

Abstract
Cyclic peptides have emerged as a research hotspot in drug development in recent years due to their excellent stability, specificity, and cell penetration. However, existing computational models face challenges in accurately predicting the three-dimensional structures of cyclic peptides containing unnatural amino acids (unAAs), thereby limiting their drug design. The release of AlphaFold 3 has significantly enhanced the modeling capability of biomolecular complexes and enabled the inclusion of unAAs through definitions provided by the Chemical Component Dictionary (CCD). Nevertheless, its training data reliance limits its ability to accurately predict cyclic peptide structures, failing to meet the demand for precise cyclic peptide structure prediction. Based on the AlphaFold 3 framework, we developed HighFold3 by introducing the Cyclic Position Offset Encoding Matrix (CycPOEM). HighFold3 comprises two submodels: HighFold3-Linear and HighFold3–Cyclic, designed for predicting the structures of linear and cyclic peptides, respectively. Our results demonstrate that HighFold3 outperforms existing models (HighFold, HighFold2, CyclicBoltz1, NCPepFold, CABS-flex, ESMFold, and HelixFold) in cyclic peptide structure prediction. It achieves atomic-level precision in predicting cyclic peptide monomers while demonstrating enhanced accuracy and generalization capability for cyclic peptide complexes containing unAAs. This offers unprecedented technical support for the structural design and optimization of cyclic peptide–based therapeutics.
Keywords: deep learning, cyclic peptides, structure prediction, AlphaFold3
Introduction
Peptides have emerged as a central focus in drug development due to their diverse functionalities and excellent pharmacological properties [1–4]. Cyclic peptides, as a distinct class of peptides, possess a unique cyclic structure that confers greater stability, enhanced protein–protein interaction capabilities, and reduced susceptibility to protease degradation compared to their linear counterparts, making them highly promising candidates for pharmaceutical research and development [5–7]. To optimize their pharmacokinetic properties, increase structural rigidity, and mitigate enzymatic degradation, researchers frequently incorporate unnatural amino acids into cyclic peptide sequences, thereby enhancing their potential as viable drug candidates [5, 8].
In recent years, protein structure prediction technologies have garnered significant attention for accelerating the development of complex peptides. The advent of AlphaFold 2 revolutionized the modeling of protein structures and their interactions [9]. Subsequently, AlphaFold 3, with its novel architecture and optimized training strategies, further improved the accuracy of predicting biomolecular complex structures [10]. However, accurately predicting the three-dimensional structures of cyclic peptides containing unAAs remains a formidable challenge in computational biology [11]. Moreover, most deep learning-based structure prediction tools rely on existing protein structure databases during training, which are predominantly composed of linear protein structures [12]. When the same peptide sequence adopts either a linear or cyclic conformation depending on environmental conditions or design objectives [13], current models such as AlphaFold 3 struggle to flexibly adapt to this conformational diversity [14].
Notably, novel tools for cyclic peptide structure prediction are continually emerging. AFCyCDesign innovatively incorporates cyclic relative positional encoding into AlphaFold, demonstrating the feasibility of leveraging AlphaFold for handling cyclic structures [15]. NCPepFold, built upon RoseTTAFold All-Atom, integrates cyclic positional matrix algorithm with the Transformer framework, effectively predicting the structures of cyclic peptides containing unAAs [14]. CyclicBoltz1, developed from the open-source AlphaFold 3 variant Boltz-1, focuses on predicting cyclic peptide models encompassing all types of unAAs and exhibits competitive performance compared to other tools [16, 17]. In addition to structural modeling, the development of advanced functional prediction models has further driven the integration of deep learning approaches into multiple stages of peptide design [18–20]. These evolving tools are unlocking new possibilities for cyclic peptide–based drug discovery.
Building on this foundation, we developed the HighFold series of models to enhance the accuracy and applicability of cyclic peptide structure prediction. The first-generation model, HighFold, modifies the positional encoding matrices of AlphaFold and AlphaFold-Multimer, enabling high-accuracy predictions of cyclic peptides and their complexes constrained by head-to-tail cyclization and disulfide bonds [21]. HighFold2 builds upon this by incorporating fine-tuned training and atomic-level feature extraction, supporting the structural modeling of cyclic peptides containing 23 commonly occurring unAAs, significantly improving the precision of modeling cyclic peptides with unAAs [11]. Drawing on the successes of these prior models, this study integrates cyclization constraints into AlphaFold 3, optimizing and introducing two submodels tailored for distinct applications: HighFold3-Linear and HighFold3-Cyclic, designed for predicting the three-dimensional structures of linear and cyclic peptides containing unAAs, respectively. Furthermore, HighFold3 leverages the Chemical Component Dictionary (CCD) [22] to encompass all known unAA types, demonstrating substantial improvements in prediction accuracy and generalization capability compared to existing tools, thereby providing robust support for the design of complex cyclic peptide–based therapeutics.
Material and methods
Datasets
Cyclic peptide monomer dataset
The dataset includes 63 cyclic peptide structures (sourced from the AFCyCDesign) to evaluate the predictive performance of HighFold3 against HighFold, CyclicBoltz1, ESMFold [23], and HelixFold [24]. In addition, 21 cyclic peptide structures (also sourced from the AFCyCDesign) are used to evaluate the predictive performance of HighFold3 against CABS-flex.
Cyclic peptide complex dataset
The dataset is divided into two parts: an internal dataset containing 17 cyclic peptide–complex structures (sourced from the ADCP) for comparing the predictive performance of HighFold3 with HighFold, and an external dataset containing 15 cyclic peptide–complex structures (sourced from CyclicBoltz1) for comparison with CyclicBoltz1.
Cyclic peptide dataset containing unAAs
The dataset includes 34 cyclic peptide structures with unAAs (sourced from HighFold2), encompassing both monomers and complexes, used to benchmark performance against HighFold2. In addition, 33 cyclic peptide structures with unAAs (sourced from HighFold2 and NCPepFold) are used to evaluate the predictive performance of HighFold3 against NCPepFold.
