screen in Supplementary Fig. 17c). To create the cell line, the top-performing PE (LM1589), pegRNA (AHK209), and ngRNA (AHK205) combination was re-transfected into low passage HEK 293T cells. Transfected cells were grown for approximately 72 hours prior to dilution plating into 96-well plates. Wells containing single colonies were identified and grown until confluent, and then transferred into 48-well plates with some cell mass reserved to extract genomic DNA (gDNA) for genotyping via PCR and NGS to verify introduction of RHO P23H.
We also created a HEK 293T cell line encoding the HBB E7V mutation causative of sickle cell disease. Briefly, the E7V cell line was generated similar to as described above via prime editing using 70 ng pCMV-T7-PEmax (Addgene plasmid ID 174820) or pCMV-PEmax-P2A-hMLH1dn (Addgene plasmid ID 174828)76, 38 ng of mpknot or evopreQ1 epegRNA78 expression plasmids, 12.5 ng of nicking sgRNA expression plasmid, and 0.79 μL of TransIT-X2 (Mirus) in a total volume of 20 μL Opti-MEM. Cells were grown for approximately 72 hours prior to extracting gDNA to assess editing efficiency in bulk transfected cells (by next-generation sequencing (NGS) as described below, using PCR primers in Supplementary Table 3; see resulting data from pegRNA screen in Supplementary Figs. 15d). To create the cell line, the top-performing PE (pCMV-PEmax-P2A-hMLH1dn), pegRNA (LLH439), and ngRNA (LLH50) combination was re-transfected into low passage HEK 293T cells. Single cell clones were genotyped via NGS (Supplementary Table 3).
Assessment of nuclease, base editor, and prime editor activities in human cells
Genome editing efficiencies of nucleases and base editors were determined by targeted amplicon sequencing as previously described3. Briefly, a 2-step PCR-based protocol was utilized to construct Illumina-competent NGS libraries (using PCR primers in Supplementary Table 3). On-target genome editing activities were analyzed using CRISPResso293 in pooled mode with the following custom input parameters for nucleases: --min_reads_to_use_region 100 -w 3; for base editors: --min_reads_to_use_region 100 --quantification_window_size 10 --quantification_window_center -10 --base_editor_output --min_frequency_alleles_around_cut_to_plot 0.05; and for prime editors: --min_reads_to_use_region 100 -w 10.
Base editing of patient-derived B cell lines
Epstein Barr virus transformed B cell lines (BCLs) were established from a patient with Chronic Granulomatous Disease harboring the CYBB T362I mutation as previously described94,95 (the patient was consented via NIH protocol 05-I-0213). BCLs were maintained in RPMI + 10% fetal bovine serum. SpCas9 sgRNAs targeting the CYBB T362I mutation (spacer sequence in Supplementary Table 5) were synthesized (Synthego). mRNAs encoding the ABE8e-KWRQLC and ABE8e-SpG were produced by in vitro transcription incorporating 100% substitution of the UTP content with pseudoUTP (CELLSCRIPT™). mRNAs were post-translationally capped to >95% and poly(A) tailed to >200 A’s and subsequently purified for removal of double-stranded RNA content (CELLSCRIPT™). Base editing experiments were performed in BCLs by electroporation (EP) (MaxCyte ATx, Program BCL#3) to deliver the ABE mRNA and synthetic sgRNA (Synthego). BCLs were washed with EP buffer (MaxCyte) and resuspended at ~2x107 cells/mL EP buffer. Approximately 0.25-0.5 x106 BCLs per sample were combined with base editor mRNA (~0.04 μg/μL final concentration), sgRNA (~0.192 μg/μL final concentration), and ScriptGuard RNase inhibitor (1.6 U/μL final concentration; CELLSCRIPT™) in a volume of 25 μL. After EP, cells were transferred to 12-well tissue culture plate and cultured at 0.5-1.0x106/mL for a further two days before harvesting genomic DNA (DNeasy kit; Qiagen) for analysis of editing by targeted sequencing.
Specificity assessment using GUIDE-seq-2
Approximately 20,000 HEK 293T cells were seeded per well in 96-well plates ~ 20 hours prior to transfection, performed using 29 ng of nuclease expression plasmid, 12.5 ng of sgRNA expression plasmid, 1 pmol of the GUIDE-seq double-stranded oligodeoxynucleotide tag (dsODN; oSQT685/686)96, and 0.3 μL of TransIT-X2 (Mirus). Genomic DNA was extracted ~72 hours post transfection using the DNAdvance Kit (Beckman Coulter) according to manufacturer’s instructions, and then quantified by Qubit (Thermo Fisher). On-target dsODN integration was assessed by PCR amplification, library preparation, and next-generation sequencing as described above, with data analysis via CRISPResso293 run in non-pooled mode by supplying the target site spacer, the reference amplicon, and both the forward and reverse dsODN-containing amplicons as ‘HDR’ alleles with custom parameters: -w 25 -g GUIDE --plot_window_size 50. The fraction of alleles bearing an integrated dsODN was calculated as the number of reads mapped to the forward dsODN amplicon plus the number of reads mapped to the reverse dsODN amplicon divided by the sum of the total reads mapped to all three amplicons.
GUIDE-seq-2 reactions were performed essentially as described55 with minor modifications. Briefly, the Tn5 transposase was prepared by combining 36 μL hyperactive Tn5 (1.85 mg/mL, purified as previously described97), 15 μL annealed i5 adapter oligos encoding 8 nucleotide (nt) barcodes and 10-nt unique molecular indexes (UMIs) (Supplementary Table 3), with 52 μL 2x Tn5 dialysis buffer (100 mM HEPES-KOH pH 7.2, 200 mM NaCl, 0.2 mM EDTA, 2 mM DTT, 0.2% Triton X-100, and 20% glycerol) for 60 minutes at 24 °C. Tagmentation reactions were performed in 40 μL reactions for 7 minutes at 55 °C, containing approximately 250 ng of genomic DNA, 8 μL of the assembled Tn5/i5 -transposome, and 8 μL of freshly prepared 5x TAPS-DMF buffer (50 mM TAPS-NaOH, 25 mM MgCl2, and 50% dimethylformamide (DMF)). Tagmentation reactions were halted using 5 μL of a 50% proteinase K (NEB) solution (mixed with H2O) with incubation at 55 °C for 15 minutes, purified using SPRI-guanidine magnetic beads, and analyzed via TapeStation with High Sensitivity D5000 tapes (Agilent). Separate PCR reactions were performed using dsODN sense- and antisense-specific primers (Supplementary Table 3) using Platinum Taq (Thermo Fisher), with a thermocycler program of 95 °C for 5 minutes, followed by 15 cycles of temperature cycling (95 °C for 30 s, 70 °C (−1 °C per cycle) for 120 s, and 72 °C for 30 s), 20 constant cycles (95 °C for 30 s, 55 °C for 60 s, and 72 °C for 30 s), an a final extension at 72 °C for 5 minutes. PCR products were purified using SPRI beads and analyzed via QIAxcel (Qiagen) prior to sample pooling to form single sense- and antisense-libraries. Libraries were purified using the Pippin Prep (Sage Science) DNA size selection system to achieve a size range of 250-500 base pairs. Sense- and antisense-libraries were quantified using Qubit (Thermo Fisher) and pooled in equal amounts to achieve a final concentration of 2 nM. The library was sequenced using NextSeq1000/2000 P3 kit (Illumina) with cycle settings of 146, 8, 18, 146. Demultiplexed sequencing reads were down sampled to ensure equal numbers of reads for samples being compared using the same sgRNA. Data analysis was performed using an updated version of the open-source GUIDE-seq-2 analysis software98 (https://github.com/tsailabSJ/guideseq/tree/V2) with max_mismatches parameter set to 6 (GUIDE-seq-2 data is available in Supplementary Table 6).
