

Abstract
Purpose
The current study was designed to develop and validate a population pharmacokinetic (PPK) model of methotrexate (MTX) in pediatric patients with acute lymphoblastic leukemia (ALL). We aimed to develop a PPK model to evaluate the effects of potential covariates and explore dosing regimen.
Patients and Methods
We retrospectively analyzed data from 214 pediatric patients with ALL who received high-dose methotrexate (HD-MTX) therapy, incorporating a total of 1672 plasma concentration measurements. Plasma samples were assayed using Enzyme-Multiplied Immunoassay Technique (EMIT). The PPK model was developed using a nonlinear mixed-effects model approach utilizing the NONMEM 7.4 software. Monte Carlo simulation was conducted to optimize the dosage regimen.
Results
A two-compartment model with a 1-year age cutoff was found to adequately describe the PK disposition of MTX. The population typical values for clearance (CL) and volume of distribution (V) were 4.46 L/h and 15.9 L, respectively. Estimated glomerular filtration rate (eGFR) was identified as the most significant covariate, with body weight and blood urea nitrogen (BUN) also emerging as primary factors influencing CL. The model exhibited satisfactory predictive performance, with bootstrap analysis showing a 93.6% success rate. For external validation, the median prediction error (MPE) and median absolute prediction error (MAPE) were −3.99% and 22.4%, respectively. Additionally, 46.36% of prediction errors fell within ±20%, and 64.55% within ±30%, confirming the model’s acceptable predictive performance. Monte Carlo simulations showed that optimized loading doses significantly improved steady-state MTX levels and reduced delayed elimination, especially in patients with renal impairment (eGFR < 100 mL/min/1.73m²).
Conclusion
The PPK model established in this study can well predict the MTX exposure level in children with ALL, and it clearly identifies renal function status as a key basis for adjusting the loading dose. Combined with the results of Monte Carlo simulations, we propose that for patients with mild to moderate renal insufficiency, increasing the loading dose and prolonging the infusion time can improve the steady-state concentration compliance rate while reducing the risk of delayed excretion, providing a more targeted reference for clinical decision-making.
Keywords: methotrexate, acute lymphocytic leukemia, population pharmacokinetics, nonlinear mixed-effect model, dosing regimen
Introduction
Acute lymphoblastic leukemia (ALL) is the most common subtype of leukemia in the pediatric population. Epidemiological studies indicate that it accounts for approximately 75–80% of childhood leukemia cases.1–3 Methotrexate (MTX), a classic antimetabolite antineoplastic agent, exerts its therapeutic effects primarily by competitively inhibiting the enzymatic activity of dihydrofolate reductase.4,5 This disruption interferes with the de novo synthesis of purine and pyrimidine nucleotides, ultimately inhibiting the proliferation of tumor cells.6 In the clinical treatment of ALL, MTX plays a pivotal role, particularly high-dose methotrexate (HD-MTX), which has emerged as a crucial therapeutic strategy for improving long-term survival rates and prognosis in pediatric patients.7
Owing to the polyglutamylation of MTX within cells, its intracellular retention time is significantly prolonged. This process not only enhances the antitumor activity of MTX but also increases cellular toxicity.8 Metabolic pathway analysis indicates that the liver is the primary organ responsible for MTX metabolism. The major metabolite formed is 7-hydroxymethotrexate, which has been reported to exhibit even greater cytotoxicity than MTX itself.5,8,9 Regarding the excretion mechanisms, MTX and its metabolites are primarily eliminated through renal clearance, with the majority being excreted unchanged in urine.10–12 However, due to the low solubility of MTX and its metabolites in acidic urine, crystallization within renal tubules may occur, leading to nephrotoxicity and impaired drug excretion.13,14 Therefore, in clinical practice, strict adherence to supportive measures such as adequate hydration, urinary alkalinization, and leucovorin rescue is essential during HD-MTX administration to reduce the risk of nephrotoxicity and other systemic toxicities.8,15
Population pharmacokinetic (PPK) modeling provides a reliable approach for identifying covariates that influence the pharmacokinetics of MTX, enabling the prediction of drug exposure levels in patients and reducing the risk of adverse reactions.
In PPK studies of MTX for treating childhood ALL, although multiple models have been developed to analyze drug variability and optimize dosing regimens, there are still significant limitations.16–18 Firstly, despite the clear recognition that the kidney is the core pathway for MTX excretion, most studies have insufficiently explored the association between renal function and clearance, failing to systematically evaluate the predictive value of different renal function markers.19,20 Secondly, research on the infant population has shortcomings, particularly neglecting the significant differences in physiological characteristics between infants and older children, which leads to inadequate accuracy in depicting the pharmacokinetic characteristics of MTX in younger patients and restricts the formulation of individualized dosing regimens.
These limitations highlight the importance of refining age stratification (such as focusing on the 1-year age cutoff) and integrating multi-dimensional renal function markers to enhance the predictive ability of the models.
Based on the aforementioned research limitations, this study collects MTX plasma concentration data from children with ALL over the past five years. By establishing a PPK model of MTX, it focuses on exploring the refined stratification effect with 1 year of age as the cutoff and the impact of multi-dimensional renal function markers such as estimated glomerular filtration rate (eGFR) and blood urea nitrogen (BUN) on MTX pharmacokinetics, clarifying the mechanism of action of key covariates. Ultimately, it aims to provide a more accurate theoretical basis and practical reference for formulating individualized MTX dosing regimens for younger children and patients with renal dysfunction.
Materials and Methods
Study Population
The study protocol and retrospective clinical data collection were approved by the Ethics Committee of Anhui Provincial Children’s Hospital (Approval No. EYLL-2025-025). A retrospective analysis was conducted on pediatric patients (age ≤ 18 years) diagnosed with ALL at our institution from May 2021 to November 2024. Inclusion criteria were as follows: a confirmed pathological diagnosis of ALL, receipt of HD-MTX therapy, and at least one plasma concentration measurement during treatment. The study strictly adhered to the principles of the Declaration of Helsinki and its subsequent amendments to ensure scientific integrity and ethical compliance.