Linear peptide dataset
The dataset (including peptide monomers and their complexes) is derived from HighFold2 and contains 36 peptide structures with unAAs, utilized to compare predictive performance with HighFold2.
All structures in these datasets were determined through nuclear magnetic resonance (NMR) or X-ray crystallography experiments to ensure evaluation reliability.
Cyclic and linear peptide sequence dataset
We selected a representative set of peptide sequence samples from the CyclicPepedia [25] cyclic peptide database and from the linear peptide collections within the CPPsite [26] and ParaPep [27] databases. This set includes 100 cyclic peptide sequences and 100 linear peptide sequences, covering a range of sequence lengths and amino acid compositions. Detailed sequence information is provided in Table S1 and Table S2.
Experimental settings
Experiments were conducted on a workstation running Ubuntu 20.04.2, equipped with an Intel Xeon Gold 6430 CPU (64 cores), four NVIDIA A800 80 GB GPUs (total GPU memory 320 GB), 500 GB of RAM, and 40 TB of storage space.
HighFold3 was developed based on the open-source AlphaFold 3 code and consists of two submodels: HighFold3-Cyclic and HighFold3-Linear, designed for predicting the structures of cyclic and linear peptides, respectively. Users can toggle between models using the Boolean parameter “head_to_tail” to accommodate different conformational requirements.
Metrics
Root mean square deviation calculation
This study employs Root Mean Square Deviation (RMSD) to assess the deviation between predicted and experimental structures, with the default selection being the highest-confidence conformation (Top-1) from the model predictions. Key metrics include RMSD of the Cα coordinates (RMSDCα), all atom RMSD (RMSDall-atom), and RMSD of unAA (RMSDunAA). The calculation method is as follows: based on Cα atom coordinates, the Kabsch algorithm is used for rigid superposition. For peptide monomers, RMSDCα or RMSDall-atom is calculated directly across the entire chain; for complexes, chains with fewer than 40 residues are defined as peptide chains, and the same method is applied. For unAA, atomic coordinates within a 10 Å radius are extracted for local superposition and calculation, with the average taken if multiple unAAs are present.
HighFold3 framework
HighFold3 is an enhanced model developed on the AlphaFold 3 framework, designed to accurately predict the three-dimensional structures of peptides containing unAAs (including monomers and their protein complexes) while handling special topological features such as head-to-tail cyclization and disulfide bond constraints. The overall architecture of HighFold3 is illustrated in Fig. 1A. Based on the frozen AlphaFold3 pretrained model, the CycPOEM was introduced, and an innovatively designed “Cyclization Switch” module was employed to dynamically select either a linear or cyclic positional encoding matrix within the model. For linear peptides, the sequence interval between adjacent amino acid residues is defined as 1, and the distance between the N-terminus and C-terminus is defined as the peptide chain length minus 1 (Fig. 1B). For cyclic peptides, CycPOEM directly connects the N-terminus and C-terminus of the peptide chain, constructing a closed-loop distance matrix (Fig. 1C). By adjusting the input features, the model is enabled to correctly recognize the closed-loop topology while inherently avoiding the risk of overfitting that could arise from training on small-sample cyclic peptide datasets. When predicting cyclic peptide–protein complexes, the model’s input distance matrix is explicitly divided into two parts: a linear positional encoding matrix for the target protein and a CycPOEM for the cyclic peptide ligand. This design ensures the model can flexibly adapt to diverse conformational demands, accurately modeling the binding of cyclic peptide ligands to protein receptors.
Figure 1.

Overview of HighFold3. (A) The basic workflow of HighFold3. The “cyclization switch” determines whether a linear or cyclic positional matrix is used based on user input, with features subsequently processed through the AlphaFold 3 module (comprising a Pairformer stack and diffusion module) to generate predicted structures for linear or cyclic peptides. (B) Positional information matrix for linear peptides, alongside the predicted structure of a linear peptide. (C) Positional information matrix for cyclic peptides, alongside the predicted structure of a cyclic peptide.
Construction of the cyclic position offset encoding matrix
HighFold3 adopts an improved Floyd-Warshall algorithm [28] (Floyd for Cyclic Peptides, FCP) to compute the shortest path distances between any two residues in a cyclic peptide sequence, incorporating information on peptide bond directionality to accurately reflect the cyclic topology of the peptide.
Specifically, for a cyclic peptide sequence S of length N, an N × N distance matrix D is first constructed, where D[i,j] represents the initial distance between amino acids i and j. Set D[i,i] = 0, indicating the distance from any residue to itself is zero; for adjacent amino acids i and i + 1 in the sequence, set D[i,i + 1] = 1 and D[i + 1,i] = 1; for the head-to-tail connection in cyclic peptides, set D[0,N − 1] = 1 and D[N − 1,0] = 1. Subsequently, the FCP algorithm is applied, traversing all amino acid pairs (i,j) through a triple loop with intermediate node k to update the shortest path using the formula:
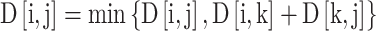 |
Here, D[i,j] denotes the shortest path distance between amino acids i and j. Following this process, the shortest path distance matrix for the cyclic peptide is obtained. Additionally, based on the directionality of peptide bond connections, positive, or negative signs are assigned to matrix elements as follows:
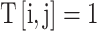 , if the N-terminus of ai connects to the C-terminus of aj;
, if the N-terminus of ai connects to the C-terminus of aj;
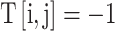 , if the C-terminus of ai connects to the N-terminus of aj;
, if the C-terminus of ai connects to the N-terminus of aj;
 , if ai and aj are connected via a disulfide bond.
, if ai and aj are connected via a disulfide bond.
Through this assignment strategy, the CycPOEM matrix not only contains distance information between residues but also encodes the directional information of peptide chain cyclization (see detailed algorithm in Algorithm 1).
Construction of the Disulfide bond combination matrix
In cyclic peptides or proteins, disulfide bonds are formed between the sulfur atoms of two cysteine side chains, playing a critical role in maintaining molecular stability [29]. When multiple cysteines are present in a sequence, various disulfide bond pairing combinations are possible. For example, with three cysteine residues A, B, and C, theoretically possible disulfide bond pairings include (A, B), (A, C), or (B, C). To generate all possible disulfide bond connection combinations, HighFold3 adopts the Complete Set of Disulfide Bridges (CSDB) method [21], constructing a disulfide bond combination matrix to ensure the model accounts for all potential disulfide bond topologies during prediction.
