

Abstract
To determine the enzymatic function of the starch-related R1 protein it was heterologously expressed in Escherichia coli and purified to apparent homogeneity. Incubation of the purified protein with various phosphate donor and acceptor molecules showed that R1 is capable of phosphorylating glucosyl residues of α-glucans at both the C-6 and the C-3 positions in a ratio similar to that occurring naturally in starch. Phosphorylation occurs in a dikinase-type reaction in which three substrates, an α-polyglucan, ATP, and H2O, are converted into three products, an α-polyglucan-P, AMP, and orthophosphate. The use of ATP radioactively labeled at either the γ or β positions showed that solely the β phosphate is transferred to the α-glucan. The apparent Km of the R1 protein for ATP was calculated to be 0.23 μM and for amylopectin 1.7 mg⋅ml−1. The velocity of in vitro phosphorylation strongly depends on the type of the glucan. Glycogen was an extremely poor substrate; however, the efficiency of phosphorylation strongly increased if the glucan chains of glycogen were elongated by phosphorylase. Mg2+ ions proved to be essential for activity. Incubation of R1 with radioactively labeled ATP in the absence of an α-glucan showed that the protein phosphorylates itself with the β, but not with the γ phosphate. Autophosphorylation precedes the phosphate transfer to the glucan indicating a ping-pong reaction mechanism.
Starch is the storage carbohydrate most widely distributed in the plant kingdom. In storage organs it serves as a long-term carbon reserve, whereas in photosynthetically competent tissues it is transiently accumulated to provide both reduced carbon and energy during periods unfavorable for photosynthesis. Starch consists of essentially linear (amylose) and highly branched (amylopectin) glucose polymers that are arranged as semicrystalline particles, the starch granules. Amylopectin from many sources contains phosphate-monoesters that are covalently bound at the C6 and C3 positions of the glucosyl residues (1). In potato tuber starch ≈0.1–0.5% of the glucose moieties are phosphorylated. The amount of phosphate monoesters in starch strongly influences its physicochemical properties (2) and, therefore, affects the ability of different starches to be used by industry.
In potato tubers the starch-bound phosphate comprises a significant proportion of the total tuber phosphate content, but its function in starch metabolism is not clear. The same holds true for the biochemical reaction(s) leading to the formation of the starch phosphate monoesters. A protein (designated as R1) has been recently identified using a proteomic approach, and circumstantial evidence suggests that it is involved in the phosphorylation of starch (2, 3). The C-terminal sequence of the R1 protein shows some homology to bacterial PEP synthases (pyruvate, water dikinase EC 2.7.9.2), which transfer phosphate from ATP in a dikinase reaction to pyruvate and water. Antisense repression of R1 in potato leads to a strong reduction in the amount of starch-bound phosphate, whereas the expression of R1 in Escherichia coli resulted in an increase in the phosphorylation of the bacterial glycogen (2). In addition, a mutation in a homologous gene in Arabidopsis leads also to a reduction in the phosphate content of leaf starch (3).
Interestingly, the degradability of starch was strongly impaired in these transgenic and mutant plants leading to a starch-excess phenotype in leaves and a repression of cold-sweetening in potato tubers. It cannot be ruled out, therefore, that the R1 protein is involved in starch degradation and that the decrease in starch phosphorylation seen in plants with reduced R1 protein amounts is the result of some pleiotropic effect. This view is supported as the R1 protein binds to leaf starch granules during periods of net starch degradation, but not during synthesis (4). Furthermore, in various plant species R1 levels did not correlate with the degree of starch phosphorylation (5).
In the present communication we describe the purification of the heterologously expressed potato R1 protein to apparent homogeneity and its functional analysis in vitro. The results clearly show that R1 catalyses the phosphorylation of starch-like glucans.
Materials and Methods
Materials.
The following commercial biochemicals were used: amylopectin from potato and corn (Sigma), glycogen from bovine liver (Fluka), and amylose from potato (Merck), myokinase (EC 2.7.4.3) from rabbit muscle (Sigma), pyruvate kinase (EC 2.7.1.40) from rabbit muscle (Roche), and phosphorylase a (EC 2.4.1.1) from rabbit muscle (Sigma). [β-33P]ATP [1 mCi/ml; 3000 Ci/mmol (1 Ci = 37 GBq)] and [ γ-33P]ATP (10 mCi/ml, 3000 Ci/mmol) were purchased from NEN.
Glucose-3-phosphate was synthesized in 45% overall yield from β-d-(1,2)(4,6)-diacetone glucose (Aldrich) following a procedure described by Perich and Johns (6, 7), which includes 3-phosphorylation using dibenzyl diisopropyl-phosphoramidite and subsequent deprotection. The product was identified to be (α, β)-d-glucose-3-phosphate by NMR and mass spectrometry. The following characteristics were obtained. Rf (MeOH): 0.52; matrix-assisted laser desorption ionization (MALDI)-MS (positive ion mode, matrix: THAP): 299.1 (M+K), 283.1 (M+Na), 261.1 (M+H); 1H NMR (300 MHz, D2O) δ-: 5.19 - 5.20 (d, J = 3.2, 1 H, H-1, α), 4.62–4.65 (d, J = 7.9, 1 H, H-1), 4.22–4.26 (q, J = 8.6, 1 H, H-3), 4.02–4.06 (q, J = 8.5, 1 H, H-3), 3.31–3.85 (m, 10 H, H-2, 4, 5, 6); 13C NMR (75 MHz, D2O) δ-: 100.0 C-1 (α), 96.5 C-1 (β), 86.2, 86.1 C-3, 84.0, 83.9, 79.9, 77.8, 75.5, 73.2, C-2,4,5, 65.12, 64.9 C-6). 31P-NMR [121 MHz, citrate buffer (pH 4.5), D2O, 85% H3PO4 as external standard] resulted in a chemical shift (δ-: 2.05 ppm) as described by Blennow et al. (8).