Homology modeling of protein structures
Amino acid substitutions in SpCas9-derived PAMmla predicted enzymes were homology modeled in Coot (v0.9.8.93)99 using the mutate and rotamer selection functions. Most amino acid and PAM DNA base substitutions were modeled using the structure for SpG (PDB: 8U3Y)6, except T1337R, T1337K, and T1337C substitutions or the MRRWMR enzyme variant, which were modeled using the structure for VRER (PDB: 5FW3)100. Homology models were visualized using ChimeraX (v1.8)101.
In silico directed evolution
In an ISDE campaign, a starting sequence is computationally mutated to generate a small sub-library of ~1,000-100,000 sequences bearing random amino acid substitutions with a defined hamming distance from the original sequence. PAM predictions are then generated for each member of the sub-library using PAMmla. A customizable fitness function is used to score each variant according to the desired properties, and a selection step is performed where the most “fit” enzymes are isolated according to the chosen fitness metric. The resulting ISDE enzymes are then used as starting sequences for subsequent rounds of evolution, iterating the process until the fitness function plateaus. For NGTG-targeting RHO variants, ISDE was performed using a custom python script “evolve_vars.py” (https://github.com/RachelSilverstein/PAMmla). Evolution was initiated from the starting amino acids D1135, S1136, G1218, E1219, R1335, and T1337 (wild-type SpCas9) with the following custom parameters; starting mutations per variant: 4; variants per round of evolution: 1000; n best variants to keep after each round: 10; decay mutation rate after n rounds plateau: 3; PAM to maximize: NGTG; and additional PAM cutoffs of NGGG < −3.7 (for MRRWMR trajectory) or NGGG < −4 (for KRHWMR trajectory). ISDE functionality is also implemented on the PAMmla web app: https://pammla.streamlit.app/Evolve_Variants.
Sub-retinal injections, in vivo electroporation, and retinal cell collection
A DNA solution containing 1.6-2 μg of pCBh-Cas9-P2A-mTagBFP2 plasmids that express Cas9 PAM variant enzymes (WT SpCas9, RAS3575; SpG, RAS3583; SpCas9-MRRWMR, RAS3579; SpCas9-KRHWMR, RAS3594), and 0.8-1 μg sgRNA plasmids (AHK383) were injected into the subretinal space of neonatal pups (P0-P2) using established methods63. Briefly, pups were anesthetized by hypothermia. The fused upper and lower eyelids were separated. A small incision was made at the limbus using a 30-gauge needle, and 0.5 μl of plasmid DNA mix was injected into the sub-retinal space of right eye through the limbal incision using a Hamilton syringe with a 33-gauge blunt-ended needle. Left eyes were used as a negative control. The injected DNA plasmid was electroporated into retinal cells using a 7 mm diameter tweezer-type electrode (Model 520, BTX-Harvard Apparatus, Holliston, MA), and the electroporation parameters were set at five 90 V square pulses, 50 ms duration with 950 ms intervals (ECM830, BTX).
Two-three weeks post injection, the mice were euthanized. The retinas were dissected out through a corneal incision, placed into a drop of BGJB culture medium (ThermoFisher, # 12591038) in a petri dish, and examined for BFP expression under a fluorescent microscope. For dissociation, retinas were transferred into a tube containing 400 μl of solution (1mg/ml pronase and 2mM EGTA in BGJB medium) and incubated at 37 °C for 30 minutes. Retinal samples were broken into single cells by pipetting up and down 20 times. Another 400 μl solution containing 100 u/mL DNase I, 0.5% BSA, 2mM EGTA in BGJB medium was added and incubated at room temperature for 10 minutes. Cell suspensions were filtered through a cell strainer (Falcon, #352235) and sorted for BFP positive cells using MA900 Multi-Application Cell Sorter. Transfection efficiency was ~0.1–3%, measured by the percentage of BFP+ cells compared to all retinal cells. Only samples with more than 1,000 BFP+ sorted cells were used for analysis. Collected BFP+ cells were spun down at 15,000 x g for 15 minutes. Genomic DNA was extracted from cell pellet using QuickExtract (Biosearch Technologies, #SS000773), incubated at 60 °C overnight, and heat inactivated at 98 °C for 3 minutes. The humanized Rho P23H region was amplified using primers oRAS1384/oRAS1385, and then sequenced and analyzed as described above for human cell samples.
Natural sequence models analysis
Five iterations of jackhmmer with bitscore 0.9 querying the UniRef100 database with the wildtype spCas9 sequence was performed to build a multiple sequence alignment. Any sequence that had more than 30% gaps in the alignment was excluded, and any columns in the alignment with more than 30% gaps were also excluded. A theta reweighting of 0.8 was used such that anything more than 80% similarity is down weighted relative to other sequences. This alignment was used to train an EVCouplings73 model, and the delta hamiltonians from this model were used as the sequence scores to approximate protein fitness. Finally, a hybrid LLM and variational autoencoder (VAE) called TranceptEVE72 was used to score sequences. To score the full Cas9 sequence, the sliding window method of length 1024 was used. The Large Tranception model was used for the LLM component and four separately trained EVE models71 were averaged for the VAE component. The EVE models were trained with the same alignment used for EVCouplings with the same sequence and column coverage cutoffs and theta reweighting parameter.
Extended Data
Extended Data Fig. 1 |. Targeting range and characterization of previous engineered SpCas9 PAM variant enzymes.

(a) Quantification of pathogenic and likely pathogenic single nucleotide variants (SNVs) from ClinVar102 that are theoretically revertible using ABE or CBE based on their proximity to an NGG PAM. SNVs were considered editable if a GG dinucleotide PAM was available at the appropriate distance upstream on the correct DNA strand, positioning the SNV anywhere between positions 5-9 of the spacer sequence (counting from the PAM-distal end of the spacer; typically called the ‘edit window’ of base editors). (b-d) Heatmap representations of the PAM profiles of SpCas9 enzymes determined using the HT-PAMDA assay3,47, for wild-type SpCas9 (panel b), for enzymes with altered PAM requirements (e.g. SpCas9-VRQR14,43, SpCas9-VRER14, and xCas92; panel c), and for enzymes with relaxed PAM requirements (e.g. SpCas9-NG1, SpG and SpRY3, and SpCas9-NRRH/NRCH/NRTH4); panel d). The log10 rate constants (k) are the mean of n = 2 replicate HT-PAMDA experiments performed using two distinct spacer sequences. Because the HT-PAMDA assay measures the relative depletion of substrates encoding various PAMs, it may underestimate rate constants for enzymes with highly relaxed PAM requirements such as SpRY and Cas9-NRRH.