The specific steps are as follows: First, scan the input sequence S to identify the positional indices of all cysteines and store them in a vector Vc. For instance, given the sequence “CXXCXCXX” (where C denotes cysteine and X denotes any noncysteine amino acid), Vc = [1, 4, 6], indicating a total of M = 3 cysteines in the first chain. Next, calculate the maximum possible number of disulfide bonds Kmax based on M. If M is odd, Kmax = M − 1. Then, use a double loop to iterate over the possible number of paired residues, generating all unique disulfide bond combinations. The outer loop iterates over the total number of residues k involved in pairing (from two to Kmax, with a step size of 2, representing the number of cysteines involved in pairing), while the inner loop selects k cysteines from M to form candidate subsets. For each candidate subset, enumerate all possible pairwise combinations within the subset (e.g. for the subset {1, 1, 4, 6}, pairings include (1, 1, 4), (1, 1, 6), and (1, 4, 6)), and eliminate invalid combinations with duplicate residues. Finally, record all valid disulfide bond combinations in the combination matrix. Each row of the matrix represents a possible disulfide bond combination, each column corresponds to a disulfide bond pair, and the number of columns depends on the maximum possible number of disulfide bond pairs.
As shown in Fig. 2A, using the sequence “CXXCXCXX” as an example, HighFold3 can flexibly select different disulfide bond pairing combinations during prediction (setting the distance between residues forming a disulfide bond to one in the initial distance matrix) and model them in conjunction with linear or cyclic positional offset matrices, demonstrating the model’s flexibility in handling multiple disulfide bond pairings and peptide chain topologies.
Figure 2.

Additional workflow details of HighFold3. (A) Workflow of HighFold3 flexibly handling disulfide bond pairing in structure prediction. (B) Workflow for predicting the complex of a target protein with multiple cyclic peptide ligands. Here, C1, C2, and C3 represent the cyclic positional matrices of the cyclic peptide ligands, and P represents the linear positional matrix of the target protein.
Additional extensions of HighFold3
Unlike existing cyclic peptide structure prediction models, HighFold3 offers a unique functional extension: the ability to simultaneously predict the complex structures of multiple cyclic peptide ligands with the same protein receptor. Specifically, the HighFold3 framework allows users to specify a target protein represented by a linear positional matrix and simultaneously input multiple cyclic peptide ligands encoded by CycPOEM matrices. The model combines the linear positional matrix of the target protein with the CycPOEM of multiple cyclic peptide ligands to predict the three-dimensional structure of a multimolecular complex. For ligands with disulfide bond cyclization, users can specify parameters such as [[1, 3, 5], [2, 7, 9]], enabling the prediction of complex structures with multiple disulfide-bonded cyclic peptide ligands.
Fig. 2B provides a schematic of this workflow, where the model overlays multiple positional matrices to generate a complete prediction of a multiligand complex structure.
ZDOCK
For protein-peptide docking, we employed the online server ZDOCK 3.0.2 [30]. ZDOCK utilizes a rigid-body fast Fourier transform algorithm to perform a global search of rotational and translational space between the protein and peptide, even in the absence of prior binding site information. All generated docking poses are subsequently ranked using its built-in scoring function to evaluate and prioritize potential binding modes.
Results and discussion
High-precision prediction of cyclic peptide monomer structures
We evaluated the model on 63 cyclic peptide monomer structures from the AFCyCDesign dataset, using RMSDCα to quantify the deviation between predicted and experimentally determined structures. Results from HighFold and CyclicBoltz1 served as benchmarks to highlight the superiority of HighFold3. As shown in Fig. 3A, HighFold3 exhibits outstanding prediction accuracy: its median RMSDCα is 0.918 Å, with an average of 1.338 Å, compared to HighFold’s median of 1.058 Å and average of 1.478 Å, reflecting improvements of 13.23% and 9.54%, respectively. Notably, HighFold3 achieves an RMSDCα below 1.5 Å in 44 samples, exceeding HighFold (37 samples) and CyclicBoltz1 (38 samples). The Wilcoxon signed-rank test yielded 27 positive ranks (HighFold3 minus HighFold) and 36 negative ranks (HighFold minus HighFold3), with a Z-score of 1.533. Although the P-value did not reach conventional significance thresholds, the results still indicate an overall trend favoring HighFold3. These findings indicate that HighFold3 approaches experimental accuracy in most test samples, closely rivaling high-resolution X-ray crystallography or NMR results.
Figure 3.

Results of cyclic peptide monomer structure prediction. (A) RMSDCα distribution of HighFold and HighFold3 in the cyclic peptide monomer dataset. (B) Correlation between peptide pLDDT and RMSDCα for HighFold and HighFold3 in the cyclic peptide monomer dataset. (C) RMSDCα results of predicted disulfide bridge structures in HighFold and HighFold3. (D) RMSDCα distribution of CABS-flex and HighFold3 in the cyclic peptide monomer dataset. (E) RMSDCα distribution of ESMFold and HighFold3 in the cyclic peptide monomer dataset. (F) RMSDCα distribution of HelixFold and HighFold3 in the cyclic peptide monomer dataset. (G) Comparison of HighFold3 predicted structure (PDB ID: 2LWU) with the native structure. Green indicates predicted structures, light gray indicates natural structures. (Below, light gray denotes natural structures.) (H) Comparison of HighFold3 predicted structure (PDB ID: 2 M79) with the native structure. Pink indicates the predicted structure. (I) Comparison of HighFold3 predicted structure (PDB ID: 6U7R) with the native structure. Blue indicates predicted structure.