SDS/PAGE and Immunoblotting.
SDS/PAGE and immunoblotting (using the polyclonal anti R1 holoprotein antibody) were performed as described (4).
Analytical Techniques.
Proteins were quantified according to Bradford (9), using the Bio-Rad protein assay and BSA as a standard. Glucose, ADP, and AMP were measured as described (10). Pi was determined according to Parvin and Smith (11). glucose-6-phosphate (G6P) and glucose-3-phosphate (G3P) were analyzed by high-performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD) as described (5). However, a CarboPac PA-100 column (Dionex) was used. Alternatively, G6P was analyzed enzymatically (12).
Purification of Recombinant R1 Protein.
Potato R1 was heterologously expressed in E. coli BL21(DE 3) carrying the plasmid pET21d R1-tp as described (4). However, R1 expression was induced by addition of isopropyl β-d-thiogalactoside (IPTG) to give a final concentration of 1 mM. The cells were lysed and extracted in 50 mM Tris⋅HCl (pH 7.5), 2.5 mM EDTA, 2.5 mM dithioerythritol (DTE), 0.5 mM PMSF, and 2 mM benzamidine. Following centrifugation, the soluble protein fraction was subjected to anion-exchange chromatography (2). The R1-containing fractions were pooled and concentrated using ultrafiltration (Diaflo PM30, Amicon). Subsequently, the concentrate was passed through a PD10 column (Pharmacia) equilibrated with buffer A [100 mM 4-morpholinepropanesulfonic acid (Mops)-KOH (pH 7.6)/1 mM EDTA/2 mM DTE/0.5 mM PMSF]. The eluate (3.5 ml containing 3–4 mg protein) was then loaded onto a column (1.6 × 8.5 cm) filled with native starch granules from potato tubers (Merck). The granules had been washed twice in deionized water and once in buffer A before they were filled into the column. Following sample application, the starch column was washed with 20 ml of buffer A and, subsequently, with 30 ml buffer A supplemented with 50 mg/ml of malto-oligosaccharides (Glc2–Glc6; Merck) at a flow rate of 0.5 ml/min. Those fractions apparently containing only R1 were combined and concentrated to a volume of 1.5 ml by ultrafiltration. The buffer in the R1 preparation was changed by passage through a HiTrap desalting column (Pharmacia) equilibrated with buffer B [50 mM Hepes-KOH (pH 7.5)/1 mM EDTA/1 mM DTE/0.5 mM PMSF/10% glycerol]. Aliquots of the R1 preparation were stored at −80°C until use.
Synthesis of a Glucan Substrate for R1 by Chain Elongation of Glycogen (“Elongated Glycogen”).
Twenty milligrams of glycogen dissolved in 18 ml of 50 mM Hepes-KOH (pH 7.5), 1 mM EDTA, 40 mM glucose-1-phosphate (G1P), and 10% glycerol were incubated with 22 units of phosphorylase for 20 min at 25°C. Phosphorylase was then inactivated by heating for 10 min at 100°C. The polyglucan was precipitated by adding EtOH to a final concentration of 60%. Following centrifugation, the polyglucan was washed twice by resuspension in 24 ml of water and precipitation as above. This glucan was used in the nonradioactive “postelongation assay” (see below).
The maximum chain lengths (detected using MALDI-MS) in debranched samples of the untreated glycogen and the elongated glycogen were 33 and 55, respectively.
R1 Activity Assay.
If not otherwise stated activity assays (final volume 0.5 ml) were performed in buffer C [50 mM Hepes-KOH (pH 7.5)/1 mM EDTA/6 mM MgCl2/10% glycerol] that was supplemented with polyglucans and different potential phosphate donors as indicated. Reactions were started by adding purified R1 (20–40 μl in buffer B). In control samples R1 was replaced by an equal volume of buffer B.
Nonradioactive Assays.
Two different nonradioactive assays were used. (i) In the postelongation assay R1 was reacted with 1.4 mg “elongated glycogen” (see above). (ii) In the coelongation assay R1 was incubated with 0.5 mg of glycogen, 40 mM G1P, and 0.6 units of phosphorylase.