Extended Data Fig. 2 |. Structure-informed saturation mutagenesis and bacterial positive selections for SpCas9 PAM variant enzymes.

(a) Structural representation of the PAM-interacting (PI) domain of SpCas9 showing amino acid residues interacting with a canonical NGG PAM (from PDB ID: 4UN310). (b) Schematic of the bacterial positive selection assay. A plasmid encoding the SpCas9(6AA) library (with randomized NNS codons at SpCas9 positions D1135, S1136, G1218, E1219, R1335, and T1337), a sgRNA expression cassette, and chloramphenicol resistance gene is transfected into an E. coli strain harboring a selection plasmid encoding an inducible toxic gene and the Cas9 target site (with protospacer adjacent to a non-canonical 4 nt PAM of interest). Selections were performed similar to previously described14,45,75, where the ccdB gene (encoding a DNA gyrase toxin) on the selection plasmid is induced by plating on arabinose-containing media. Bacterial colonies survive the selection when they harbor a plasmid that expresses an SpCas9 enzyme variant capable of cleaving the selection plasmid (by recognizing a non-canonical PAM). (c) Summary of the SpCas9 enzymes that survived the bacterial positive selections using selection plasmids encoding each of the 16 NGNN PAMs. The heatmaps depict the percent of SpCas9 enzymes from each of the 16x selections that contain each possible amino acid substitution at each of the six SpCas9(6AA) library positions. Each heatmap is labeled based on the PAM utilized in that set of bacterial selections; the number of enzymes selected from each set of selections is indicated. The bottom panel represents a summary of the composition of amino acid residues at each of the six positions of the SpCas9(6AA) library.
Extended Data Fig. 3 |. Machine learning models to predict PAM profile from amino acid sequence.

a, Comparison of machine learning model architectures (linear regression, random forest, and neural network) and amino acid encodings (one-hot, one-hot plus all pairwise amino acid combinations, and Georgiev48). The R2 value is shown between the experimentally determined k (via HT-PAMDA) and the predicted k (via each ML model) for an internal 5-fold cross-validation on the training set. Each validation set is sub-divided according to the minimum hamming distance (HD) of each variant to the nearest neighbor in the corresponding training set; thus, validation sets become more challenging as HD increases. b, Performance of the optimal PAM machine learning algorithm (PAMmla; comprised of a neural network with one hot encoding) on two additional 80%/20% random train-test splits. c, Proportion of test set SpCas9 enzymes that have a predominant preference for A, C, G, T in the 3rd position of the PAM, or are inactive (based on HT-PAMDA data). d, Comparison of test set ks broken down by nucleotide preference of each test variant at the 3rd position of the PAM (comparing ks experimentally determined by HT-PAMDA versus predicted by PAMmla). Nucleotide preference is defined as the 3rd position nucleotide of each enzyme variant’s most preferred PAM by HT-PAMDA. e, Proportion of test set SpCas9 enzymes that have a predominant preference for A, C, G, T in the 4th position of the PAM, or are inactive (based on HT-PAMDA data). f, Comparison of test set ks broken down by preference of each test set variant at the 4th position of the PAM (comparing ks experimentally determined by HT-PAMDA versus predicted by PAMmla). Nucleotide preference is defined as 4th position nucleotide of each enzyme variant’s most preferred PAM by HT-PAMDA. g, Effect of random over-sampling by most active PAM. The PAMmla model was trained with and without randomly over-sampling the training set to balance the number of enzyme variants with different PAM preferences. R2 values for the two models were compared on subsets of variants within the test set with different preferences at the 3rd and 4th positions of the PAM. Over-sampling improved performance particularly for under-represented PAM classes (see panels c and e). h, Pearson’s correlations between HT-PAMDA replicates performed with distinct spacer sequences for a set of 28 inactive versus 28 active enzymes within the test set. Dashed line = data median. True labels for active versus inactive enzymes were determined using a cutoff value for maximum k on any PAM of 10−4.3. Enzymes separated into active and inactive classes based on these criteria showed correlation between replicates only for active enzymes, indicating HT-PAMDA data for enzymes with maximum ks below this cutoff are likely due to non-reproducible noise in the HT-PAMDA assay. i, Correlation between ks experimentally determined by HT-PAMDA versus predicted by PAMmla for inactive variants (maximum HT-PAMDA k < 10−4.3) within the test set; PAMmla is not predictive for background noise in the HT-PAMDA determined PAM profiles of inactive enzymes. For all panels that utilize HT-PAMDA data, the log10 rate constants (k) are the mean of n = 2 replicate HT-PAMDA experiments using two distinct spacer sequences. For all scatterplots, each datapoint represents the rate constant activity of one enzyme variant against on one of 64 possible NNNN PAMs.
Extended Data Fig. 4 |. PAMmla feature importance for enzymes targeting different PAM classes.

SHapely Additive exPlanations (SHAP)50 analysis to investigate the impact of amino acid substitutions (i.e. PAMmla features) on model output for each of the 16 NGNN PAMs. SHAP values are shown for 200 enzymes sampled from the training set. Top 10 features with highest mean absolute SHAP values (greatest absolute impact on model output) are plotted for each PAM.
Extended Data Fig. 5 |. Homology models of PAMmla predicted PAM-altering mutations.