To further validate HighFold3’s reliability, we analyzed the correlation between its confidence scores (the predicted local-distance difference test, pLDDT) and prediction accuracy. As shown in Fig. 3B, across the 63 cyclic peptide–monomers from the AFCyCDesign dataset, RMSDCα exhibits a strong negative correlation with pLDDT (Pearson correlation coefficient of −0.67), suggesting that higher confidence predictions typically correspond to lower structural deviations. Under high-confidence conditions (pLDDT >0.90), 43 samples achieve an RMSDCα below 1.5 Å, further confirming the reliability of the model’s predictions. Detailed prediction results are available in Table S3.
Disulfide bonds play a critical role in stabilizing cyclic peptide conformations, yet traditional models often overlook their influence, focusing solely on head-to-tail cyclization. The HighFold series of models overcomes this limitation, effectively predicting disulfide bond connectivity in cyclic peptide structures. As shown in Fig. 3C, HighFold3 excels in predicting cyclic peptide structures with a single disulfide bond pair, achieving an average RMSDCα of 0.820 Å, compared to HighFold’s 1.226 Å, an improvement of 33.12%. This demonstrates that HighFold3 enhances prediction accuracy for disulfide bonds.
We compared HighFold3 with several recent structure prediction models, including CABS-flex, ESMFold, and HelixFold (the latter two are alignment-free models). The results showed that HighFold3 achieved higher accuracy: its average RMSDCα was 1.482 Å, better than 4.022 Å for CABS-flex. In comparison with ESMFold, HighFold3 achieved an average of 1.338 Å, while ESMFold reached 2.982 Å; for HelixFold, HighFold3 showed 1.338 Å compared to 2.643 Å for HelixFold (Figs. 3D, 3E, 3F). These results indicate that HighFold3 produces lower structural deviations when modeling complex cyclic peptide topologies. This advantage may be partly attributed to the evolutionary information provided by multiple sequence alignment (MSA) and the introduction of CycPOEM. In addition, it is worth noting that ESMFold does not support closed-loop structures, and none of the three models can handle structures containing unAAs. The detailed results are provided in Supplementary Table S4, Table S5, and Table S6.
To further assess the prediction stability of HighFold3, we conducted additional experiments using different random seed values (1, 4, 16, 64) on the cyclic peptide monomer dataset. The obtained RMSDCα values were 1.338 Å, 1.493 Å, 1.334 Å, and 1.257 Å, respectively. The fluctuations among different seeds were minimal, demonstrating that HighFold3 yields stable and consistent predictions across independent runs. The detailed results are provided in Supplementary Table S7.
Finally, through visualization analysis, we intuitively showcased HighFold3’s superior performance. For instance, the predicted RMSDCα for the PawS-derived peptide (PDB ID: 2LWU) is only 0.208 Å (Fig. 3G), for Theta defensin (PDB ID: 2 M79) it is 0.488 Å (Fig. 3H), and for the SFTI derivative (PDB ID: 6U7R) it is 0.245 Å (Fig. 3I). These results indicate that HighFold3 can precisely recapitulate the atomic-level conformations of natural cyclic peptides, providing a reliable structural foundation for drug design.
High-precision prediction of cyclic peptide complex structures
To develop effective cyclic peptide binders, modeling the monomer alone is often insufficient. Predicting the complete target protein–cyclic peptide complex structure is essential to elucidate the interaction mechanisms between ligands and targets. Here, HighFold3 specifies the topology of cyclic peptide ligands using CycPOEM and evaluates ligand prediction accuracy with RMSDCα.
We tested HighFold3 on the internal dataset (17 complexes) and the external dataset (15 complexes), comparing its performance against HighFold and CyclicBoltz1. As shown in Fig. 4A, on the internal dataset, HighFold3 achieves a median RMSDCα of 0.266 Å and an average of 0.305 Å, outperforming HighFold’s 0.280 Å and 0.359 Å (improvements of 5% and 15.04%, respectively). The Wilcoxon signed-rank test yielded seven positive ranks and 10 negative ranks, with a Z-score of 1.041. In the external dataset of 15 samples, HighFold3 outperforms CyclicBoltz1 in 14 samples (Fig. 4B), demonstrating higher structural modeling precision. These results indicate that HighFold3 can more accurately construct the three-dimensional structures of cyclic peptide-target protein complexes.
Figure 4.

Results of cyclic peptide complex structure prediction. (A) RMSDCα distribution of HighFold and HighFold3 in the internal cyclic peptide complex dataset. (B) RMSDCα distribution of CyclicBoltz1 and HighFold3 in the external cyclic peptide complex dataset. (C) Comparison of HighFold3 predicted structure (PDB ID: 3AVB) with the native structure. Blue indicates the predicted structure. (D) Comparison of HighFold3 predicted structure (PDB ID: 6VXY) with the native structure. Pink indicates the predicted structure. (E) Redocking validation of the cyclic peptide complex structure (PDB ID: 5LSO) using ZDOCK. Yellow indicates the ZDOCK result, and green indicates the HighFold3 prediction. (F) Redocking validation of the cyclic peptide complex structure (PDB ID: 6D3X) using ZDOCK.
We further analyzed the ligand pLDDT scores, finding a negative correlation with RMSDCα (Pearson correlation coefficient of −0.32 and − 0.26, see Fig. S1), suggesting consistency and stability in HighFold3’s complex predictions. Detailed prediction results are available in Table S8 and Table S9.
Moreover, visualization results show that the ligand RMSDCα for the HIV integrase-cyclic peptide inhibitor complex (PDB ID: 3AVB) is 0.272 Å (Fig. 4C), and for the beta-trypsin-inhibitor complex (PDB ID: 6VXY), it is 0.459 Å (Fig. 4D). These findings confirm that the model can precisely reconstruct the binding conformations of cyclic peptide ligands, aligning closely with experimentally determined structures. Accurately elucidating such ligand-receptor binding modes is critical for drug discovery and optimization, further highlighting HighFold3’s potential in practical drug development.
Finally, we evaluated the practical applicability of HighFold3 in cyclic peptide complex structure prediction. Molecular docking analyses were performed using ZDOCK on representative complexes containing short- and medium-length cyclic peptides. The docking results were highly consistent with the HighFold3 predictions, accurately reproducing the ligand binding pockets and generating similar interaction patterns as observed experimentally (Fig. 4E and F). This validation further supports the broad potential of HighFold3 in drug design and structural biology applications.