Following incubation at 25°C (as indicated; see Results), the reaction was terminated by heating for 5 min at 95°C. Aliquots (0.3–0.5 ml) were withdrawn and the polyglucans precipitated by adding cold EtOH to give a final concentration of 70%. After incubation for 1–2 h on ice and centrifugation (20,000 × g, 10 min at 4°C) the pellet was resuspended in 0.5 ml of water and precipitation was repeated once (as above). The pellet was dried in a speed vac for 5 min and subsequently resuspended in 150–250 μl of 0.7 M HCl. Following incubation at 80°C for 15 min, an aliquot (50–70 μl) was removed and neutralized with 2.8 N KOH, and G6P was determined enzymatically. The remaining solution was then hydrolyzed at 95°C for 2.5 h. Following neutralization, G6P and glucose were determined enzymatically. Because the phosphorylase preparation may contain small amounts of contaminating phosphoglucomutase, we checked for the presence of free (not glucan bound) G6P in the solution. To calculate for glucan bound G6P residues the amount of G6P determined before hydrolysis at 95°C was subtracted from the value determined following hydrolysis (free G6P was observed only in the coelongation assay).
Radioactive Assays.
Two elucidate whether the γ-P or the β-P of ATP is transferred to the glucan we used two different labeled ATP preparations in the initial radioactive experiment (results in Table 3). (i) [γ-33P]ATP and (ii) a mixture of [γ-33P]ATP and [β-33P]ATP. The latter was obtained by mixing 1 μl of [γ-33P]ATP with 350 μl of a buffer that contained 50 mM Hepes-KOH (pH 7.5), 1 mM EDTA, 10% glycerol, 5 mM MgCl2, 5 mM KCl, 0.1 mM ATP, and 0.3 mM AMP. The formation of [β-33P]ADP was initiated by the addition of 2.4 units of myokinase. Following the incubation for 25 min at 37°C, the reaction was terminated by heating for 10 min at 95°C and filtration through a 10-kDa filter (Microcon YM-10, Amicon). [β-33P]ADP was then converted to [β-33P]ATP by adding 0.86 mM PEP and 2 units of pyruvate kinase. After 20 min incubation at 37°C the reaction was heat terminated.
Table 3.
The β-P of ATP is transferred to the glucan
| Substrate | Phosphate donor
|
|||
|---|---|---|---|---|
| [γ-33P]ATP
|
[β-33P]ATP (+ [γ-33P]ATP)
|
|||
| − R1, cpm | + R1, cpm | − R1, cpm | + R1, cpm | |
| Amylopectin | 43 | 49 | 31 | 1,512 |
| Elongated glycogen | 60 | 42 | 161 | 10,423 |
| Glycogen + G1P + Pho | 59 | 233 | 45 | 110,749 |
Buffer C was used supplemented with either 2 mg of potato amylopectin, 1.4 mg of postelongated glycogen (see Materials and Methods), or 0.5 mg of glycogen, G1P, and phosphorylase (coelongation assay) and 0.5 mM ATP. Either 1.1 μCi [γ-33P]ATP or 1.1 μCi of a preparation in which [γ-33P]ATP had been partly converted to [β-33P]ATP (see Materials and Methods) were added. Reactions were started by addition of R1 (2.6 μg each) and terminated after 15 h. The radioactivity incorporated into the glucan was counted.
In all other radioactive assays pure [β-33P]ATP was used; 0.2–1 μCi were applied in each assay (0.5 ml sample). The mixture was incubated at 25°C as indicated and was then terminated by heating for 10 min at 95°C. The polyglucan was separated from the soluble components (including the labeled ATP) by use of centrifugal filter units (Microcon YM-10; see above). 230 μl of the suspension were mixed with 270 μl of 2 mM cold ATP and passed through the 10-kDa filter by centrifugation (30 min, 14,000 × g). The polyglucan that remained on top of the filter was further washed four times by resuspension in 450 μl of cold 2 mM ATP solution and subsequent centrifugation. Finally the filter unit was placed in a scintillation vial, scintillation fluid was added (Ready Safe, Beckman), and the vial was vortexed vigorously for 10 s before the incorporated radioactivity was determined in a scintillation counter.
In Vitro Phosphorylation of the Purified R1 Protein.
R1 was incubated with labeled ATP in buffer C (further details are given in the legend to Fig. 5). Following denaturation, samples were subjected to SDS/PAGE. Gels were either dried or proteins were blotted to nitrocellulose before autoradiography. Treatments with heat, alkali, or acid were adapted from Rosenberg (13). However, the final concentrations of NaOH and HCl were decreased to 0.5 M because 1 M NaOH caused significant hydrolysis of the R1 protein, as revealed by protein staining on the membrane.
Figure 5.

Autocatalytic phosphorylation of R1. (A) The β phosphate of ATP is transferred to R1, forming a heat-labile linkage. R1 was incubated with either 1 μCi [β-33P]ATP or [γ-33P]ATP for 25 min. Incubation was terminated by adding SDS sample buffer and denaturation either for 5 min at 95°C or for 20 min 30°C. Equal protein amounts were subjected to SDS/PAGE and autoradiography. 1, [γ-33P]ATP, 95°C; 2, [β-33P]ATP, 95°C; 3, [γ-33P]ATP, 30°C; 4, [β-33P]ATP, 30°C. (B) The phosphorylated R1 is alkali-stable and acid-labile. R1 (35 μg) was incubated with 35 μCi of a preparation in which [γ-33P]ATP had been partly converted to [β-33P]ATP (see Materials and Methods) for 1 h at room temperature. The sample was then divided and either NaOH (1) or HCl (2) were added to give final concentrations of 0.5 M. Following incubation for a further 30 min, the samples were neutralized, mixed with SDS-sample buffer, and denatured for 20 min at 30°C. Equal amounts of protein were analyzed by SDS/PAGE and autoradiography.