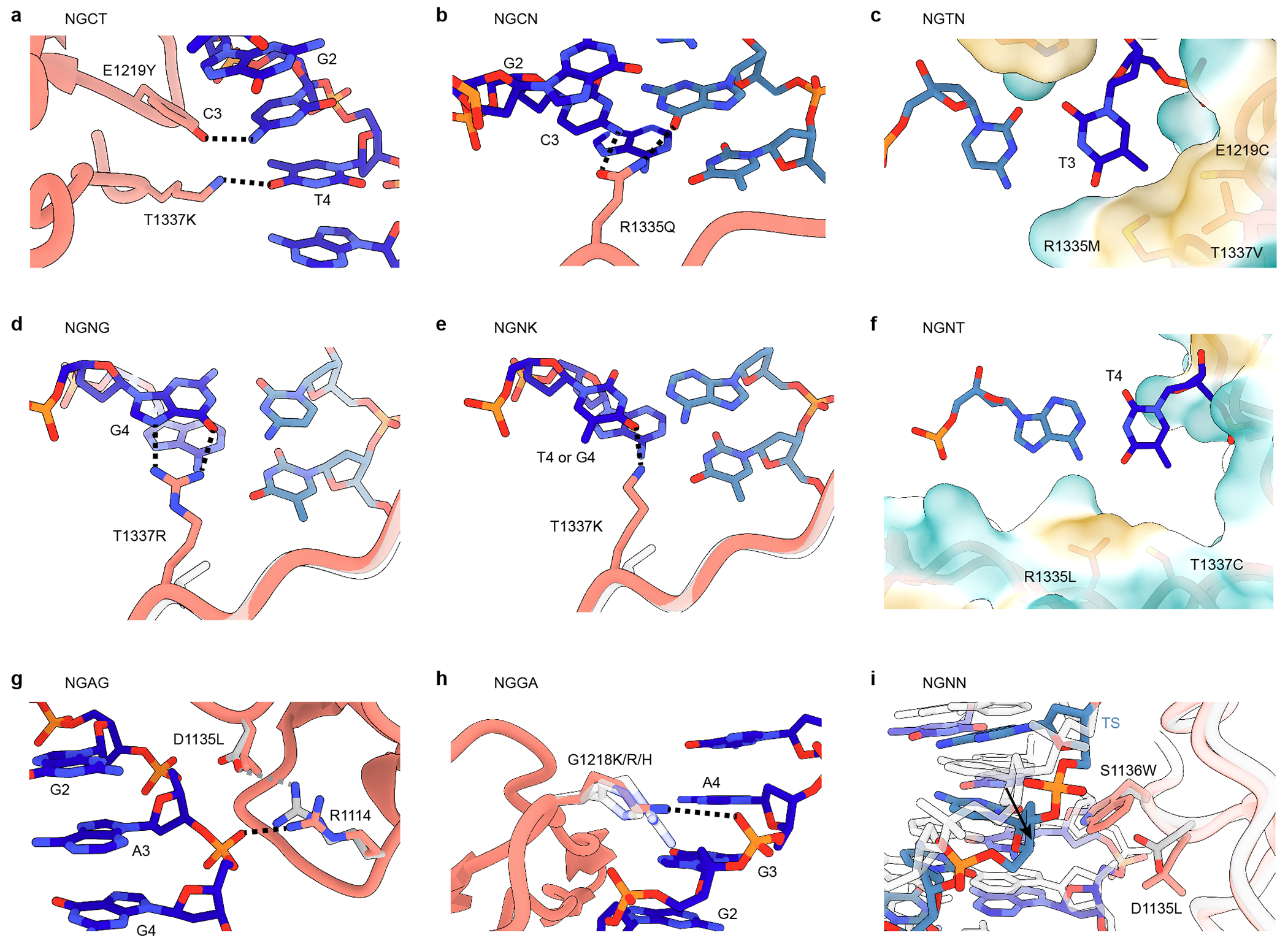
a, An E1219Y substitution may facilitate interaction with the amino group of bases in the 3rd position of the PAM. b, R1335Q permits major groove readout of both bases of a C-G pair in the 3rd position of the PAM. c, E1219C, R1335M, and T1337V substitutions form a hydrophobic pocket to promote van der Waals interactions with the methyl group of thymine in the 3rd position of the PAM. Representation of the protein surface is colored by lipophilicity potential. d, T1337R results in direct major groove readout of guanine in the 4th position of the PAM. e, T1337K facilitates major groove readout of oxygen group of bases in the 4th position the PAM. f, R1335L and T1337C substitutions form a hydrophobic pocket to promote recognition of thymine in the 4th position of the PAM. Protein surface is colored by lipophilicity potential. g, D1135L disrupts coordination with R1114, enabling improved flexibility of the R1114 side chain to contact the NTS backbone. WT SpCas9 is overlaid in grey. h, Substitution of G1218 to a positive residue establishes additional non-specific contacts with the NTS backbone. i, S1136W and D1135L result in a shift of the NTS and TS backbone towards the PAM-interacting domain, enabling novel base specific interactions in nearby regions. WT SpCas9 is overlaid in grey. For panels a-i, amino acid and PAM DNA base substitutions were modeled on the structure of SpG (PDB: 8U3Y)6 using Coot107, except for substitutions T1337R, T1337K, and T1337C which were modeled using SpCas9-VRER (PDB: 5FW3)52. Homology models were visualized using ChimeraX104.
Extended Data Fig. 6 |. Genome editing in human cells with PAMmla-predicted enzymes.
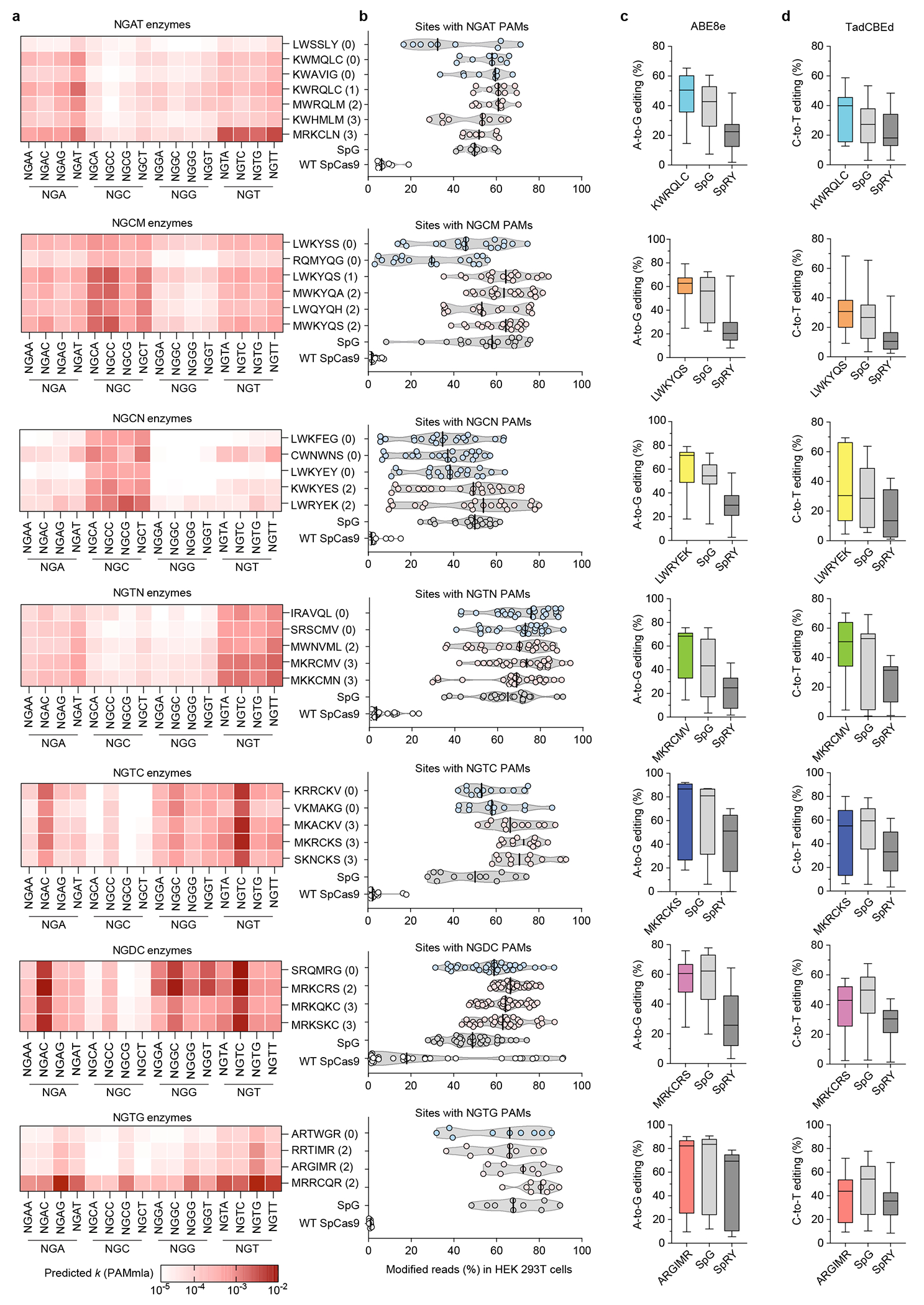
(a) PAMmla predicted ks for NGNN PAMs for enzymes targeting seven PAM categories. Hamming distances to the most similar enzyme in the training set are indicated in parentheses for each enzyme. (b) Nuclease-mediated genome editing efficiencies for each of the enzymes in panel a at endogenous target sites in HEK 293T cells harboring the PAMs they are predicted to target by PAMmla. Editing efficiencies were assessed by targeted amplicon sequencing and analyzed using CRISPResso2; data points are the mean of n = 3 biological replicates for enzymes from the training set (hamming distance = 0, shown with blue dots), enzymes predicted by PAMmla (shown in pink), SpG (gray), and wild-type (WT) SpCas9 (white); 3 to 10 genomic target sites were selected for characterization, where the black line represents median editing across all target sites for that enzyme; results at individual loci are shown in Supplementary Figs. 12a–g. (c,d) Base editing efficiencies for one PAMmla enzyme compared to SpG and SpRY, in the context of ABE8e and TadCBEd architectures (panels c and d, respectively). Base editing efficiencies were assessed by targeted amplicon sequencing for each enzyme at 3 endogenous target sites in HEK 293T cells; all edits at bases where any enzyme was observed to edit >5% efficiency are shown; Box minima, center and maxima represent data 25th, 50th, and 75th percentiles respectively; whiskers represent the range of the data. A-to-G and C-to-T base editing results at individual loci are shown in Supplementary Figs. 13a–g and Supplementary Figs. 14a–g, respectively.