High-precision prediction of cyclic peptide structures containing unnatural amino acids
We evaluated HighFold3 on 34 cyclic peptide monomer and complex structures containing unAAs from the HighFold2 dataset, including those cyclized via head-to-tail and disulfide bonds. To comprehensively assess the model’s accuracy in predicting complex cyclic peptide structures, we used RMSDCα, RMSDall-atom, and RMSDunAA as evaluation metrics, comparing results with HighFold2. As shown in Fig. 5A, HighFold3 achieves a median RMSDCα of 1.711 Å and an average of 1.907 Å, outperforming HighFold2’s 1.891 Å and 2.152 Å (improvements of 2.7% and 10.97%, respectively). The Wilcoxon signed-rank test yielded 15 positive ranks and 19 negative ranks, with a Z-score of 0.308. For RMSDall-atom, the median and average are 2.787 Å and 2.929 Å, respectively, compared to HighFold2’s 2.872 Å and 3.436 Å (improvements of 1.26% and 13.97%, Fig. 5B). The Wilcoxon signed-rank test yielded 15 positive ranks and 19 negative ranks, with a Z-score of 1.453. For RMSDunAA, the median and average are also 2.301 Å and 2.372 Å, surpassing HighFold2’s 2.579 Å and 2.965 Å (improvements of 1.26% and 13.97%, Fig. 5C). The Wilcoxon signed-rank test yielded 12 positive ranks and 22 negative ranks, with a Z-score of 1.743. Notably, CyclicBoltz1, when evaluated on this dataset, only considered head-to-tail cyclized samples, indicating its limited capability in predicting disulfide-bonded cyclic structures. These results demonstrate that HighFold3 offers higher accuracy and broader applicability in predicting cyclic peptide monomers with unAAs, outperforming other known models comprehensively.
Figure 5.

Results of cyclic peptide structure prediction containing unAAs. (A) RMSDCα distribution of HighFold2 and HighFold3 in the dataset. (B) RMSDall-atom distribution of HighFold2 and HighFold3 in the dataset. (C) RMSDunAA distribution of HighFold2 and HighFold3 in the dataset. (D) Correlation between peptide pLDDT and RMSDCα for HighFold and HighFold3. (E) Correlation between peptide pLDDT and RMSDall-atom for HighFold and HighFold3. (F) Correlation between peptide pLDDT and RMSDunAA for HighFold and HighFold3. (G) Comparison of HighFold3 predicted structure (PDB ID: 2 PM5) with the native structure. Yellow indicates the predicted structure. (H) Comparison of HighFold3 predicted structure (PDB ID: 3WNG) with the native structure. Pink indicates the predicted structure.
Across the 34 cyclic peptide monomers in the HighFold2 dataset, RMSDCα, RMSDall-atom, and RMSDunAA exhibit negative correlations with pLDDT, with coefficients of −0.52, −0.34, and − 0.78 (Figs 5D–F). At a more relaxed confidence threshold (pLDDT > 0.90), HighFold3 achieves an RMSDCα below 2 Å in 20 samples, compared to only eight for HighFold2, highlighting HighFold3’s enhanced adaptability in modeling complex cyclic peptides. Detailed prediction results are available in Table S10.
In addition, we compared HighFold3 with NCPepFold, which was fine-tuned for cyclic peptides containing unAAs. The results showed that HighFold3 achieved an average RMSDCα of 1.931 Å, better than 2.014 Å for NCPepFold (Fig. S2). It is noteworthy that HighFold3 did not require any retraining of model parameters. By adjusting the input encoding alone, it achieved better performance while avoiding potential overfitting risks that may occur due to the limited number of cyclic peptide samples. Detailed prediction results are available in Table S11.
Visualization results show that for the Human alpha-defensin 1 derivative cyclic peptide monomer (PDB ID: 2 PM5), the RMSDCα is 0.321 Å, RMSDall-atom is 0.643 Å, and RMSDunAA is 0.751 Å (Fig. 5G). For the HIV-1 cyclic peptide complex, the ligand RMSDCα is 0.846 Å, RMSDall-atom is 1.309 Å, and RMSDunAA is 2.3 Å (Fig. 5H). These findings indicate that the model can accurately reconstruct the three-dimensional structures of complex cyclic peptides containing unAAs, expanding the application scope of cyclic peptide structure prediction.
High-precision prediction of linear peptide structures
In addition to excelling in cyclic peptide structure prediction, we assessed HighFold3’s capability in predicting linear peptide structures containing unAAs, comparing it with HighFold2. Evaluation metrics included RMSDCα, RMSDall-atom, and RMSDunAA. On the linear peptide dataset, HighFold3 achieves an average RMSDCα of 1.631 Å, outperforming HighFold2’s 2.080 Å (an improvement of 21.59%, see Fig. S3A). The Wilcoxon signed-rank test yielded 23 positive ranks and 28 negative ranks, with a Z-score of 0.862. For RMSDall-atom, HighFold3 achieves an average of 2.383 Å, surpassing HighFold2’s 3.037 Å (an improvement of 21.53%; see Fig. S3B). The Wilcoxon signed-rank test yielded 21 positive ranks and 30 negative ranks, with a Z-score of 1.640. For RMSDunAA, HighFold3 achieves an average of 2.128 Å, better than HighFold2’s 2.826 Å (an improvement of 24.7%; see Fig. S3C). The Wilcoxon signed-rank test again yielded 21 positive ranks and 30 negative ranks, with a Z-score of 1.350.
Furthermore, RMSDCα, RMSDall-atom, and RMSDunAA exhibit negative correlations with pLDDT (correlation coefficients of −0.64, −0.66, and − 0.59, see Figs S2D–F), with 15 samples achieving an RMSDCα below 1.5 Å under high-confidence conditions, reflecting the model’s high consistency in generalization and structural prediction accuracy. Detailed prediction results are available in Table S12.