Treatment Regimen
Before October 2021, pediatric patients were treated according to the CCLG - ALL - 2018 protocol. Since October 2021, the CCCG - ALL - 2020 protocol has been adopted. Based on risk stratification, patients were categorized into low-risk (LR), intermediate and high-risk (I/HR) groups. The MTX dosing regimen for the LR group was 3 g/m², while the I/HR groups received 5 g/m². All patients received HD-MTX via a 24-hour intravenous infusion using a loading dose strategy: 10% of the total dose was administered over 0.5 h, followed by continuous infusion of the remaining 90% over 23.5 h. Routine plasma concentration monitoring was performed at 20–24 hours, 44–48 hours, and 68–72 hours after the start of infusion, and continues until the concentration of MTX falls to ≤ 0.2 μmol/L.
In this study, demographic data and laboratory test results of pediatric patients were collected via the electronic medical record system, including age, sex, height (HT), weight (WT), body surface area (BSA), serum creatinine (Scr), BUN, alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin (ALB), total bilirubin (TBIL), and alkaline phosphatase (ALP). The eGFR was calculated using the bedside Schwartz equation.21
Biological Analysis
The plasma concentration of MTX was determined using an enzyme multiplied immunoassay technique (EMIT, Viva-ProE, Siemens, Munich, Germany). The calibration range for this assay was between 0.3 and 2600 μmol/L. Previous studies had demonstrated that the accuracy of this method consistently remains within an acceptable range (%CV < 4%) when the concentrations exceed 0.17 µmol/L.22 Therefore, 0.17 µmol/L was set as the lower limit of quantification (LLOQ), and values below this threshold were excluded from the analysis.
Population Pharmacokinetic Model Development
PPK analysis was conducted using the nonlinear mixed-effects model approach with NONMEM software (version 7.4, ICON Development Solutions, Ellicott City, MD, United States) and PsN (Perl-speaks-NONMEM, Version 4.6.0). R software (version 3.4.0, http://www.r-project.org/) was used to visualize the outputs. The first-order conditional estimation method with η-ε interaction (FOCE-I) was employed to calculate pharmacokinetic (PK) parameters. A two-compartment model with first-order absorption and elimination (ADVANS3 TRANS4) was selected as the base model. Exponential models were used to describe inter-individual variability, while proportional models were applied to characterize residual variability.
Covariate Model
The covariates examined during this study were classified as continuous or categorical variables. Continuous variables included age, WT, BSA, Scr, eGFR, AST, ALT and BUN. The categorical variables were sex and the number of methotrexate chemotherapy cycles (MTXNUM). Potentially significant continuous covariates were normalised by the median and examined using the exponential model (Eq. 1). Eq. 2 was used to examine the binary variables.
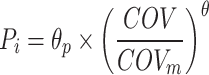 |
(1) |
 |
(2) |
where Pi is the population parameter value of the individual, θp is the typical value of the parameter, COV is the covariate value of the individual, COVm is the median value of the covariate, θ is the fixed effect parameter scaling the effect of COV on Pi, θcov is the parameter indicating the covariate effect, and CATEGORICAL is a categorical variable which can take on values of zero or one.
Covariates that were more commonly used clinically or that had the highest correlation with PK parameters were selected for inclusion in the model through base model estimation. NONMEM designates the −2-fold log-likelihood value as the OFV, and it is used as an overall indicator of fit. The effects of covariates on PK parameters were examined using a stepwise method.
During the forward inclusion process, the covariates were individually added to the model. Assuming that the test level α = 0.05, if the decrease in OFV exceeded 3.84 (χ2 test; P < 0.05; df = 1) after adding a covariate, then we added the covariate to the model. Otherwise, it was excluded. The process was repeated until the OFV showed no significant change. At that time, a full regression model was established. The covariates in the full regression model were individually eliminated using the one-by-one backward elimination method. The test level was set at a more stringent α = 0.001, and the factor was retained in the model if the change in the OFV was more than 10.83 (χ2 test; P < 0.001; df = 1). The covariates in the model were selected based on the physiological plausibility of the parameter estimates, GOF plots, and statistical significance. The covariates evaluated in this study included age, WT, sex, BSA, Scr, eGFR, AST, ALT, BUN, and MTXNUM.
Model Evaluation
Internal validation of the model was performed using goodness-of-fit (GOF) plots, bootstrap analysis, and prediction-corrected visual predictive check (pc-VPC). About 20% of the pediatric patient date was randomly selected for external validation, and predictive performance was assessed by calculating the prediction error (PE), absolute prediction error (APE), median prediction error (MPE), and median absolute prediction error (MAPE). Additionally, the percentages of PE within ± 20% (F20) and ± 30% (F30) were calculated as overall indicators of predictive performance. The model was considered to have acceptable predictive performance when MPE was ≤ ± 20%, MAPE was ≤ 30%, F20 was > 35%, and F30 was > 50%.
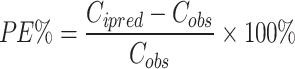 |
(3) |
 |
(4) |
 |
(5) |
 |
(6) |
Model Application
Literature review revealed that there were differences in the loading dose strategies used in clinical protocols for HD-MTX treatment of ALL. For example, Susanna et al23 administered 20% of the total dose as a loading dose over 1-hour; Faganel et al24 and Christine et al25 used 10% of the total dose infused over 1-hour, and Beechinor et al26 used 5% of the total dose infused within 20 minutes. To evaluate the impact of loading doses on treatment outcomes, this study utilized Monte Carlo simulation to assess the effects of different loading doses (0%, 5%, 10%, 15%, 20%) infused over 0.5-hour and 1-hour on the steady-state drug concentrations in patients with varying levels of renal function (eGFR: 160, 100, and 80 mL/min/1.73m²). The simulation was conducted using a 5-year-old child as the model subject, with baseline characteristics of 19 kg in weight and 0.77 m² in BSA. Dosing was determined based on BSA: 3 g/m² for the IR group and 5 g/m² for the I/HR groups.