Results
Purification of R1.
R1 from potato was heterologously expressed in E. coli and then purified to apparent homogeneity. As a first step, anion exchange chromatography proved to be efficient (Fig. 1, lane 2). For further purification we took advantage of the interaction of R1 with starch granules (2, 4). The concentrated protein fraction obtained by anion-exchange chromatography was passed through a column filled with native starch granules from potato tubers. When the starch column was eluted with buffer A (without malto-oligosaccharides), contaminating proteins were eluted first, whereas release of most of the R1, because of the interaction with the granules, required a larger buffer volume (data not shown). However, under these conditions elution of R1 results in a considerable dilution. To prevent the dilution, a two-step procedure was established that consists of elution with 20 ml of buffer A to remove contaminating proteins and, subsequently, with a malto-dextrin mixture dissolved in buffer A. Those R1 molecules that had remained on the column until then were eluted as a sharp peak (data not shown). The resulting R1 preparation apparently lacks contaminating proteins and the identity of R1 was confirmed by Western blotting (Fig. 1, lanes 3 and 4).
Figure 1.

Purification of recombinant R1. Proteins were separated by SDS/PAGE and stained with Coomassie (lanes M and 1–3) or subsequently transferred to nitrocellulose and probed with an antibody raised against R1 (lane 4). M, molecular weight marker; 1, crude E. coli extract following induction (10 μg); 2, protein fraction derived from anion-exchange chromatography (10 μg); 3, purified R1 following passage through the starch column (2.5 μg); 4, Western blot of the purified R1-fraction (0.7 μg).
R1 Catalyzes an ATP-Dependent Phosphorylation of Polyglucans.
No starch-related phosphotransferase activity has ever been demonstrated and neither the phosphate donor nor the actual phosphate acceptor molecule for this type of reaction has been identified. In potato tubers starch phosphorylation appears to occur during biosynthesis of the polysaccharide (12). To mimic the process of starch biosynthesis we used a system composed of glycogen, G1P, and phosphorylase from rabbit muscle (coelongation assay; see Materials and Methods). Glycogen served as primer for the glucosyl transfer from G1P catalyzed by phosphorylase. If purified R1 and different possible phosphate donors are added to this mixture, elongating α-glucan chains of the glycogen can act as acceptors for a putative phosphotransferase reaction. Following overnight incubation, the polyglucans were precipitated and hydrolyzed to release glucose and, if phosphorylation has occurred, glucose-phosphates as well. G6P and glucose were determined enzymatically. As shown in Table 1, R1 catalyzes glucan phosphorylation in the presence of ATP. No phosphorylation was observed in the presence of PEP, PPi, UTP, or GTP (or in the absence of ATP). Thus, among all compounds tested only ATP functions as a phosphate donor. As a control experiment, we transformed E. coli with an empty vector and subjected the extracted proteins to the purification procedure described for R1. No phosphorylating activity was detectable in the resulting protein preparation. This finding demonstrates that the enzyme activity described here was neither due to any contaminating bacterial protein that copurifies with R1 nor derived from the native starch granules applied for affinity chromatography.
Table 1.
The phosphorylation of glucans catalyzed by R1 depends on ATP
| Substrates | Phosphate incorporation, nmol G6P/μmol Glc |
|---|---|
| ATP, PEP | 9.31 (9.29, 9.32) |
| ATP, PPi | 8.60 (9.52, 7.67) |
| ATP | 8.55 (7.81, 9.28) |
| PEP, PPi | ND |
| PEP | ND |
| PPi | ND |
| UTP | ND |
| GTP | ND |
| None | ND |
Activity of R1 was determined using the coelongation assay. R1 (7.2 μg each) was incubated for 14 h with 0.5 mg glycogen, 40 mM G1P, 0.6 units of phosphorylase, and different possible phosphate donors (5 mM ATP, PEP, UTP, and GTP; 2.5 mM PPi). Following precipitation and hydrolysis of the polyglucan, G6P and glucose (Glc) were determined enzymatically. Values are means of two independent experiments. Values in parentheses are the individual measurements for experiments 1 and 2 (each run in duplicate). In controls (lacking R1) no phosphate incorporation was observed (not shown). ND, not detectable.
During incubation the amount of polyglucan per reaction mixture increased from 0.5 mg (amount of glycogen added) to about 2 mg (as calculated from the glucose quantification following acid hydrolysis). It should be noted that no phosphate incorporation was detected by using the nonradioactive assay when R1 was incubated with 2 mg of glycogen and ATP but phosphorylase was omitted (data not shown).
R1 Phosphorylates both the C-6 and C-3 Positions.
