


Abstract
Recently discovered CRISPR-associated transposons (CASTs) are natural RNA-guided DNA transposition systems capable of single-step genomic integration of large DNA cargo. Wild-type CASTs exhibit low integration activity in heterologous systems; therefore, engineering efforts are required to develop therapeutically relevant tools. Here we developed a high-throughput dual genetic screen capable of accurately quantifying the relative activity and specificity of a large pool of CAST variants. Under the conditions of our screen, we discovered that the wild-type V-K CAST system can consistently achieve between 88% and 95% on-site targeting specificity. We used site-saturation mutagenesis of the conserved core transposition machinery (TnsB, TnsC, and TniQ) to reveal novel mechanistic insights into the function of these transposon proteins. Furthermore, we found that different components have varying trade-offs between activity and specificity, a critical aspect overlooked in conventional screening pipelines. These findings provide clear engineering principles for further optimization of CASTs. Finally, we identified several mutations that, together, enhance CAST activity up to four-fold while minimally impacting targeting specificity. These methods are a powerful tool to characterize the sequence-function landscape across multiple functional parameters while also providing a robust platform for developing enhanced genome-editing tools.
Graphical Abstract
Graphical Abstract.

Introduction
Large-cargo genomic integration offers opportunities for a variety of applications, from efficient strain engineering to therapeutics for genetic diseases with mutational heterogeneity. Conventional approaches using CRISPR–Cas nucleases rely on generating cytotoxic DNA double-strand breaks (DSBs), followed by homology-directed repair (HDR) [1]. However, the inherently low efficiency of HDR in non-dividing cells limits broad application of this method [2]. Recently, engineered systems coupling the Bxb1 recombinase with prime editors have successfully integrated large DNA cargo without inducing DSBs [3–5], but they require a multi-step process that could produce undesired, incomplete edits. Tn7-like transposons provide an attractive alternative system, by enabling one-step integration and highly specific targeting. Target specificity is imparted either by a sequence-specific DNA binding protein, such as TnsD in canonical Tn7, or by a CRISPR-like effector in CRISPR-associated transposons (CASTs) [6, 7]. The latter are particularly exciting because they are RNA-guided and thus programmable; DNA integration occurs within a fixed distance downstream of the target site [8, 9] due to the architecture of the integration complex [10, 11].
CASTs are promising tool for precise manipulation of the microbiome [12, 13] and could potentially solve open problems in clinical genome editing. However, many technical challenges remain, with different subtypes offering unique advantages and disadvantages. For example, the I-F3 subtype is highly specific [14] and has demonstrated activity in human cells [15]. However, its intrinsic complexity and large genetic size—stemming from its multi-subunit CRISPR-like effector—hamper delivery and limit utility. In contrast, the V-K subtype has shown only marginal activity in human cells [16] with lower specificity in bacteria (10%–70% on-target events) [17], as their core transposition machinery can exhibit substantial integration activity even in the absence of the CRISPR effector [8, 17]. Aside from these limitations, V-K CAST offers compelling advantages: they have a single-subunit CRISPR-like effector, Cas12k, and their smaller genetic size simplifies delivery; moreover, our extensive mechanistic understanding [10] provides a strong foundation for engineering efforts.
V-K CAST initiates RNA-guided DNA integration with the Cas12k effector binding to target DNA via its associated guide RNA (gRNA). Host ribosomal protein S15 and transposon protein TniQ (TnsD homolog) associate with Cas12k, and the AAA+ regulator TnsC forms a filamentous oligomeric assembly around target DNA (Fig. 1A, left) [18]. Donor-bound transposase, TnsB, is recruited by TnsC to the target site, where it forms an active tetrameric nucleoprotein assembly [19]. The recruited TnsB catalyzes donor DNA integration, forming a stable post-integration complex called the strand-transfer complex (STC) [19]. Previous efforts to engineer V-K CAST focused on making genetic fusions in order to simplify the system and generate simple insertion products [16]. While this approach avoids potentially toxic co-integration products, the designs didn’t substantially improve integration activity of V-K CAST. It is plausible that simple fusions may not cooperatively promote transpososome formation, which occurs via precise protein–protein interactions on target DNA [10]. Thus, enhancing the function of such multi-component systems would benefit from more careful consideration of their structure and mechanism.
Figure 1.