The analysis focused on the influence of loading dose regimens on the probability of target attainment (PTA) of steady-state concentrations and the incidence of delayed elimination. According to current treatment protocols, the target steady-state concentration is 26–60 μmol/L for LR patients and 52–100 μmol/L for I/HR patients, with a requirement that the 44-hour plasma concentration remains below 1 μmol/L. During the simulations, 1000 virtual individuals were generated for each treatment subgroup.
Results
Patient Characteristics
A total of 214 pediatric patients and 1672 plasma concentration data points were included in the PPK analysis. The modeling group consisted of 171 pediatric patients with a total of 1342 concentration data, while the external validation group included 43 pediatric patients with 330 concentration data. The demographic information and clinical characteristics of the study subjects were detailed in Table 1.
Table 1.
Baseline Demographic and Clinical Characteristics
| Variable | Modeling Data | External Validation Data | ||
|---|---|---|---|---|
| Mean | Median (Range) | Mean | Median (Range) | |
| No. of patients (Male/Female) | 171 | 43 | ||
| Male | 105 | 26 | ||
| Female | 66 | 17 | ||
| Samples | 1342 | 330 | ||
| Age, year | 5.52 | 5 (0.65–14) | 6.35 | 6 (0.86–14) |
| Body Weight, kg | 22.43 | 19.50 (7–62.50) | 26.20 | 21 (7.50–71) |
| Height, cm | 114.98 | 111 (21.60–180) | 121.48 | 123 (71–173) |
| Body Surface Area, m2 | 0.83 | 0.77 (0.36–1.71) | 0.92 | 0.84 (0.39–1.83) |
| Alanine Aminotransferase, U/L | 53.33 | 29.50 (3.30–1268.6) | 52.80 | 29 (5.90–503.20) |
| Aspartate Aminotransferase, U/L | 71.15 | 36.30 (10.20–1258) | 70.70 | 37 (5.90–782.80) |
| Blood Urea Nitrogen, mmol·L−1 | 3.15 | 2.95 (0.40–16.70) | 3.04 | 2.90 (0.80–7.50) |
| Serum Creatinine, mg·dL−1 | 0.32 | 0.29 (0.07–2.09) | 0.33 | 0.29 (0.14–2.02) |
| Estimated Glomerular Filtration Rate, mL·min−1/1.73m2 | 162.40 | 160 (21.90–405.65) | 164.20 | 161.20 (31.78–304.24) |
Population Pharmacokinetic Modelling
A two-compartment model with first-order absorption and elimination adequately described the PK characteristics of MTX in ALL pediatric patients. Covariate analysis identified that eGFR had the most significant effect on clearance (CL). Incorporating eGFR into the model resulted in a substantial reduction in the objective function value (OFV) by 170.45, accompanied by a marked improvement in GOF plots, underscoring its critical role in model development.
Age was also identified as a statistically significant covariate influencing CL. Modeling the relationship between age and CL using a population PK model with a 1-year age cutoff further reduced the OFV by 55.825, indicating that age was an important covariate influencing MTX clearance.
Additionally, WT, BUN, and BSA demonstrated significant effects on CL and volume of distribution (V), respectively, and were therefore retained in the final model. Although both BUN and eGFR are renal function predictors, the scatter plot, and correlation analysis (correlation coefficient ρ = −0.1192, variance inflation factor (VIF) = 1.007457) revealed no significant collinearity between them, indicating a weak correlation. The complete covariate screening workflow was delineated in Table S1. Final population PK parameter estimates were summarized in Table 2. The final model was represented as follows:
Table 2.
NONMEM Estimates and Bootstrap Analysis of the Final Model
| Parameters | NONMEM | Bootstrap | ||
|---|---|---|---|---|
| Estimate | RSE (%) | Median | 2.5th–97.5th Percentile |
|
| θCL age>1 (L/h) | 4.46 | 3 | 4.63 | 4.23–4.86 |
| θCL age ≤1 (L/h) | 1.69 | 10 | 1.76 | 0.65–2.73 |
| θQ (L/h) | 0.149 | 5 | 0.147 | 0.124–0.174 |
| θV1 (L) | 15.90 | 4 | 16.01 | 13.84–17.89 |
| θV2 (L) | 7.23 | 9 | 7.27 | 4.89–9.57 |
| θGFR | 0.537 | 6 | 0.512 | 0.354–0.721 |
| θBSA | 1.10 | 10 | 1.09 | 0.76–1.45 |
| θWT | 0.45 | 11 | 0.45 | 0.35–0.55 |
| θBUN | −0.0823 | 24 | −0.0779 | −0.1553–-0.0093 |
| Interindividual variability ωCL (%) | 21.24 | 14 | 20.73 | 17.61–24.33 |
| Interindividual variability ωQ (%) | 27.66 | 31 | 27.60 | 9.38–38 |
| Interindividual variability ωV2 (%) | 41.47 | 27 | 40.24 | 27.39–51.86 |
| Residual unexplained variability (%) | 44.16 | 5 | 43.82 | 40.74–47.32 |
Abbreviations: NONMEM, Nonlinear Mixed Effects Modeling; RSE, Relative Standard Error; CL, Clearance; V1, the central volume of distribution; V2, the peripheral volume of distribution; Q, inter-compartmental (central-peripheral) clearance; GFR, Glomerular Filtration Rate; BSA, Body Surface Area; WT, Weight; BUN, Blood Urea Nitrogen.