In vivo starch phosphorylation results in esters linked to both the C-6 and C-3 of glucosyl moieties (1). To study whether the same phosphorylation pattern is achieved by the in vitro action of R1, the polyglucan fraction (obtained in the coelongation assay) was subjected to acid hydrolysis and the resulting monomers were analyzed by HPAEC-PAD. As shown in Fig. 2, both G6P and G3P were clearly detectable in the R1-containing reaction mixture, but absent in the control lacking R1. The identity of the two glucose-phosphates was confirmed by comparison with authentic G6P and G3P (Fig. 2) and by disappearance of the respective peaks if the hydrolyzed samples were treated with alkaline phosphatase (not shown). This shows that under in vitro conditions R1 catalyzes the phosphorylation of both C-6 and C-3 position of the glucose residues. The ratio of G6P to G3P residues is comparable to that obtained in native potato amylopectin (not shown).
Figure 2.

G6P and G3P residues are products of in vitro phosphorylation by R1. Following the coelongation assay (7.2 μg R1,5 mM ATP, 14 h incubation) the synthesized polyglucan was precipitated and hydrolyzed. Samples containing equal amounts of glucose were then analyzed by HPAEC-PAD. The elution profile of authentic G6P and G3P (4 nmol each) is also shown.
R1 Is a Dikinase.
In principle, either a kinase or a dikinase reaction could transfer a phosphate group from ATP to the α-glucan. If R1 acts as a kinase, the incorporation of phosphate and the formation of ADP are expected to occur in a 1 to 1 stoichiometry. However, because a sequence stretch of R1 displays some homology to bacterial PEP synthases (2, 3) it has already been speculated that R1 might act as a starch, water dikinase (3). In this case, phosphate incorporation, AMP production, and the release of orthophosphate should be equimolar. To identify the reaction catalyzed by R1, we determined the stoichiometry of glucan phosphorylation, and the production of ADP, AMP, and orthophosphate (Pi). The coelongation assay is not appropriate for these measurements because the phosphorylase also releases Pi. However, by preincubation of glycogen with G1P and phosphorylase and the subsequent removal of the enzyme, G1P and the Pi formed we were able to generate a polyglucan (postelongation assay; see Materials and Methods) that functioned effectively as a phosphate acceptor for R1. This polyglucan (designated as elongated glycogen) was then incubated with R1 and ATP. Following in vitro phosphorylation, the polyglucan was hydrolyzed and the amount of glucosyl-6 phosphate residues was determined enzymatically. The formation of G3P was confirmed by HPAEC-PAD (not shown; cf. Fig. 2). Because small amounts of ADP, AMP, and Pi were also formed in the absence of the elongated glycogen, we subtracted these values from those measured in the complete reaction mixture (Table 2). The amount of ADP produced was about six times lower than the observed phosphate incorporation (a minimum and maximum estimation based on enzymatic G6P determination and HPAEC-PAD-analysis is given in Table 2). However, both AMP and Pi increased by nearly the same amounts compared with the total phosphate incorporation. These results strongly suggest that R1 is a dikinase that transfers phosphate from ATP to starch (or starch like glucans) and water as paired acceptors and, thereby, generates starch-P, AMP, and Pi.
Table 2.
R1 is a dikinase
| A (without glucan) | B (+ glucan) | B − A | |
|---|---|---|---|
| P-incorporation (G6P) | — | 8.8 (9.3, 8.2) | 8.8 |
| P-incorporation (G6P + G3P) | — | 12–16 | 12–16 |
| ADP | 2.4 (2.8, 2.0) | 4.8 (4.6, 4.9) | 2.4 |
| AMP | 3.0 (2.4, 3.6) | 17.6 (18.5, 16.7) | 14.6 |
| Pi | 7.6 (7.3, 7.8) | 23.1 (22.6, 23.6) | 15.6 |
In a total volume of 0.5 ml with or without 1.4 mg of glucan substrate (“elongated glycogen,” see Materials and Methods), 2.6 μg of R1 were incubated with 250 nmol of ATP for 15 h. Phosphate incorporation and the increase in ADP, AMP, and Pi were determined and given as nmol. Values are means of two independent experiments. Values in parentheses are the individual measurements for experiments 1 and 2 (each run in duplicate). The glucan substrate was free of the analyzed metabolites (not shown).
R1 Transfers the β-P of ATP to the Glucan.
The assumption that R1 catalyzes a dikinase-type reaction was further confirmed using radioactive assays. Two differently labeled [33P]ATP preparations were used: one was [γ-33P]ATP, and the other was a mixture of [β-33P]ATP and [γ-33P]ATP (see Materials and Methods). Various glucan substrates (potato amylopectin, post- and co-elongated glycogen) were incubated in the presence or absence of R1 with either [γ-33P]ATP or the mixture of [β-33P]ATP and [γ-33P]ATP. The glucans were then separated from the labeled adenine nucleotides and the radioactivity incorporated into the α-glucan was quantified (Table 3). Two observations can be made: First, labeling of all of the glucans was detectable only if R1 and [β-33P]ATP were added. Thus, R1 phosphorylates α-glucans according to the following equation:
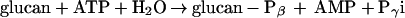 |
Secondly, the efficiency of phosphorylation strongly depends on the type of the α-glucan. Phosphate incorporation using the coelongation assay (glycogen + G1P + phosphorylase) proved to be most effective.