Ablation study
To thoroughly investigate the contribution of the CycPOEM module to HighFold3’s performance, we conducted an ablation study to assess its criticality. Specifically, we removed the CycPOEM module while keeping all other HighFold3 modules and parameters unchanged and evaluated structure prediction performance on two cyclic peptide complex datasets. As shown in Figs. 6A and 6B, the removal of CycPOEM led to a decline in prediction accuracy. In the first dataset, the average RMSDCα increased from 0.305 Å to 0.804 Å. The Wilcoxon signed-rank test yielded one positive ranks and 16 negative ranks, with a Z-score of 3.266. In the second dataset, it rose from 0.612 Å to 1.032 Å. The Wilcoxon signed-rank test yielded nine positive ranks and six negative ranks, with a Z-score of 0.256. This drop in accuracy underscores the essential role of CycPOEM in maintaining prediction precision. Detailed results are provided in Table S13 and Table S14.
Figure 6.

Ablation study analysis. (A) Comparison of prediction performance between the full model and the model without CycPOEM on the internal cyclic peptide complex dataset. “w/o” denotes without. (B) Comparison of prediction performance between the full model and the model without CycPOEM on the external cyclic peptide complex dataset. (C) Comparison of the predicted structure (PDB ID: 5TU6) by the model without CycPOEM with the native structure. Pink indicates the predicted structure. (D) Comparison of the predicted structure (PDB ID: 5TU6) by the full model with the native structure. Blue indicates the predicted structure.
Further analysis revealed the specific impact of omitting CycPOEM. Without this module, the model failed to effectively recognize the head-to-tail closure characteristic of cyclic peptides, resulting in some samples being erroneously predicted as linear conformations. We selected the structure with the most significant RMSD deviation (PDB ID: 5TU6) for comparison: as shown in Fig. 6C, the model without CycPOEM failed to correctly predict its cyclic topology, with RMSD increasing by 4.336 Å, whereas the full model (Fig. 6D) accurately recapitulated the experimental structure. This stark contrast validates the indispensable role of CycPOEM in precisely modeling cyclic peptide structures, highlighting its central contribution to HighFold3.
Advantages of HighFold3 in structure prediction
Although AlphaFold 3 has achieved groundbreaking progress in biomolecular structure prediction, its performance in cyclic peptide structure prediction remains suboptimal, particularly for unseen cyclic peptide sequences. This deficiency is attributed to AlphaFold 3’s lack of specific constraints for closed-loop structures, which limits its generalization capability. To test this hypothesis, we compared the performance of HighFold3 and AlphaFold 3 on cyclic peptide sequences outside their training sets.
We first selected a recently resolved head-to-tail cyclic peptide (PDB ID: 9HVC) [31]. Results revealed that AlphaFold 3 predicted the sequence as a linear conformation, with the N-to-C terminal distance exceeding 20 Å (Fig. 7A). In contrast, HighFold3 not only accurately recapitulated its closed-loop conformation but also precisely captured the topological features of key α-helical regions characteristic of the native structure, aligning with the experimentally determined conformation (Fig. 7B). This indicates that AlphaFold 3 lacks the necessary topological constraints for cyclic structures, while HighFold3 effectively addresses this gap through its cyclization mechanism. To further validate its generalization capability, we screened 100 sequences from a cyclic peptide database, varying in length and amino acid composition, all derived from experimental studies but lacking crystal structures. As shown in Table 1, AlphaFold 3 achieved a head-to-tail cyclization success rate of only 21% for these sequences, while HighFold3 attained a 100% success rate. This comparison clearly reveals AlphaFold 3’s limitations in cyclic peptide prediction, whereas HighFold3 demonstrates exceptional adaptability and reliability.
Figure 7.

Advantages of HighFold3 in structure prediction. (A) Structure predicted by AlphaFold 3 (PDB ID: 9HVC). (B) Structure predicted by HighFold3 (PDB ID: 9HVC). (C) Structure predicted by HighFold3-linear (sequence: KLARLLT). (D) Structure predicted by HighFold3-cyclic (sequence: KLARLLT).
Table 1.
Model accuracy on the cyclic peptide sequence database
| Model | Head-to-tail cyclization success rate |
|---|---|
| AlphaFold 3 | 21% |
| HighFold3-Cyclic | 100% |
Additionally, we evaluated both models’ performance on a linear peptide dataset. Results indicated that neither AlphaFold 3 nor HighFold3 erroneously predicted linear sequences as cyclic conformations (Table S15), confirming that HighFold3 maintains accuracy in linear structure prediction while avoiding conformational misclassification. This stability further underscores the flexibility of its design.
Notably, peptide conformation (linear or cyclic) is not solely determined by the amino acid sequence but is profoundly influenced by synthesis conditions, enzymatic reactions, or artificial regulation [32, 33]. For instance, cyclic peptides are often catalyzed by nonribosomal peptide synthetases (NRPS) or postmodification enzymes to form stable closed-loop structures via head-to-tail linkage or side-chain crosslinking [34, 35]. Conversely, cyclic peptides may be cleaved by proteases or remain linear due to insufficient synthesis conditions [8, 36]. This dynamic property inspired the core design philosophy of HighFold3: enabling flexible differentiation and prediction of linear or cyclic conformations through user-controlled input directives.
To explore HighFold3’s practical research potential, we tested a known peptide sequence existing in both linear and cyclic forms. Williams et al. designed the linear peptide KLARLLT and its cyclic derivative Cyclo (KLARLLT), experimentally verifying differences in Epidermal Growth Factor Receptor binding affinity and serum stability, and constructed their 3D structures using traditional molecular modeling and simulation13. We applied HighFold3-Linear and HighFold3-Cyclic modules for prediction: KLARLLT was predicted as a linear conformation with an N-to-C terminal distance of ~10 Å (Fig. 7C), while Cyclo (KLARLLT) exhibited a closed-loop conformation with an amide bond length of ~1.35 Å (Fig. 7D). These results align closely with experimental expectations, demonstrating HighFold3’s utility in real-world scenarios. In the future, we plan to integrate molecular dynamics (MD) simulations to further dissect the dynamic details of postcyclization peptide-target interactions.