If age >1 years old CL (L/h) =
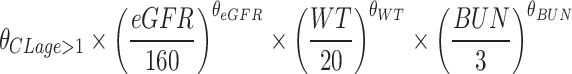 |
If age ≤1 years old CL (L/h) =
 |
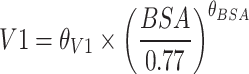 |
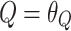 |
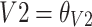 |
Model Evaluation
The GOF plots (Figure 1) demonstrated that the model adequately described the observed data. Both observed concentration (DV) vs population predicted concentration (PRED) and DV vs individual predicted concentration (IPRED) displayed no apparent structural bias, with data points uniformly distributed around the reference line y = x. Additionally, the conditional weighted residuals (CWRES) were symmetrically distributed within the range of y = ± 2 without significant trend changes. Bootstrap analysis yielded parameter estimates deviating by less than 16% from the final model, with a 93.6% success rate, indicating robust model stability. The prediction-corrected visual predictive check (pc-VPC) plot (Figure 2A) showed that the 5th, 50th, and 95th percentiles of the observed data generally fell within the 95% confidence intervals (CIs) of the predicted percentiles, demonstrating that the model adequately described the characteristics of the original data.
Figure 1.

Goodness of fit plots of the final MTX PPK model. (A) Individual predicted concentration (IPRED) vs observed concentration (DV); (B) Population predicted concentration (PRED) vs DV; (C) PRED vs conditional weighted residuals (CWRES); (D) Time vs CWRES.
Figure 2.

Prediction-corrected visual predictive check (pc-VPC). (A) Internal validation. (B) External evaluation. The shaded regions respectively indicate the 90% CIs of the 5th percentile, median, and 95th percentile of PRED. Hollow dots represent observed data. The lines outline the best fits of the 5th percentile (dashed), median (solid), and 95th percentile (dashed) of the data.
To further evaluate the model’s performance, the study conducted external validation and calculated the prediction error. The results showed that the MPE% and MAPE were −3.99% and 22.4%, while F20% and F30% reached 46.36% and 64.55%, respectively. These findings indicated that the prediction error of the model was within an acceptable range. The pc-VPC for external validation (Figure 2B) demonstrated that the simulated data exhibited distribution characteristics similar to those of the observed data, suggesting a high level of consistency.
Analysis of Probability of Target Attainment
Simulation results (Figure 3) demonstrated that increasing the loading dose (5–20%) significantly improved the PTA for steady-state concentrations in both LR and I/HR groups, though the magnitude of improvement varied depending on renal function and infusion duration. When eGFR was 160 mL/min, a loading dose of 10% or higher could achieve a PTA of over 90% in the LR group with a 0.5-hour infusion. However, in the I/HR group, PTA slightly decreased when the loading dose exceeded 10%. At eGFR levels of 100 and 80 mL/min, increasing the loading dose from 0% to 20% resulted in the PTA of the steady-state concentration increasing by 30.8% and 31%, respectively, in the LR group after a 0.5-hour infusion. Similarly, in the I/HR group, PTA of the steady-state concentration rose by 31% and 35.6%, respectively, with the same infusion duration. These findings suggested that patients with impaired renal function might require higher loading doses to achieve target concentrations.
Figure 3.

Effects of different loading doses infused within 0.5-hour and 1-hour on the probability of target attainment of steady-state concentration target. The red dashed lines represent the lower and upper limits of the target steady-state concentrations for LR (26, 60 μmol/L) and I/HR (52, 100 μmol/L), respectively. The gray dotted line indicates the 90% probability of target attainment of steady-state concentration target. In each panel, 0% (blue), 5% (Orange), 10% (green), 15% (red), and 20% (purple) represent different loading doses.
At the same loading dose, a 0.5-hour infusion results in higher PTA than a 1-hour infusion, but it may increase the risk of delayed excretion, especially in patients with renal insufficiency (Table 3). When the eGFR was 80 mL/min and the loading dose increased from 0% to 20%, the incidence of delayed excretion in the LR group decreased by 4.4% (0.5-hour infusion) and 5.3% (1-hour infusion), respectively. In the I/HR group, even with a 20% loading dose, the incidence of delayed excretion remained as high as 99%, confirming that adjusting the loading dose alone was ineffective for this subgroup. For such patients, a multi-dimensional intervention strategy was required. In addition to optimizing the loading dose and infusion duration, the total dose should be individualized based on renal function, and hydration and urine alkalinization should be intensified to accelerate drug excretion. Meanwhile, therapeutic drug monitoring (TDM) should be used to closely track plasma concentrations at 44 hours and 68 hours, and the rescue dose of leucovorin should be dynamically optimized to maximize therapeutic efficacy while minimizing toxicity risks.
Table 3.
The Incidence of Delayed Excretion of MTX (44-Hour Concentration > 1 μmol/L)
| Time (h) |
Loading Dose (%) |
LR/eGFR (mL/min/1.73 m2) | I/HR/eGFR (mL/min/1.73 m2) | ||||
|---|---|---|---|---|---|---|---|
| 160 | 100 | 80 | 160 | 100 | 80 | ||
| 0.5 | 0 | 25.4% | 67.0% | 84.3% | 50.1% | 86.5% | 99.5% |
| 5 | 22.9% | 65.8% | 83.9% | 48.4% | 87.3% | 99.6% | |
| 10 | 21.7% | 64.3% | 83.1% | 46.7% | 86.4% | 99.4% | |
| 15 | 20.4% | 62.9% | 81.6% | 45.4% | 85.7% | 99.4% | |
| 20 | 21.5% | 62.1% | 79.9% | 44.2% | 84.6% | 99.5% | |
| 1 | 0 | 25.4% | 67.0% | 84.3% | 50.1% | 86.5% | 99.5% |
| 5 | 20.8% | 63.6% | 82.5% | 46.1% | 86.3% | 99.6% | |
| 10 | 19.6% | 62.5% | 81.0% | 45.0% | 85.3% | 99.5% | |
| 15 | 19.0% | 60.9% | 80.5% | 43.9% | 84.5% | 99.4% | |
| 20 | 20.3% | 59.9% | 79.0% | 43.3% | 83.4% | 99.4% | |
Abbreviations: LR, low-risk; I/HR, intermediate and high-risk; eGFR, estimated Glomerular Filtration Rate.