To test whether the phosphorylation reaction is reversible (as is the phosphorylation of pyruvate by PEP synthase) we incubated R1 (4 μg) with 5 mM Pi, 2 mM AMP, and potato amylopectin that had been prelabeled with 33P (either by in vitro phosphorylation or by feeding potato plants with 33P). However, there was no release of radioactivity during a 7-h incubation. Thus, the phosphorylation reaction is irreversible, at least under our experimental conditions.
Kinetics of Amylopectin Phosphorylation.
The coelongation assay does not allow for a detailed kinetic analysis of the glucan phosphorylation because during the R1 action the glucan substrate is not constant. Therefore, we used potato amylopectin for a more detailed analysis of in vitro phosphorylation. In these experiments pure [β-33P]ATP served as phosphate donor to allow for quantification of phosphate incorporation. As shown in Fig. 3, the phosphorylating reaction continued over the entire incubation period of 18 h; however, the rate decreased with time. This may be caused by a limited stability of the enzyme activity or by decreasing accessibility of the phosphate acceptor sites.
Figure 3.

Time dependence of potato amylopectin phosphorylation. Potato amylopectin (4 mg⋅ml−1) was incubated with 0.5 mM ATP containing 1 μCi [β-33P]ATP and R1 (2.6 μg) at 25°C for the indicated times. Two experiments were performed with very similar results. Data from one experiment are shown.
Under the assay conditions used extremely low concentrations of ATP were required to saturate the velocity of potato amylopectin phosphorylation (Fig. 4A). The apparent Km was 0.23 μM (as calculated from a Lineweaver–Burk plot). The rate was unchanged in the range of 0.01 mM to 5 mM ATP (data not shown). At a fixed ATP concentration (25 μM), the rate of glucan phosphorylation was determined using varying amylopectin concentrations (Fig. 4B). The half maximum rate of phosphorylation was observed at an amylopectin concentration of 1.7 mg⋅ml−1. The apparent maximum velocities were 3.2 and ≈5 nmol⋅min−1⋅mg−1 in Fig 4 A and B, respectively. Slightly different Vmax values were obtained because two separate R1 preparations were used in the two sets of experiments.
Figure 4.

Analysis of potato amylopectin phosphorylation. Experiments were performed using [β-33P]ATP. Incubation time, 5 min. (A) Fixed amylopectin concentration (10 mg⋅ml−1) and different ATP concentrations. In the range of 1 μM to 25 μM ATP, 2.6 μg of R1 were used. 0.5 μg R1 were used in the samples containing 0.05, 0.1, 0.5, 1, and 10 μM ATP. (B) R1 (1 μg) was incubated with 25 μM ATP and different amylopectin concentrations. The phosphorylation rate remained essentially unchanged in the range of 10 mg⋅ml−1 to 50 mg⋅ml−1 (data not shown).
Incubation of R1 with saturating concentrations of ATP and potato amylopectin at different pH values revealed a pH optimum for R1 activity of 7.0 (data not shown).
Mg2+ proved to be essential for the phosphorylation reaction. If MgCl2 was omitted from the assay mixture activity decreased by 90–95% (data not shown). No significant inhibition (or stimulation) of the phosphorylation reaction was observed in the presence of Pi (5 mM) or AMP (1 mM). Likewise, G1P (present in the coelongation assay), pyruvate, and dithioerythritol (DTE) did not significantly influence the in vitro phosphorylation of potato amylopectin (data not shown).
Autophosphorylation of R1 Precedes the Phosphotransfer to the Glucan.
A phosphohistidine (containing the β-P) is an intermediate in the dikinase-type reaction catalyzed by PEP-synthase (14). To analyze whether R1 follows a similar mechanism, the purified protein was incubated with either [γ-33P]ATP or [β-33P]ATP in the absence of glucan. The reaction was terminated by adding SDS-sample buffer and denaturation at either 30°C or 95°C. Following SDS/PAGE, radioactivity was visualized by autoradiography. As shown in Fig. 5, R1 is capable of autocatalytic phosphorylation. The β-P is transferred to the protein and the linkage proved to be heat-labile (Fig. 5A). This finding is consistent with a phosphohistidine being formed because phosphohistidine is heat-sensitive in contrast to phosphoserine, phosphotyrosine, and phosphothreonine (13). Moreover, the phosphorylated R1 was stable if treated with strong alkali but labile if treated with acid (Fig. 5B), thereby fulfilling another criterion that indicates a phosphoramidate linkage suggestive of histidine phosphorylation (13, 14). In any case, the autophosphorylation of R1 precedes the phosphotransfer to glucosyl residues (Table 4). R1 was preincubated with [β-33P]ATP in the absence of any α-glucan. Nonlabeled ATP (in large excess) and amylopectin were then sequentially added and the suspension was incubated for a further 10 min. As a control, amylopectin was omitted. Under these conditions labeling of amylopectin was detectable (Table 4A), whereas no incorporation was detectable on simultaneous addition of labeled and unlabeled ATP (Table 4B). A comparison of the samples containing or lacking amylopectin revealed that under conditions of preincubation with [β-33P]ATP (Table 4A) labeled protein also contributes to the total radioactivity retained on the filter units used to separate free and bound label (see Materials and Methods). However, this contribution was insignificant if labeled and nonlabeled ATP were added simultaneously as usually done in the activity assays (data not shown).