In summary, AlphaFold 3 exhibits limited generalization capability for structure prediction beyond its training data, whereas HighFold3, with its innovative cyclization constraints and flexible design, provides a comprehensive and efficient solution for peptide conformation prediction.
Conclusion
Elucidating the interaction mechanisms between proteins and cyclic peptide ligands based on structural information is critical for accelerating the development of cyclic peptide–based therapeutics. To this end, this study introduces CycPOEM into the AlphaFold 3 framework, developing the HighFold3-Linear and HighFold3-Cyclic submodels tailored for linear and cyclic peptides, respectively. Evaluation results demonstrate that HighFold3 outperforms existing methods in predicting the structures of cyclic peptide monomers, complexes, and peptides containing unAAs, while exhibiting robust generalization capability.
In addition, HighFold3 demonstrates significant practical value in peptide drug design and structural biology. First, by leveraging the Chemical Component Dictionary, it supports structural modeling of peptides containing all known types of unAAs, meeting the design demands of novel and complex peptides in current drug development. Second, the flexible Cyclization Switch and CycPOEM mechanisms greatly expand the design space for new molecular structures. Third, by simply adjusting input features, HighFold3 efficiently adapts to diverse systems while avoiding the risk of overfitting. Fourth, the high accuracy and broad applicability of HighFold3 provide reliable structural information for target binding site identification, lead optimization, and conformation screening, thereby reducing experimental validation costs and offering broad application prospects in drug discovery, molecular design, and structural biology research.
Nevertheless, HighFold3 has certain limitations: (i) The dynamic behavior of cyclic peptides in physiological environments is crucial to their biological function, yet the current model primarily focuses on static structure modeling, limiting its applicability in simulating realistic drug-target binding mechanisms. (ii) While HighFold3 offers high prediction accuracy for cyclic peptides, critical structural details have yet to be validated through biological experiments. (iii) HighFold3 remains reliant on AlphaFold 3’s framework and pretrained parameters, and its structure prediction capability may exhibit instability for certain extreme sequences. In the future, we plan to integrate HighFold3 with MD simulations to further reveal the dynamic interaction mechanisms of peptide drugs under physiological conditions; concurrently, we aim to enhance the model’s credibility and practical value by incorporating biological experiments to validate key predicted structural details. To address adaptability issues in the current framework, we will explore fine-tuning strategies based on downstream tasks to improve the model’s broad compatibility.
Key Point
We developed HighFold3, a cyclic peptide structure prediction model based on AlphaFold3, introducing the CycPOEM and Cyclization Switch modules.
HighFold3 consists of two submodels: (i) HighFold3-Linear, designed for predicting linear peptide structures; (ii) HighFold3-Cyclic, specifically designed for peptides with cyclization, including those with disulfide bonds and unAAs.
Among existing cyclic peptide structure prediction models, HighFold3 demonstrates the best overall performance.
Supplementary Material
Contributor Information
Sen Cao, Faculty of Applied Sciences, Macao Polytechnic University, R. de Luís Gonzaga Gomes, Macao 999078, China.
Cheng Zhu, College of Pharmaceutical Sciences, Zhejiang University of Technology, Chaowang Road, Gongshu District, Hangzhou 310014, China.
Qingyi Mao, College of Pharmaceutical Sciences, Zhejiang University of Technology, Chaowang Road, Gongshu District, Hangzhou 310014, China.
Jingjing Guo, Faculty of Applied Sciences, Macao Polytechnic University, R. de Luís Gonzaga Gomes, Macao 999078, China.
Ning Zhu, Faculty of Applied Sciences, Macao Polytechnic University, R. de Luís Gonzaga Gomes, Macao 999078, China.
Hongliang Duan, Faculty of Applied Sciences, Macao Polytechnic University, R. de Luís Gonzaga Gomes, Macao 999078, China.
Acknowledgment
This research was supported by an internal grant from Macao Polytechnic University (RP/FCA-07/2024) and the Macao Science and Technology Development Fund (Grant No. 0151/2024/RIA2).
The manuscript was approved by Macao Polytechnic University with the submission code (fca.0ba1.9a94.d).
Author contributions
Sen Cao (Conceptualization, Data curation, Methodology, Software, Writing—original draft), Cheng Zhu (Methodology), Qingyi Mao (Methodology), Jingjing Guo (Methodology), Ning Zhu (Supervision) and Hongliang Duan: Supervision. All authors (Writing—review & editing)
Conflict of interest: The authors declare no competing interests.
Data availability
All data this work uses is available at (https://github.com/hongliangduan/HighFold3) or the Supporting data.