Based on the findings of this study, we propose the following optimized recommendations for the loading dose and infusion duration of HD-MTX in patients with different renal function statuses (Table 4). For patients with the eGFR ≥ 160 mL/min, it is recommended to complete the infusion of 10–15% of the loading dose within 0.5-hour to improve the rate of achieving the steady-state concentration. For LR and I/HR patients with 100 mL/min ≤ eGFR < 160 mL/min, as well as LR patients with 80 mL/min ≤ eGFR < 100 mL/min, to balance efficacy and safety and reduce the risk of delayed clearance, a 1-hour infusion protocol is suggested, with the loading dose controlled at 20% and combined with real-time TDM to guide subsequent administration. It is particularly important to note that for I/HR patients with 80 mL/min ≤ eGFR < 100 mL/min, the total dose should first be adjusted individually according to their renal function status, and then further research on optimizing the loading dose and infusion duration needs to be carried out to ensure the safety and accuracy of the treatment.
Table 4.
Optimizing Loading Dose Regimen Based on Renal Function
| eGFR (mL/min/1.73m2) |
Loading Dose (%) | Time (h) |
|
|---|---|---|---|
| LR | I/HR | ||
| ≥160 | 15% | 10% | 0.5 |
| 100–160 | 20% | 20% | 1 |
| 80–100 | 20% | NA | 1 |
Note: 100–160 mL/min (eGFR: ≥ 100 and < 160 mL/min); 80–100 mL/min (eGFR: ≥ 80 and < 100 mL/min).
Abbreviations: LR, low-risk; I/HR, intermediate and high-risk; eGFR, estimated Glomerular Filtration Rate; NA, Not applicable.
Discussion
In recent years, significant progress has been made in the treatment of ALL, with dose optimization of MTX and improved supportive care enhancing therapeutic efficacy. This study focused on pediatric patients newly diagnosed with ALL within the past five years and developed a PPK model for MTX in ALL treatment. Model fitting and validation indicated that WT, BSA, eGFR, BUN, and AGE were key factors influencing MTX PK parameters. Specifically, WT and BSA significantly affected MTX CL and V, respectively. In detail, increased body weight correlated with elevated CL, while V showed a positive correlation with BSA. These findings were consistent with previous studies.27–30 Compared with published literature,23,24,27,31,32 the estimated of V from this study’s model aligned with reported values, confirming the reliability of the model in characterizing MTX distribution.
Covariate analysis indicated that eGFR was a key factor affecting the CL of MTX, consistent with MTX’s PK profile of primarily renal excretion.30,33–35 Notably, this study innovatively incorporated both BUN and eGFR into the clearance model. Among them, BUN can indirectly reflect the accumulation risk of MTX and its metabolites (such as 7-hydroxymethotrexate). An increase in BUN indicates an elevated renal excretion load, which may be accompanied by the deposition of MTX crystals in renal tubules, thereby exacerbating kidney injury. Studies have found that BUN and MTX serum concentrations ≥ 2 μmol/L at 48–72 hours have a synergistic effect. Combined monitoring can improve the predictive efficacy for acute kidney injury.36 In addition, BUN has more advantages in assessing long-term kidney damage (such as the progression of chronic kidney disease), as it is associated with changes in the expression of urea transporters (such as UT-A1 and UT-B1). Abnormalities in these transporters can affect the renal tubular reabsorption of MTX and urine concentration function.37 By capturing multi-dimensional information on renal excretion function, BUN complements eGFR. In this study, after introducing BUN and eGFR, the OFV decreased significantly by 170 and 17.57, respectively (P<0.001). Additionally, the GOF plots demonstrated improved agreement between predicted and observed values. These results suggest that eGFR and BUN may reflect renal function status from different perspectives. Their synergistic effect enables a more comprehensive and accurate assessment of the impact of renal function on drug clearance. This conclusion was corroborated by the study of Pricilla et al,38 which confirmed that the combined use of Scr and BUN significantly enhances model predictive performance. Our study highlighted the important value of using multiple renal function markers in PPK modeling.
In published MTX PPK models, age was often incorporated as a continuous variable.34,39,40 However, infants and children exhibited significant differences in hepatic and renal function during growth, leading to distinct impacts on drug metabolism. This study innovatively introduced 1 year of age as a cutoff point in the model. Infants under 1 year old had incompletely developed hepatic and renal functions, resulting in lower drug clearance rates, while children over 1 year old gradually approach adult-level organ function, exhibiting significantly higher clearance rates.41,42 Studies have confirmed that infants over 12 months of age exhibit a significant upward trend in MTX clearance, with their drug clearance efficiency approaching that of older children.43,44 This age-based distinction using 1 year as the cutoff more precisely reflects the leap in clearance capacity brought about by the maturation of renal function (such as GFR and renal tubular function) during infancy. It breaks through the limitation of previously analyzing infants as a homogeneous group. Model analysis revealed that using a piecewise function at 1 year of age significantly improved the GOF plots (ΔOFV = −55.825), compared to the traditional continuous variable approach (ΔOFV = −12.535), further confirming the impact of age on the CL of MTX.
This study used Monte Carlo simulations to primarily analyze the impact of loading dose and infusion duration on the efficacy and safety of HD-MTX, providing important evidence for clinical individualized treatment. The simulation threshold of 1 μmol/L at 44 hours was selected primarily based on consensus from clinical protocols and evidence-based guidelines. The Chinese Evidence-Based Guidelines for High-Dose Methotrexate Use clearly states that the C48h ≤ 1 μmol/L is generally recognized as the standard for normal clearance. The risk of adverse events is significantly reduced at this threshold.45 Clinical data also confirm that the C48h > 1μmol/L is associated with increased incidence of multi-system toxicities, among which a concentration > 5 μmol/L indicates early delayed excretion, and >10 μmol/L is directly linked to a high risk of toxicity.8,46,47 The selection of 44 hours as the monitoring time point both aligns with the routine clinical monitoring protocol of our institution and is close to the recognized 48-hour standard, enabling earlier prediction of clearance trends for timely intervention adjustments. In summary, the choice of this threshold and time point provides a clinically relevant basis for evaluating the impact of loading dose strategies on drug exposure and treatment safety.