Table 4.
Autophosphorylation of R1 precedes the phosphotransfer to amylopectin
| Sample | Addition
|
43 min radioactivity, cpm | ||
|---|---|---|---|---|
| 0 min | 30 min | 33 min | ||
| A | R1 + [β-33P]ATP | + Nonlabeled ATP | + Amylopectin | 58, 63 |
| + Buffer | 28, 34 | |||
| B | R1 | + [β-33P]ATP, + nonlabeled ATP | + Amylopectin | 4, 12 |
| + Buffer | 7, 0 | |||
R1 (5 μg) was preincubated with 0.15 μCi [β-33P]ATP (nonlabeled ATP was absent) in buffer C for 30 min in a total volume of 110 μl. Subsequently 10 μl of 0.1 M nonlabeled ATP was added and the suspension was incubated for a further 3 min. Fifty microliters each were then withdrawn and mixed with 5 mg of amylopectin dissolved in 450 μl of buffer C or in buffer C alone (A). Following incubation for 10 min the reaction was terminated by heating at 95°C for 15 min. The sample was divided and 230 μl each were filtered through microcon filter units (see Materials and Methods) and the radioactivity retained on the filter was counted. Another sample was treated identically; however, [β-33P]ATP was omitted during the preincubation and was instead simultaneously applied together with nonlabeled ATP (B). The background counts (scintillation fluid alone, 43 ± 7) were subtracted.
Discussion
In higher plants, R1 deficiency results in two significant changes in phenotype: a strongly reduced starch phosphorylation (below the limit of detection in plants lacking R1) and an enhanced starch accumulation resulting in a starch-excess phenotype. Both effects have been observed in transgenic potato plants expressing an R1 antisense construct (2) and in the sex1 mutant of Arabidopsis (3). By complementation of the Arabidopsis mutant with the wild-type R1 gene, phosphorylation of starch was restored and starch accumulation was adjusted to that of the wild type. On a biochemical basis, these two effects were difficult to understand because the enzymology of glucan phosphorylation was not known.
The data presented in this study show that R1 is an α-glucan, water dikinase and, therefore, provide a biochemical explanation for the effect on starch phosphorylation, but not yet for the starch excess phenotype. The dikinase-type reaction is in accordance with the reported homology of the C-terminal region of R1 to sequence regions of both PEP synthase (2) and pyruvate, phosphate dikinase (PPDK, EC 2.7.9.1) containing the ATP-binding site (3). PEP synthase and PPDK catalyze the transfer of the β-P of ATP to pyruvate and the transfer of the γ-P to water or orthophosphate, respectively. Likewise R1 transfers the β-P to the glucan and the γ-P to water. Furthermore, the autocatalytic phosphorylation of R1 (Fig. 5) supports the occurrence of a phosphohistidine intermediate (containing the β-P) as reported for both PEP-synthase (13) and PPDK (15). Recently, a conserved stretch of amino acids has been identified in the potato R1 protein and in the R1 homologue of Arabidopsis (SEX1) that is similar to the phosphohistidine domains of PEP synthase and PPDK (3). In any case, autophosphorylation of R1 precedes the phosphate transfer to the α-glucan, indicating a ping pong mechanism.
R1 strongly discriminates between various types of polyglucans. Glycogen from bovine liver was an extremely poor substrate (6% 33P incorporation compared with potato amylopectin, data not shown). Phosphate incorporation dramatically increased if the glucan chains of glycogen were elongated and was most effective if phosphorylase and R1 were added simultaneously (coelongation assay). However, it should be noted that the glycogen-derived polysaccharides obtained in the co- or postelongation assay differ. During the coelongation assay, a glucan of low water solubility was formed (presumably because of the generation of very long chains) that to some extent precipitated during incubation. In contrast, the postelongated glycogen (under the conditions chosen) remained water soluble. Thus, currently we do not know whether the activity of R1 increases when the glucan chains are simultaneously elongated. In any case, ongoing chain elongation is no prerequisite for R1 activity because the postelongated glycogen and potato amylopectin were also phosphorylated.
Increased activity of R1 during, or following, glucan chain elongation concurs with the fact that in vivo phosphate groups are mainly located in relatively long chains of the amylopectin fraction (chain length 30–100 glucose units) (16, 17). However, it is possible that following in vivo phosphorylation the chains are further elongated by starch synthases or modified by branching enzyme activity (18). Amylopectin from wild-type potato plants contains a relatively large amount of phosphate esters, but it can be further phosphorylated by the recombinant R1 protein and, therefore, possesses additional phosphorylation sites. In contrast, amylopectin from R1 antisense potato plants (in which the starch phosphate is decreased by about 85%) and also corn amylopectin (that does not contain detectable amounts of covalently bound phosphate) proved to be less effective as phosphate acceptors. In vitro phosphate incorporation in corn amylopectin was less than 15% compared with wild-type potato amylopectin (data not shown). The same holds true for potato amylose, concurring with amylose being essentially free of phosphate esters in vivo (19).