Extended Data Fig. 7 |. Genome-wide off-target analysis of PAMmla predicted enzymes.

a, Quantification of GUIDE-seq2 double-stranded oligodeoxynucleotide (dsODN) tag integration at the on-target site, in nuclease-based experiments with SpG, SpRY, and PAMmla predicted enzymes targeting endogenous target sites in HEK 293T cells. SpCas9 variant enzymes are named based on their amino acids at each of the six positions in the SpCas9(6AA) library. dsODN integration efficiency was assessed by targeted amplicon sequencing and modified reads were analyzed using CRISPResso2; mean, standard deviation, and individual datapoints shown for n = 3 technical replicates. b, Venn diagram representations of the GUIDE-seq-2 detected off-target sites that are shared between or unique to PAMmla generated, SpG, and SpRY nucleases. c, Nucleotide composition of PAMs adjacent to off-target spacers detected in GUIDE-seq-2 experiments, not including the on-target reads. The y-axis represents the fraction of total off-target GUIDE-seq-2 reads containing each nucleotide at each position of the PAM. d, Quantification of GUIDE-seq-2 double-stranded oligodeoxynucleotide (dsODN) tag integration at the on-target site, in nuclease-based experiments with KWRQLC and SpG when using the CYBB T362I sgRNA but targeting the wild-type genome of HEK 293T cells. SpCas9 variant enzymes are named based on their amino acids at each of the six positions in the SpCas9(6AA) library. dsODN integration efficiency was assessed by targeted amplicon sequencing and modified reads were analyzed using CRISPResso2; mean, standard deviation, and individual datapoints are shown for n = 3 technical replicates. e, GUIDE-seq-2 genome-wide specificity outputs for KWRQLC and SpG nucleases using the CYBB T362I targeted sgRNA; note that HEK 293T cells harbor the wild-type copy of the CYBB gene and are therefore an imperfect match to the sgRNA. Mismatched positions in the spacers of the off-target sites are highlighted in color; GUIDE-seq read counts from consolidated unique molecular events for each variant are shown to the right of the sequence plots.
Extended Data Fig. 8 |. Design and validation of in silico directed evolution.

a, Schematic of in silico directed evolution (ISDE) pipeline to rapidly identify bespoke SpCas9 enzymes with user-specifiable PAM profiles. b-d, Effect of ISDE parameter values on the identification of optimized PAMmla predicted enzymes, including varying the number of starting mutations per round (m) (panel b), random variants generated per round (panel c) and number of additional evolution rounds performed once a plateau is reached before decreasing m (panel d). Proof-of-concept PAMmla-ISDE runs were performed to identify enzymes with maximal activity against NGAT, NGCC, or NGTA PAMs. Aside from the parameter being tested, ISDE was run with default parameters of 1,000 random starting sequences, m = 4 starting mutations per enzyme, s = 1,000 sampled enzymes per round, n = 10 top variants to keep per round, and p = 1 additional round of evolution after a plateau is reached. The number of true top 10 predicted enzymes, determined by exhaustive sorting of PAMmla predictions, recovered by ISDE are shown. Top bar graphs represent the number of replicates in which the most optimal enzyme was recovered.
Extended Data Fig. 9 |. Characterization of PAMmla-ISDE generated enzymes in human cells.

a, Nuclease-mediated genome editing at endogenous target sites in HEK 293T cells harboring different PAMs for wild-type (WT) SpCas9, SpG, and MRRWMR. b, Nuclease-mediated genome editing of the wild-type RHO or mutant RHO P23H alleles in a heterozygous RHO P23H HEK 293T cell line using wild-type SpCas9, SpG, and various PAMmla generated enzymes. For reads containing indels that span the P23H mutation (and therefore could not be identified as WT or mutant), counts were distributed between WT and mutant alleles with the same ratio as WT:mutant ratio observed for the identifiable edited reads. c, Nuclease-mediated genome editing of the RHO target site in wild-type HEK 293T cells using wild-type SpCas9, SpG, and various PAMmla generated enzymes. d, Unidentifiable sequencing reads that were either P23H or WT due to deletions spanning the mutation for data shown in heterozygous P23H HEK 293T cells from data in Fig. 5f; edited reads were distributed based on the balance in identifiable reads. e, Ratio of editing efficiencies observed on mutant (P23H) versus WT RHO alleles, for each editor tested in Fig. 5f. Editing efficiencies in panels a-c,e were assessed by targeted amplicon sequencing and modified reads were analyzed using CRISPResso2; mean, standard deviation, and individual datapoints shown for n = 3 independent biological replicates.
Extended Data Fig. 10 |. Specificity assessment of PAMmla-derived enzymes.