The study found that the benefits of loading doses were closely related to the patient’s renal function status. For patients with normal renal function, increasing the loading dose could significantly improve the PTA of steady-state concentrations. However, for patients with reduced renal function, while higher loading doses increase PTA, they also elevate the risk of drug accumulation. The infusion duration also exhibited a dual regulatory effect. Although short-term infusion could improve the PTA of steady-state concentration, it may increase the risk of delayed excretion, especially for patients with poor renal function. Prolonging the infusion duration was the key to balancing efficacy and safety.
However, this study had several limitations. Firstly, as a retrospective study, there may be some deviations in the sampling time points. Secondly, the data were obtained from a single-center which primarily comprises Han Chinese children treated at our institution. This homogeneity may constrain the external generalizability of our findings to other ethnic groups or patient populations across different clinical centers. To address this, future multicenter studies incorporating ethnically diverse cohorts are warranted to validate the broader applicability of the proposed model. Thirdly, the external validation cohort of this study, consisting of 43 patients, is relatively small, which may to some extent limit the generalizability of the research findings. Due to the retrospective design of this study and the limited number of eligible external cases that met the inclusion criteria, a formal power analysis was not performed for the sample size of the external validation cohort. Subsequent studies should include larger and more diverse external validation populations to further enhance the robustness and applicability of the model. Finally, a notable limitation of the present study is the absence of pharmacogenetic data, which precluded including key variants such as SLCO1B1, ABCC2, and MTHFR in covariate analysis. These variants are known regulators of MTX transport and metabolic pathways. Given their well-established role in mediating inter-individual variability in MTX pharmacokinetics, further prospective studies should integrate pharmacogenetic profiling to refine characterization of individual differences and enhance precision of personalized dosing strategies.
Conclusion
This study has established and validated a PPK model of MTX for pediatric patients with ALL in the study institution. The model incorporates the novelty of the 1-year age cutoff and dual use of eGFR and BUN as renal markers. Body weight is also included as a key covariate to evaluate its impact on MTX clearance. Based on simulations from the final model, we provide a theoretical basis for personalized MTX dosing in ALL children, particularly offering important guidance for dose adjustment in children with renal insufficiency. It is worth noting that this study still has certain limitations, such as its retrospective design and the absence of genetic data. Subsequent investigations should prioritize multicenter validation and pharmacogenomic integration to further optimize the model and strengthen its clinical utility.
Acknowledgments
The authors would like to thank An Zhang from Anhui Provincial Children’s Hospital, for his expert assistance in data extraction.
Funding Statement
This research was financially supported by the Foundation of Health Commission of Anhui Province (Grant No. AHWJ2023BAa20159).
Ethics Approval
Studies were performed in accordance with ethical principles based on the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards, International Council for Harmonisation Guideline for Good Clinical Practice, and applicable laws and regulations. The study protocol was approved by the Ethics Committee of Anhui Provincial Children’s Hospital (Approval number:EYLL-2025-025). All data were de-identified prior to analysis to ensure patient confidentiality.
Consent to Participate
Authorization to access patient records had been obtained from the Ethics Committee of Anhui Provincial Children’s Hospital. As this was a retrospective study using anonymized data, the requirement for individual informed consent was waived by the ethics committee.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Stanulla M, Schrappe M. Treatment of childhood acute lymphoblastic leukemia. Semin Hematol. 2009;46(1):52–63. doi: 10.1053/j.seminhematol.2008.09.007 [DOI] [PubMed] [Google Scholar]
- 2.Wu C, Li W. Genomics and pharmacogenomics of pediatric acute lymphoblastic leukemia. Crit Rev Oncol Hematol. 2018;126:100–111. doi: 10.1016/j.critrevonc.2018.04.002 [DOI] [PubMed] [Google Scholar]
- 3.Tasian SK, Loh ML, Hunger SP. Childhood acute lymphoblastic leukemia: integrating genomics into therapy. Cancer. 2015;121(20):3577–3590. doi: 10.1002/cncr.29573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bleyer WA. Methotrexate: clinical pharmacology, current status and therapeutic guidelines. Cancer Treat Rev. 1977;4(2):87–101. doi: 10.1016/s0305-7372(77)80007-8 [DOI] [PubMed] [Google Scholar]
- 5.Maksimovic V, Pavlovic-Popovic Z, Vukmirovic S, et al. Molecular mechanism of action and pharmacokinetic properties of methotrexate. Mol Biol Rep. 2020;47(6):4699–4708. doi: 10.1007/s11033-020-05481-9 [DOI] [PubMed] [Google Scholar]