Taken together, the data indicate that R1 phosphorylates distinct glucan targets. Accordingly, the modification of the starch structure by antisense repression of starch synthases or branching enzymes significantly affect the starch phosphate content (20–23). The actual target structure(s) that are recognized by R1 as a phosphorylation site(s) remain(s) to be identified.
The identification of R1 as an α-glucan, water dikinase clearly shows that the strongly impaired starch degradability in the R1 antisense potato plants and in the sex1 mutant of Arabidopsis is the consequence of reduced starch phosphorylation. Because of an increased hydrophilicity, the phosphorylated starch may be more easily accessible for degrading enzymes such as amylases. Alternatively, the activity and/or binding of starch degrading enzymes, yet to be identified, may directly depend on phosphate residues within starch. The regulatory link between phosphorylation and degradability of starch is the subject of our current work.
Acknowledgments
We thank Kerstin Pusch and Anke Scharf for excellent experimental support and Dr. Ruth Lorberth for providing the R1 expression vector. We also thank Sandra Techritz for performing the matrix-assisted laser desorption ionization (MALDI) analyses. Financial support by the Deutsche Forschungsgemeinschaft is gratefully acknowledged.
Abbreviations
- G1P
glucose-1-phosphate
- G6P
glucose-6-phosphate
- G3P
glucose-3-phosphate
- HPAEC-PAD
high-performance anion exchange chromatography with pulsed amperometric detection
References
- 1.Lim S-T, Kasemsuwan T, Jane J-L. Cereal Chem. 1994;71:488–493. [Google Scholar]
- 2.Lorberth R, Ritte G, Willmitzer L, Kossmann J. Nat Biotechnol. 1998;16:473–477. doi: 10.1038/nbt0598-473. [DOI] [PubMed] [Google Scholar]
- 3.Yu T-S, Kofler H, Häusler R E, Hille D, Flügge U-I, Zeeman S C, Smith A M, Kossmann J, Lloyd J, Ritte G, et al. Plant Cell. 2001;13:1907–1918. doi: 10.1105/TPC.010091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ritte G, Lorberth R, Steup M. Plant J. 2000;21:387–391. doi: 10.1046/j.1365-313x.2000.00683.x. [DOI] [PubMed] [Google Scholar]
- 5.Ritte G, Eckermann N, Haebel S, Lorberth R, Steup M. Starch/Stärke. 2000;52:179–185. [Google Scholar]
- 6. Perich, J. W. & Johns, R. B. (1988) Synthesis, 142–144.
- 7.Perich J W, Johns R B. Tetrahedron Lett. 1988;29:2369–2372. [Google Scholar]
- 8.Blennow A, Bay-Schmidt M A, Olsen C E, Møller B L. J Chromatogr A. 1998;829:385–391. [Google Scholar]
- 9.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 10.Stitt M, Lilley R M, Gerhardt R, Heldt H W. Methods Enzymol. 1989;174:518–552. [Google Scholar]
- 11.Parvin R, Smith R A. Anal Biochem. 1969;27:65–72. doi: 10.1016/0003-2697(69)90219-x. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen T H, Wischmann B, Enevoldsen K, Møller B L. Plant Physiol. 1994;105:111–117. doi: 10.1104/pp.105.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg I M. Protein Analysis and Purification. Boston: Birkhäuser; 1996. pp. 242–244. [Google Scholar]
- 14.Narindrasorasak S, Bridger W A. J Biol Chem. 1977;252:3121–3127. [PubMed] [Google Scholar]
- 15.Dunaway-Mariano D. Methods Enzymol. 1999;308:149–176. doi: 10.1016/s0076-6879(99)08009-x. [DOI] [PubMed] [Google Scholar]
- 16.Takeda Y, Hizukuri S. Carbohydr Res. 1982;102:321–327. [Google Scholar]
- 17.Blennow A, Bay-Schmidt M A, Wischmann B, Olsen C E, Møller B L. Carbohydr Res. 1998;307:45–54. [Google Scholar]
- 18.Viksø-Nielsen A, Blennow A, Nielsen T H, Møller B L. Plant Physiol. 1998;117:869–875. doi: 10.1104/pp.117.3.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hizukuri S, Tabata S, Nikuni Z. Starch/Stärke. 1970;22:330–343. [Google Scholar]
- 20.Kossmann J, Lloyd J. Crit Rev Plant Sci. 2000;19:171–226. [Google Scholar]
- 21.Abel G J W, Springer F, Willmitzer L, Kossmann J. Plant J. 1996;10:981–991. doi: 10.1046/j.1365-313x.1996.10060981.x. [DOI] [PubMed] [Google Scholar]
- 22.Kossmann J, Abel G J W, Springer F, Lloyd J R, Willmitzer L. Planta. 1999;208:503–511. doi: 10.1007/s004250050587. [DOI] [PubMed] [Google Scholar]
- 23.Schwall G P, Safford R, Westcott R J, Jeffcoat R, Tayal A, Shi Y C, Gidley M J, Jobling S A. Nat Biotechnol. 2000;18:551–554. doi: 10.1038/75427. [DOI] [PubMed] [Google Scholar]