a, Quantification of GUIDE-seq2 double-stranded oligodeoxynucleotide (dsODN) tag integration at on-target sites in nuclease-based experiments with MRRWMR, SpG, and SpRY and sgRNAs targeting two different endogenous sites in HEK 293T cells. SpCas9 variant enzymes are named based on their amino acids at each of the six positions in the SpCas9(6AA) library. dsODN integration efficiency was assessed by targeted amplicon sequencing and modified reads were analyzed using CRISPResso2; mean, standard deviation, and individual datapoints are shown for n = 3 technical replicates. b, Venn diagram representations of the GUIDE-seq-2 detected off-target sites that are shared between or unique to MRRWMR, SpG, and SpRY nucleases using the two sgRNAs targeted to sites with NGTG PAMs (similar to the RHO P23H on-target site). c, Fraction of GUIDE-seq-2 reads attributed to on- and off-target sites for MRRWMR, SpG, and SpRY from experiments using the NGTG-2 or NGTG-3 sgRNAs. d, Quantification of GUIDE-seq-2 dsODN tag integration at the on-target site for experiments in the homozygous RHO P23H cell line, when using the RHO P23H sgRNA and SpCas9-MRRWMR and -KRHWMR, SpG, and SpRY expression plasmids. dsODN integration efficiency was assessed by targeted amplicon sequencing and modified reads were analyzed using CRISPResso2; mean, standard deviation, and individual datapoints are shown for n = 3 technical replicates. e, GUIDE-seq-2 genome-wide specificity outputs for SpCas9-MRRWMR and -KRHWMR, SpG, and SpRY nucleases using the RHO P23H targeted sgRNA in homozygous RHO P23H HEK 293T cells. Mismatched positions in the spacers of the off-target sites are highlighted in color; GUIDE-seq read counts from consolidated unique molecular events for each variant are shown to the right of the sequence plots. f, Venn diagram representation of the GUIDE-seq-2 detected off-target sites that are shared between or unique to SpCas9-MRRWMR and -KRHWMR, SpG, and SpRY nucleases using the RHO P23H sgRNA. g, Unidentifiable sequencing reads unattributable to either WT or P23H alleles due to deletions spanning the base harboring the mutation, for data from heterozygous RHO P23H mice shown in Figs. 5l, h, Ratio of in vivo editing efficiencies observed on mutant (P23H) versus WT RHO alleles, for each SpCas9 nuclease tested in Fig. 5l.
Extended Data Fig. 11 |. Analysis of factors contributing to MRRWMR and KRHWMR PAM preferences.

a, Structural prediction of an alternative conformation of the S1136R mutation leading to additional hydrogen bonding with T at position 3 of the PAM. b-e, SHapely Additive exPlanations50 (SHAP) values for PAMmla predictions for MRRWMR (panels b,c) and KRHWMR (panels d,e) interacting with NGTG (panels b,d) or NGGG PAMs (panels c,e) PAMs. Feature values are shown in gray (1: mutation is present, 0: mutation is absent). Red represents features with positive impact on predicted rate constant and blue represent features with negative impact on predicted rate constant.
Supplementary Material
Acknowledgements
We thank L. Ma, E. Oliver, M. Prew, and M. Welch for assistance with plasmid cloning; J. Lemanski, M. Talkowski, and the Genomics and Technology Core in the Center for Genomic Medicine at MGH for technical support with the TapeStation; R. Mouro Pinto for access to and assistance with the Pippin Prep; J. Zhong and M. Suva for access to and assistance with the NextSeq2000; P. Chatterjee and G. Church for discussions; M. Ma at the NIH for technical assistance; Z. Hebert and M. Berkeley at the Dana-Farber Cancer Institute Molecular Biology Core Facilities for support with NextSeq500 sequencing; and W. Wang from St. Jude Protein Production Core Facility for recombinant Tn5. We acknowledge funding from Natural Sciences and Engineering Research Council of Canada (NSERC) Postgraduate Scholarship-Doctoral (PGS D – 567791 to R.A.S.), a Chan Zuckerberg Initiative Award (Neurodegeneration Challenge Network, CZI2018-191853; to D.S.M.) a Massachusetts General Hospital (MGH) Executive Committee on Research (ECOR) Fund for Medical Discovery Fundamental Research Fellowship Award (K.A.C.), Peter und Traudl Engelhorn Stiftung (M.P.), an MGH Research Scholar Award 2024-2029 (L.P.), the Fighting Blindness Foundation (Q.L.), an MGH ECOR Howard M. Goodman Fellowship (B.P.K.), the Kayden-Lambert MGH Research Scholar Award 2023-2028 (B.P.K.), the Gilbert Family Foundation’s Gene Therapy Initiative Grant No. 521004 (B.P.K.), and National Institutes of Health (NIH) grants TR01CA260415 (D.S.M.), U01AI176470 (S.Q.T.), R35HG010717 (L.P.), UM1HG012010 (L.P and B.P.K.), R01EY033107 (Q.L.), P30EY014104 (MEE core support), DP2CA281401 (B.P.K.), and P01HL142494 (B.P.K.).
Footnotes
Competing Interests
R.A.S. and B.P.K. are inventors on a patent application filed by Mass General Brigham (MGB) that describes the development of PAMmla. B.P.K. and R.T.W. are inventors on additional patents or patent applications filed by MGB that describe genome engineering technologies related to the current study. S.Q.T. is an inventor on a patent application for GUIDE-seq. S.Q.T. is a member of the scientific advisory boards of Ensoma and Prime Medicine. L.P. has financial interests in Edilytics and SeQure Dx. Q.L. is a consultant for Entrada Therapeutics. B.P.K. is a consultant for EcoR1 capital, Novartis Venture Fund, and Jumble Therapeutics, and is on the scientific advisory boards of Acrigen Biosciences, Life Edit Therapeutics, and Prime Medicine. B.P.K. has a financial interest in Prime Medicine, Inc., a company developing therapeutic CRISPR-Cas technologies for gene editing. L.P. and B.P.K.’s interests were reviewed and are managed by MGH and MGB in accordance with their conflict-of-interest policies. The other authors declare no competing interests.
Data Availability
Primary datasets for this study are available in Supplementary Table 1 (HT-PAMDA data), Supplementary Table 2 (PAMmla predictions), Supplementary Table 6 (GUIDE-seq-2 data), and Supplementary Table 7 (source data). The HT-PAMDA training datasets are also available on GitHub at https://github.com/RachelSilverstein/PAMmla. Next-generation sequencing results are available through the NCBI sequence read archive (SRA) under PRJNA1169103. PAMmla predictions for all 64 million SpCas9(6AA) enzymes can be viewed through an online webtool (https://pammla.streamlit.app/). The UniRef100 dataset used to generate multiple sequence alignments for natural sequence models can be downloaded at https://www.uniprot.org/uniref/.
Code Availability
Custom scripts, the PAMmla source code, and the in silico directed evolution code are available on GitHub at https://github.com/RachelSilverstein/PAMmla and https://github.com/RachelSilverstein/multiplex_seq_analysis.