- 6.Toksvang LN, Shawn HRL, Yang JJ, Schmiegelow K. Maintenance therapy for acute lymphoblastic leukemia: basic science and clinical translations. Leukemia. 2022;36(7):1749–1758. doi: 10.1038/s41375-022-01591-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Qiu Y, Wang Z, et al. High-Dose Methotrexate-Induced Idiopathic Intracranial Hypertension in Infant Acute Lymphoblastic Leukemia. Front Pharmacol. 2020;17(11):839. doi: 10.3389/fphar.2020.00839 [DOI] [Google Scholar]
- 8.Widemann BC, Adamson PC. Understanding and managing methotrexate nephrotoxicity. Oncologist. 2006;11(6):694–703. doi: 10.1634/theoncologist.11-6-694 [DOI] [PubMed] [Google Scholar]
- 9.Song Y, Linlin Liu BL, Liu R, et al. Interaction of nobiletin with methotrexate ameliorates 7-OH methotrexate-induced nephrotoxicity through endoplasmic reticulum stress-dependent PERK/CHOP signaling pathway. Pharmacol Res. 2021;165:105371. doi: 10.1016/j.phrs.2020.105371 [DOI] [PubMed] [Google Scholar]
- 10.Levêque D, Santucci R, Gourieux B, Herbrecht R. Pharmacokinetic drug-drug interactions with methotrexate in oncology. Expert Rev Clin Pharmacol. 2011;4(6):743–750. doi: 10.1586/ecp.11.57 [DOI] [PubMed] [Google Scholar]
- 11.Greenmyer JR, Burd L, Kobrinsky NL. Urine methotrexate concentration at 46–48 hours post-treatment reflects methotrexate-induced acute kidney injury. J Pediatr Pharmacol Therap. 2021;26(3):300–305. doi: 10.5863/1551-6776-26.3.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang W, Zhou H, Liu L. Side effects of methotrexate therapy for rheumatoid arthritis: a systematic review. Eur J Med Chem. 2018;5(158):502–516. doi: 10.1016/j.ejmech.2018.09.027 [DOI] [Google Scholar]
- 13.Xu M, Wu S, Wang Y, et al. Association between high-dose methotrexate-induced toxicity and polymorphisms within methotrexate pathway genes in acute lymphoblastic leukemia. Front Pharmacol. 2022;30(13):1003812. doi: 10.3389/fphar.2022.1003812 [DOI] [Google Scholar]
- 14.Yang S, Zhao F, Song H, Shen D, Xu X. Methotrexate associated renal impairment is related to delayed elimination of high-dose methotrexate. Sci World J. 2015;2015:751703. doi: 10.1155/2015/751703 [DOI] [Google Scholar]
- 15.Howard SC, McCormick J, Pui C-H, Buddington RK, Harvey RD. Preventing and managing toxicities of high-dose methotrexate. Oncologist. 2016;21(12):1471–1482. doi: 10.1634/theoncologist.2015-0164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei S, Zhang S, Wang D, et al. Population pharmacokinetics of high-dose methotrexate in patients with primary central nervous system lymphoma. Front Pharmacol. 2025;19(16):1578033. doi: 10.3389/fphar.2025.1578033 [DOI] [Google Scholar]
- 17.Yang Y, Liu Z, Chen J, Wang X, Jiao Z, Wang Z. Factors influencing methotrexate pharmacokinetics highlight the need for individualized dose adjustment: a systematic review. Eur J Clin Pharmacol. 2024;80(1):11–37. doi: 10.1007/s00228-023-03579-0 [DOI] [PubMed] [Google Scholar]
- 18.Cheng Y, Zhang Y, Zhang Y, Liu M, Zhao L. Population pharmacokinetic analyses of methotrexate in pediatric patients: a systematic review. Eur J Clin Pharmacol. 2024;80(7):965–982. doi: 10.1007/s00228-024-03665-x [DOI] [PubMed] [Google Scholar]
- 19.Wang S, Yin Q, Yang M, Cheng Z, Xie F. External evaluation of population pharmacokinetic models of methotrexate for model-informed precision dosing in pediatric patients with acute lymphoid leukemia. Pharmaceutics. 2023;15(2):569. doi: 10.3390/pharmaceutics15020569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi JY, Kwon H, Kim H, et al. Novel genomic variants influencing methotrexate delayed clearance in pediatric patients with acute lymphoblastic leukemia. Front Pharmacol. 2024;14(15):1480657. doi: 10.3389/fphar.2024.1480657 [DOI] [Google Scholar]
- 21.Schwartz GJ, Muñoz A, Schneider MF, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. 2009;20(3):629–637. doi: 10.1681/ASN.2008030287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chavan P, Bhat V, Karmore A, Mohite R, Waykar S, Kadu H. Establishing Syva Methotrexate Assay on Siemens dimension RxL analyzer: experience in a Tertiary Cancer Care Laboratory. J Lab Phys. 2017;9(1):67–68. doi: 10.4103/0974-2727.187923 [DOI] [Google Scholar]
- 23.Medellin-Garibay SE, Hernández-Villa N, Cecilia Correa-González MNM-B L, et al. Population pharmacokinetics of methotrexate in Mexican pediatric patients with acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2020;85(1):21–31. doi: 10.1007/s00280-019-03977-1 [DOI] [PubMed] [Google Scholar]
- 24.Kotnik BF, Grabnar I, Grabar PB, Dolžan JJ V. Association of genetic polymorphism in the folate metabolic pathway with methotrexate pharmacokinetics and toxicity in childhood acute lymphoblastic leukaemia and malignant lymphoma. Eur J Clin Pharmacol. 2011;67(10):993–1006. doi: 10.1007/s00228-011-1046-z [DOI] [PubMed] [Google Scholar]
- 25.Plard C, Bressolle F, Fakhoury M, et al. A limited sampling strategy to estimate individual pharmacokinetic parameters of methotrexate in children with acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2007;60(4):609–620. doi: 10.1007/s00280-006-0394-3 [DOI] [PubMed] [Google Scholar]
- 26.Beechinor RJ, Thompson PA, Hwang MF, et al. The population pharmacokinetics of high-dose methotrexate in infants with acute lymphoblastic leukemia highlight the need for bedside individualized dose adjustment: a report from the Children’s Oncology Group. Clin Pharmacokinet. 2019;58(7):899–910. doi: 10.1007/s40262-018-00734-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aumente D, Buelga DS, Lukas JC, Gomez P, Torres A, García MJ. Population pharmacokinetics of high-dose methotrexate in children with acute lymphoblastic leukaemia. Clin Pharmacokinet. 2006;45(12):1227–1238. doi: 10.2165/00003088-200645120-00007 [DOI] [PubMed] [Google Scholar]