References
- 1. Mannes M, Martin C, Menet C. et al. Wandering beyond small molecules: Peptides as allosteric protein modulators. Trends Pharmacol Sci 2022;43:406–23. 10.1016/j.tips.2021.10.011. [DOI] [PubMed] [Google Scholar]
- 2. Li H, Yang Y, Hong W. et al. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct Target Ther 2020;5:1–23. 10.1038/s41392-019-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lau JL, Dunn MK. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg Med Chem 2018;26:2700–7. 10.1016/j.bmc.2017.06.052. [DOI] [PubMed] [Google Scholar]
- 4. Petsalaki E, Russell RB. Peptide-mediated interactions in biological systems: New discoveries and applications. Curr Opin Biotechnol 2008;19:344–50. 10.1016/j.copbio.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 5. Ji X, Nielsen AL, Heinis C. Cyclic peptides for drug development. Angew Chem Int Ed Engl 2024;63:e202308251. 10.1002/anie.202308251. [DOI] [PubMed] [Google Scholar]
- 6. Whitty A, Zhong M, Viarengo L. et al. Quantifying the chameleonic properties of macrocycles and other high-molecular-weight drugs. Drug Discov Today 2016;21:712–7. 10.1016/j.drudis.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu S, Tan P, Tang Q. et al. Enhancing the stability of antimicrobial peptides: From design strategies to applications. Chem Eng J 2023;475:145923. 10.1016/j.cej.2023.145923. [DOI] [Google Scholar]
- 8. Dougherty PG, Sahni A, Pei D. Understanding cell penetration of cyclic peptides. Chem Rev 2019;119:10241–87. 10.1021/acs.chemrev.9b00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jumper J, Evans R, Pritzel A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596:583–9. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abramson J, Adler J, Dunger J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024;630:493–500. 10.1038/s41586-024-07487-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu C, Cao S, Shang T. et al. Predicting the structures of cyclic peptides containing unnatural amino acids by HighFold2. Brief Bioinform 2025;26:bbaf202. 10.1093/bib/bbaf202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Badaczewska-Dawid A, Wróblewski K, Kurcinski M. et al. Structure prediction of linear and cyclic peptides using CABS-flex. Brief Bioinform 2024;25:bbae003. 10.1093/bib/bbae003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Williams TM, Sable R, Singh S. et al. Peptide ligands for targeting the extracellular domain of EGFR: Comparison between linear and cyclic peptides. Chem Biol Drug Des 2018;91:605–19. 10.1111/cbdd.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mao Q, Shang T, Xu W. et al. NCPepFold: Accurate prediction of noncanonical cyclic peptide structures via cyclization optimization with multigranular representation. J Chem Theory Comput 2025;21:4979–91. 10.1021/acs.jctc.5c00139. [DOI] [PubMed] [Google Scholar]
- 15. Rettie SA, Campbell KV, Bera AK. et al. Cyclic peptide structure prediction and design using AlphaFold2. Nat Commun 2025;16:4730. 10.1038/s41467-025-59940-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wohlwend J, Corso G, Passaro S. et al. Boltz-1 democratizing biomolecular interaction Modeling. BioRxiv 2024; 2024.11.19.624167. 10.1101/2024.11.19.624167. [DOI]
- 17. Xie X, Li CZ, Lee JS. et al. CyclicBoltz1, fast and accurately predicting structures of cyclic peptides and complexes containing non-canonical amino acids using AlphaFold 3 framework. BioRxiv 2025; 2025.02.11.637752. 10.1101/2025.02.11.637752. [DOI] [Google Scholar]
- 18. Gligorijević V, Renfrew PD, Kosciolek T. et al. Structure-based protein function prediction using graph convolutional networks. Nat Commun 2021;12:3168. 10.1038/s41467-021-23303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kha Q-H, Tran T-O, Nguyen T-T-D. et al. An interpretable deep learning model for classifying adaptor protein complexes from sequence information. Methods 2022;207:90–6. 10.1016/j.ymeth.2022.09.007. [DOI] [PubMed] [Google Scholar]
- 20. Zhao Z, Gui J, Yao A. et al. Improved prediction model of protein and peptide toxicity by Integrating Channel attention into a convolutional neural network and gated recurrent units. ACS Omega 2022;7:40569–77. 10.1021/acsomega.2c05881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang C, Zhang C, Shang T. et al. HighFold: Accurately predicting structures of cyclic peptides and complexes with head-to-tail and disulfide bridge constraints. Brief Bioinform 2024;25:bbae215. 10.1093/bib/bbae215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Westbrook JD, Shao C, Feng Z. et al. The chemical component dictionary: Complete descriptions of constituent molecules in experimentally determined 3D macromolecules in the protein data Bank. Bioinforma Oxf Engl 2015;31:1274–8. 10.1093/bioinformatics/btu789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin Z, Akin H, Rao R. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023;379:1123–30. 10.1126/science.ade2574. [DOI] [PubMed] [Google Scholar]
- 24. Wang G, Fang X, Wu Z. et al. HelixFold: An efficient implementation of AlphaFold2 using PaddlePaddle. ArXiv 2022; 2207.05477. 10.48550/arXiv.2207.05477. [DOI] [Google Scholar]
- 25. Liu L, Yang L, Cao S. et al. CyclicPepedia: A knowledge base of natural and synthetic cyclic peptides. Brief Bioinform 2024;25:bbae190. 10.1093/bib/bbae190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Agrawal P, Bhalla S, Usmani SS. et al. CPPsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res 2016;44:D1098–103. 10.1093/nar/gkv1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mehta D, Anand P, Kumar V. et al. ParaPep: A web resource for experimentally validated antiparasitic peptide sequences and their structures. Database 2014;2014:bau051–1. 10.1093/database/bau051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Toroslu IH. Improving the Floyd-Warshall all pairs shortest paths algorithm. ArXiv 2021; 2109.01872. 10.48550/arXiv.2109.01872. [DOI] [Google Scholar]
- 29. Karimi M, Ignasiak MT, Chan B. et al. Reactivity of disulfide bonds is markedly affected by structure and environment: Implications for protein modification and stability. Sci Rep 2016;6:38572. 10.1038/srep38572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pierce BG, Wiehe K, Hwang H. et al. ZDOCK server: Interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 2014;30:1771–3. 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ishimoto N, Wong JLC, He S. et al. Cryo-EM structure of the conjugation H-pilus reveals the cyclic nature of the TrhA pilin. Proc Natl Acad Sci 2025;122:e2427228122. 10.1073/pnas.2427228122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mezö G, Hudecz F. Synthesis of linear, branched, and cyclic peptide chimera. Methods Mol Biol Clifton NJ 2005;298:63–76. [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y, Liu R, Jin H. et al. Straightforward access to linear and cyclic polypeptides. Commun Chem 2018;1:1–7. [Google Scholar]
- 34. Ding Y, Lambden E, Peate J. et al. Rapid peptide cyclization inspired by the modular logic of nonribosomal peptide Synthetases. J Am Chem Soc 2024;146:16787–801. 10.1021/jacs.4c04711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu H, Bai L, Jiang X. Recent progress on total synthesis of cyclic peptides. Tetrahedron Lett 2024;151:155314. 10.1016/j.tetlet.2024.155314. [DOI] [Google Scholar]
- 36. Thakkar A, Trinh TB, Pei D. Global analysis of peptide cyclization efficiency. ACS Comb Sci 2013;15:120–9. 10.1021/co300136j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data this work uses is available at (https://github.com/hongliangduan/HighFold3) or the Supporting data.