References
- 1.Nishimasu H et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science (1979) (2018) doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature (2018) doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walton RT, Christie KA, Whittaker MN & Kleinstiver BP Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science (1979) (2020) doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller SM et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat Biotechnol (2020) doi: 10.1038/s41587-020-0412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang W et al. In-depth assessment of the PAM compatibility and editing activities of Cas9 variants. Nucleic Acids Res 49, 8785–8795 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hibshman GN et al. Unraveling the mechanisms of PAMless DNA interrogation by SpRY Cas9. bioRxiv 2023.06.22.546082 (2023) doi: 10.1101/2023.06.22.546082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mojica FJM, Díez-Villaseñor C, García-Martínez J & Almendros C Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology (N Y) 155, 733–740 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Liu G, Lin Q, Jin S & Gao C The CRISPR-Cas toolbox and gene editing technologies. Mol Cell 82, 333–347 (2022). [DOI] [PubMed] [Google Scholar]
- 9.Pacesa M, Pelea O & Jinek M Past, present, and future of CRISPR genome editing technologies. Cell 187, 1076–1100 (2024). [DOI] [PubMed] [Google Scholar]
- 10.Anders C, Niewoehner O, Duerst A & Jinek M Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature (2014) doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sternberg SH, Redding S, Jinek M, Greene EC & Doudna JA DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature (2014) doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (1979) (2012) doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang W, Bikard D, Cox D, Zhang F & Marraffini LA RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology 2013 31:3 31, 233–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleinstiver BP et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature (2015) doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldberg GW et al. Engineered dual selection for directed evolution of SpCas9 PAM specificity. Nature Communications 2021 12:1 12, 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chatterjee P et al. A Cas9 with PAM recognition for adenine dinucleotides. Nature Communications 2020 11:1 11, 1–6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao L et al. PAM-flexible genome editing with an engineered chimeric Cas9. Nature Communications 2023 14:1 14, 1–8 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chatterjee P et al. An engineered ScCas9 with broad PAM range and high specificity and activity. Nature Biotechnology 2020 38:10 38, 1154–1158 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Kleinstiver BP et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 33, 1293–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang TP et al. High-throughput continuous evolution of compact Cas9 variants targeting single-nucleotide-pyrimidine PAMs. Nature Biotechnology 2022 41:1 41, 96–107 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y et al. Genome-wide analyses of PAM-relaxed Cas9 genome editors reveal substantial off-target effects by ABE8e in rice. Plant Biotechnol J 20, 1670–1682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi H et al. Rapid two-step target capture ensures efficient CRISPR-Cas9-guided genome editing. bioRxiv 2024.10.01.616117 (2024) doi: 10.1101/2024.10.01.616117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang KK, Wu Z & Arnold FH Machine-learning-guided directed evolution for protein engineering. Nature Methods Preprint at 10.1038/s41592-019-0496-6 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Wu Z, Jennifer Kan SB, Lewis RD, Wittmann BJ & Arnold FH Machine learning-assisted directed protein evolution with combinatorial libraries. Proc Natl Acad Sci U S A 116, 8852–8858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wittmann BJ, Yue Y & Arnold FH Informed training set design enables efficient machine learning-assisted directed protein evolution. Cell Syst (2021) doi: 10.1016/j.cels.2021.07.008. [DOI] [PubMed] [Google Scholar]
- 26.Thean DGL et al. Machine learning-coupled combinatorial mutagenesis enables resource-efficient engineering of CRISPR-Cas9 genome editor activities. Nature Communications 2022 13:1 13, 1–14 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hie BL & Yang KK Adaptive machine learning for protein engineering. Current Opinion in Structural Biology Preprint at 10.1016/j.sbi.2021.11.002 (2022). [DOI] [PubMed] [Google Scholar]
- 28.Yang KK, Wu Z & Arnold FH Machine learning in protein engineering Kevin. Nat Methods (2019). [DOI] [PubMed] [Google Scholar]
- 29.Wittmann BJ, Johnston KE, Wu Z & Arnold FH Advances in machine learning for directed evolution. Current Opinion in Structural Biology Preprint at 10.1016/j.sbi.2021.01.008 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Wu Z, Johnston KE, Arnold FH & Yang KK Protein sequence design with deep generative models. Current Opinion in Chemical Biology Preprint at 10.1016/j.cbpa.2021.04.004 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Biswas S, Khimulya G, Alley EC, Esvelt KM & Church GM Low-N protein engineering with data-efficient deep learning. Nature Methods 2021 18:4 18, 389–396 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Makowski EK, Chen H-T & Tessier PM Simplifying complex antibody engineering using machine learning. Cell Syst 14, 667–675 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hie BL et al. Efficient evolution of human antibodies from general protein language models.Nature Biotechnology 2023 1–9 (2023) doi: 10.1038/S41587-023-01763-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saka K et al. Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Scientific Reports 2021 11:1 11, 1–13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mason DM et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nature Biomedical Engineering 2021 5:6 5, 600–612 (2021). [DOI] [PubMed] [Google Scholar]
- 36.Gupta A et al. An improved predictive recognition model for Cys2-His2 zinc finger proteins. Nucleic Acids Res 42, 4800 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aizenshtein-Gazit S & Orenstein Y DeepZF: improved DNA-binding prediction of C2H2-zinc-finger proteins by deep transfer learning. Bioinformatics 38, II62–II67 (2022). [DOI] [PubMed] [Google Scholar]
- 38.Ichikawa DM et al. A universal deep-learning model for zinc finger design enables transcription factor reprogramming. Nature Biotechnology 2023 41:8 41, 1117–1129 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bryant DH et al. Deep diversification of an AAV capsid protein by machine learning. Nature Biotechnology 2021 39:6 39, 691–696 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Ogden PJ, Kelsic ED, Sinai S & Church GM Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science (1979) 366, 1139–1143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eid F-E et al. Systematic multi-trait AAV capsid engineering for efficient gene delivery. bioRxiv 2022.12.22.521680 (2022) doi: 10.1101/2022.12.22.521680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malbranke C et al. Computational design of novel Cas9 PAM-interacting domains using evolution-based modelling and structural quality assessment. PLoS Comput Biol 19, e1011621 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kleinstiver BP et al. High-fidelity CRISPR-Cas9 variants with undetectable genome-wide off-targets. Nature 529, 490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joung K & Kleinstiver B US20230407277A1 - Engineered CRISPR-Cas9 Nucleases with Altered PAM Specificity - Google Patents. (2023). [Google Scholar]
- 45.Chen Z & Zhao H A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Res 33, e154–e154 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quick J et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc (2017) doi: 10.1038/nprot.2017.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walton RT, Hsu JY, Joung JK & Kleinstiver BP Scalable characterization of the PAM requirements of CRISPR–Cas enzymes using HT-PAMDA. Nat Protoc (2021) doi: 10.1038/s41596-020-00465-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Georgiev AG Interpretable numerical descriptors of amino acid space. Journal of Computational Biology 16, 703–723 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Wittmann BJ, Yue Y & Arnold FH Informed training set design enables efficient machine learning-assisted directed protein evolution. Cell Syst (2021) doi: 10.1016/j.cels.2021.07.008. [DOI] [PubMed] [Google Scholar]
- 50.Lundberg SM, Allen PG & Lee S-I A Unified Approach to Interpreting Model Predictions. doi: 10.5555/3295222.3295230. [DOI] [Google Scholar]
- 51.Rees HA & Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nature Reviews Genetics 2018 19:12 19, 770–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richter MF et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol 38, 883–891 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neugebauer ME et al. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nature Biotechnology 2022 41:5 41, 673–685 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Newby GA et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature 2021 595:7866 595, 295–302 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lazzarotto CR et al. Population-scale cellular GUIDE-seq-2 and biochemical CHANGE-seq-R profiles reveal human genetic variation frequently affects Cas9 off-target activity. bioRxiv 2025.02.10.637517 (2025) doi: 10.1101/2025.02.10.637517. [DOI] [Google Scholar]