- 28.Schulte RR, Choi L, Utreja N, Van Driest SL, Stein CM, Ho RH. Effect of SLCO1B1 polymorphisms on high-dose methotrexate clearance in children and young adults with leukemia and lymphoblastic lymphoma. Clin Transl Sci. 2021;14(1):343–353. doi: 10.1111/cts.12879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi Z, Liu Y, Hongyan G, et al. Population pharmacokinetics of high-dose methotrexate in Chinese pediatric patients with medulloblastoma. Biopharm Drug Dispos. 2020;41(3):101–110. doi: 10.1007/s00280-019-03977-1 [DOI] [PubMed] [Google Scholar]
- 30.Panetta JC, Roberts JK, Huang J, et al. Pharmacokinetic basis for dosing high-dose methotrexate in infants and young children with malignant brain tumours. Br J Clin Pharmacol. 2020;86(2):362–371. doi: 10.1111/bcp.14160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao X, Qian X-W, Zhu X-H, et al. Population pharmacokinetics of high-dose methotrexate in Chinese Pediatric patients with acute lymphoblastic leukemia. Front Pharmacol. 2021;13(12):701452. doi: 10.3389/fphar.2021.701452 [DOI] [Google Scholar]
- 32.Taylor ZL, Mizuno T, Punt NC, et al. MTXPK.org: a clinical decision support tool evaluating high-dose methotrexate pharmacokinetics to inform post-infusion care and use of glucarpidase. Clin Pharmacol Ther. 2020;108(3):635–643. doi: 10.1002/cpt.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawakatsu S, Nikanjam M, Lin M, et al. Population pharmacokinetic analysis of high-dose methotrexate in pediatric and adult oncology patients. Cancer Chemother Pharmacol. 2019;84(6):1339–1348. doi: 10.1007/s00280-019-03966-4 [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Li J, Yu L, et al. High-dose methotrexate pharmacokinetics and its impact on prognosis of paediatric acute lymphoblastic leukaemia patients: a population pharmacokinetic study. Br J Haematol. 2024;204(4):1354–1366. doi: 10.1111/bjh.19365 [DOI] [PubMed] [Google Scholar]
- 35.Yin A, de Groot FA, Guchelaar HJ, et al. Population pharmacokinetic and toxicity analysis of high-dose methotrexate in patients with central nervous system lymphoma. Clin Pharmacokinet. 2025;64(1):79–91. doi: 10.1007/s40262-024-01452-6 [DOI] [PubMed] [Google Scholar]
- 36.Lin W-Y, Tsai C-K, Yeh C-M, Li C-J. Identifying risk factors for methotrexate-induced acute kidney injury despite full prevention in patients with primary central nervous system lymphoma. Toxicol Appl Pharmacol. 2025;502:117436. doi: 10.1016/j.taap.2025.117436 [DOI] [PubMed] [Google Scholar]
- 37.Weiner ID, Mitch WE, Sands JM. Urea and ammonia metabolism and the controlof renal nitrogen excretion. Clin J Am Soc Nephrol. 2015;10(8):1444–1458. doi: 10.2215/CJN.10311013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Oliveira Henz P, Pinhatti AV, José Gregianin L, et al. Population pharmacokinetic model of methotrexate in Brazilian pediatric patients with acute lymphoblastic leukemia. Pharm Res. 2023;40(7):1777–1787. doi: 10.1007/s11095-023-03544-7 [DOI] [PubMed] [Google Scholar]
- 39.Hui KH, Chu HM, Fong PS, Cheng WTF, Lam TN. Population pharmacokinetic study and individual dose adjustments of high-dose methotrexate in Chinese pediatric patients with acute lymphoblastic leukemia or osteosarcoma. J Clin Pharmacol. 2019;59(4):566–577. doi: 10.1002/jcph.1349 [DOI] [PubMed] [Google Scholar]
- 40.Wright KD, Panetta JC, Onar-Thomas A, et al. Delayed methotrexate excretion in infants and young children with primary central nervous system tumors and postoperative fluid collections. Cancer Chemother Pharmacol. 2015;75(1):27–35. doi: 10.1007/s00280-014-2614-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology — drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–1167. doi: 10.1056/NEJMra035092 [DOI] [PubMed] [Google Scholar]
- 42.Batchelor HK, Marriott JF. Paediatric pharmacokinetics: key considerations. Br J Clin Pharmacol. 2015;79(3):395–404. doi: 10.1111/bcp.12267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lönnerholm G, Valsecchi MG, De Lorenzo P, et al. Pharmacokinetics of high-dose methotrexate in infants treated for acute lymphoblastic leukemia. Pediatr Blood Cancer. 2009;52(5):596–601. doi: 10.1002/pbc.21925 [DOI] [PubMed] [Google Scholar]
- 44.Thompson PA, Murry DJ, Rosner GL, et al. Methotrexate pharmacokinetics in infants with acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2007;59(6):847–853. doi: 10.1007/s00280-006-0388-1 [DOI] [PubMed] [Google Scholar]
- 45.Song Z, Yang H, Liu S, et al. Medication therapy of high-dose methotrexate: an evidence-based practice guideline of the Division of Therapeutic Drug Monitoring, Chinese Pharmacological Society. Br J Clin Pharmacol. 2022;88(5):2456–2472. doi: 10.1111/bcp.15134 [DOI] [PubMed] [Google Scholar]
- 46.Tsurusawa M, Gosho M, Mori T, et al. Statistical analysis of relation between plasma methotrexate concentration and toxicity in high-dose methotrexate therapy of childhood nonHodgkin lymphoma. Pediatr Blood Cancer. 2015;62(2):279–284. doi: 10.1002/pbc.25305 [DOI] [PubMed] [Google Scholar]
- 47.Ramsey LB, Balis FM, O’Brien MM, et al. Consensus guideline for use of glucarpidase in patients with high-dose methotrexate induced acute kidney injury and delayed methotrexate clearance. Oncologist. 2018;23(1):52–61. doi: 10.1634/theoncologist.2017-0243 [DOI] [PMC free article] [PubMed] [Google Scholar]