- 56.Sweeney CL et al. Correction of X-CGD patient HSPCs by targeted CYBB cDNA insertion using CRISPR/Cas9 with 53BP1 inhibition for enhanced homology-directed repair. Gene Therapy 2021 28:6 28, 373–390 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Ravin SS et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med 9, (2017). [DOI] [PubMed] [Google Scholar]
- 58.Christie KA et al. Towards personalised allele-specific CRISPR gene editing to treat autosomal dominant disorders. Sci Rep (2017) doi: 10.1038/s41598-017-16279-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sung CH et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proceedings of the National Academy of Sciences 88, 6481–6485 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dryja TP et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990 343:6256 343, 364–366 (1990). [DOI] [PubMed] [Google Scholar]
- 61.Hartong DT, Berson EL & Dryja TP Retinitis pigmentosa. The Lancet 368, 1795–1809 (2006). [DOI] [PubMed] [Google Scholar]
- 62.LaVail MM et al. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: Long-term survival and late-stage therapy. Proceedings of the National Academy of Sciences 97, 11488–11493 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li P et al. Allele-Specific CRISPR-Cas9 Genome Editing of the Single-Base P23H Mutation for Rhodopsin-Associated Dominant Retinitis Pigmentosa. https://home-liebertpub-com.ezp-prod1.hul.harvard.edu/crispr 1, 55–64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hsu PD et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology 2013 31:9 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shin JW et al. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet 25, 4566–4576 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Courtney DG et al. CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Therapy 2016 23:1 23, 108–112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ciciani M et al. Automated identification of sequence-tailored Cas9 proteins using massive metagenomic data. Nature Communications 2022 13:1 13, 1–8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pedrazzoli E et al. CoCas9 is a compact nuclease from the human microbiome for efficient and precise genome editing. Nature Communications 2024 15:1 15, 1–12 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li L et al. Machine learning optimization of candidate antibody yields highly diverse sub-nanomolar affinity antibody libraries. Nature Communications 2023 14:1 14, 1–12 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weinstein JY et al. Designed active-site library reveals thousands of functional GFP variants. Nature Communications 2023 14:1 14, 1–13 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frazer J et al. Disease variant prediction with deep generative models of evolutionary data. Nature 2021 599:7883 599, 91–95 (2021). [DOI] [PubMed] [Google Scholar]
- 72.Notin P et al. TranceptEVE: Combining Family-specific and Family-agnostic Models of Protein Sequences for Improved Fitness Prediction. doi: 10.1101/2022.12.07.519495. [DOI] [Google Scholar]
- 73.Hopf TA et al. The EVcouplings Python framework for coevolutionary sequence analysis. Bioinformatics 35, 1582–1584 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruffolo JA et al. Design of highly functional genome editors by modeling the universe of CRISPR-Cas sequences. doi: 10.1101/2024.04.22.590591. [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS SECTION-ONLY REFERENCES
- 75.Kleinstiver BP, Fernandes AD, Gloor GB & Edgell DR A unified genetic, computational and experimental framework identifies functionally relevant residues of the homing endonuclease I-BmoI. Nucleic Acids Res 38, 2411 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods 2009 6:5 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 77.Alves CRR et al. Optimization of base editors for the functional correction of SMN2 as a treatment for spinal muscular atrophy. Nature Biomedical Engineering 2023 1–14 (2023) doi: 10.1038/S41551-023-01132-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nelson JW et al. Engineered pegRNAs improve prime editing efficiency. Nat Biotechnol 40, 402 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Christie KA et al. Precise DNA cleavage using CRISPR-SpRYgests. Nature Biotechnology 2022 41:3 41, 409–416 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Robichaux MA et al. Subcellular localization of mutant P23H rhodopsin in an RFP fusion knock-in mouse model of retinitis pigmentosa. DMM Disease Models and Mechanisms 15, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chan F, Bradley A, Wensel TG & Wilson JH Knock-in human rhodopsin-GFP fusions as mouse models for human disease and targets for gene therapy. Proc Natl Acad Sci U S A 101, 9109–9114 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kleinstiver BP et al. Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nature Biotechnology 2019 37:3 37, 276–282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rohland N & Reich D Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res 22, 939 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quick J et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc (2017) doi: 10.1038/nprot.2017.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tareen A & Kinney JB Logomaker: Beautiful sequence logos in Python. Bioinformatics (2020) doi: 10.1093/bioinformatics/btz921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ofer D & Linial M ProFET: Feature engineering captures high-level protein functions. Bioinformatics (2015) doi: 10.1093/bioinformatics/btv345. [DOI] [PubMed] [Google Scholar]
- 87.Chollet F keras, GitHub. Preprint at https://github.com/fchollet/keras (2015).
- 88.Pedregosa FABIANPEDREGOSA,F et al. Scikit-learn: Machine Learning in Python Gaël Varoquaux Bertrand Thirion Vincent Dubourg Alexandre Passos PEDREGOSA, VAROQUAUX, GRAMFORT ET AL. Matthieu Perrot. Journal of Machine Learning Research 12, 2825–2830 (2011). [Google Scholar]
- 89.Chen H, Lundberg SM & Lee SI Explaining a series of models by propagating Shapley values.Nature Communications 2022 13:1 13, 1–15 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song D, Xi NM, Li JJ & Wang L scSampler: fast diversity-preserving subsampling of large-scale single-cell transcriptomic data. Bioinformatics 38, 3126–3127 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McInnes L, Healy J & Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. (2018). [Google Scholar]
- 92.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019 576:7785 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nature Biotechnology 2019 37:3 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.De Ravin SS et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med 9, (2017). [DOI] [PubMed] [Google Scholar]
- 95.Amoli MM, Carthy D, Platt H & Ollier WER EBV Immortalization of human B lymphocytes separated from small volumes of cryo-preserved whole blood. Int J Epidemiol 37, i41–i45 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology 2014 33:2 33, 187–197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Picelli S et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24, 2033–2040 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tsai SQ, Topkar VV, Joung JK & Aryee MJ Open-source guideseq software for analysis of GUIDE-seq data. Nature Biotechnology 2016 34:5 34, 483–483 (2016). [DOI] [PubMed] [Google Scholar]
- 99.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. urn:issn:0907-4449 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 100.Anders C, Bargsten K & Jinek M Structural Plasticity of PAM Recognition by Engineered Variants of the RNA-Guided Endonuclease Cas9. Mol Cell (2016) doi: 10.1016/j.molcel.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goddard TD et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Science 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Landrum MJ et al. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res (2014) doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Primary datasets for this study are available in Supplementary Table 1 (HT-PAMDA data), Supplementary Table 2 (PAMmla predictions), Supplementary Table 6 (GUIDE-seq-2 data), and Supplementary Table 7 (source data). The HT-PAMDA training datasets are also available on GitHub at https://github.com/RachelSilverstein/PAMmla. Next-generation sequencing results are available through the NCBI sequence read archive (SRA) under PRJNA1169103. PAMmla predictions for all 64 million SpCas9(6AA) enzymes can be viewed through an online webtool (https://pammla.streamlit.app/). The UniRef100 dataset used to generate multiple sequence alignments for natural sequence models can be downloaded at https://www.uniprot.org/uniref/.
Custom scripts, the PAMmla source code, and the in silico directed evolution code are available on GitHub at https://github.com/RachelSilverstein/PAMmla and https://github.com/RachelSilverstein/multiplex_seq_analysis.