Overview and characterization of genetic screen and the RNA-guided transposition system studied. (A) Schematic of the assemblies in the two modes of transposition, on- and off-target. Components: Cas12k (pink), guide-RNA (gRNA, gray), S15 (tan), TniQ (orange), TnsC filament (green), TnsB (purple), and target DNA (tDNA, light blue). Inset, atomic models show the detailed interactions near TniQ. (B) Screening assay overview. Escherichia coli strain cJP003 with an arabinose-inducible (indicated by pBAD) toxic gene (ccdB, orange) is transformed with a library of pHelper plasmid variants (circles) to integrate the pDonor carrying a kanamycin resistance gene (kanR, blue). Overall integration efficiency and on-target integration efficiency can be assessed by kanamycin (Kan) or kanamycin + arabinose (Kan/Ara) screen, respectively. Each outcome is depicted: on-target integration (top), off-target integration (middle), and no integration (bottom), alongside the expected outcomes from the screen (right): smiley faces indicate survival, and skulls indicate death under the screening conditions, either Kan or Kan/Ara. pHelper variants from the surviving cells are then analyzed by next-generation sequencing. Activity scores and specificity scores are calculated based on the functions shown. freq indicates frequency of each variant within each condition. (C) Activity scores (y-axis) presented with respect to gRNA sequence (x-axis). The bar plot shows the average score from the two biological replicates, normalized to the pool-wide average. Bars are colored based on the target gene: lacZ (blue), ccdB (orange), and non-target (NT, gray). ccdB-01 gRNA is indicated by asterisk. (D) Correlation between the screening (x-axis) and individual tests (y-axis). Number of colonies from mate-in transposition assays informs on the integration activity with each ccdB-targeting gRNA (orange) or non-target gRNA (NT, gray). r = Pearson coefficient. All data points represent mean ± standard error with errors from the screen estimated by Enrich2 (see the “Materials and methods” section) and standard deviations from colony counts (n= 3). Spacers are numbered as shown in panel (C) (i.e. ccdB-01 is the first spacer from the left) (E) Bar plots of on-target integration ratio, as determined by unbiased NGS profiling. Orange and green represent data obtained using non-replicable and replicable pDonor vectors, respectively. On-target ratio values are shown above each bar. Spacers are named as described in panel (D). N/A, insufficient data due to low colony numbers.
An ideal programmable transposon would have high activity (i.e. all desired target sites contain an insertion) and high specificity (i.e. no off-target integration events). Typically, directed evolution pipelines only optimize for integration activity [20, 21], but such approaches can be problematic, depending on how these properties are related. In the case of CRISPR–Cas9 systems, extensive engineering has revealed a tradeoff between activity and specificity: mutations that increase activity also lead to promiscuous targeting, and conversely, mutations that increase target-site specificity lead to decreased activity [22]. Site-specific large serine recombinases also have such activity-specificity tradeoffs [23], but their modular nature enables specificity reprogramming without compromising activity [24]. CASTs are more complicated than either of these systems, because transposition can involve multiple proteins and potentially multiple targeting pathways [17, 25]. Therefore, it remains unclear how activity and specificity are related in Tn7-like transposition systems, and this information is critical for engineering efforts.
To address this gap in our knowledge, here we developed a high-throughput dual screen that can measure the relative activity and specificity of thousands of pooled variants of V-K CAST. Because Cas12k is primarily associated with targeting specificity (via its gRNA) and is exchangeable across subtypes, we decided to explore the sequence-function landscape for the three highly conserved core transposon components: TnsB, TnsC, and TniQ. These components can form an integration complex in the absence of Cas12k, resulting in off-target integration (Fig. 1A, right) [17]. We thus hypothesized that mutagenizing these proteins could reveal the sequence determinants governing flux between on- and off-target integration pathways, and such knowledge would be invaluable to engineer a highly active and specific tool for DNA insertion.
Materials and methods
Plasmid construction and E. coli strain engineering
Polymerase chain reaction (PCR) for molecular cloning was performed using Q5 Hot Start High-Fidelity DNA polymerase (NEB, M0494L) and all primers used in this research were purchased from Integrated DNA Technologies. To enable efficient modification of each gene encoding ShCAST proteins, we separated the open reading frames of TnsC and TniQ from the original entry helper plasmid, pHelper_ShCAST_sgRNA (Addgene #127921) [8]. This new construct, pHelper_ShCAST, was used as pHelper throughout this study. pDonor-Kan plasmid containing kanamycin resistance gene was also a gift from Feng Zhang (Addgene #127 924) [8]. Replicable pDonor-Kan was constructed by replacing the R6K origin of replication in pDonor-Kan with the p15A origin of replication and the chloramphenicol resistance gene. Further information on the plasmids used in this study can be found in Supplementary Table S7.
To generate the recipient strain cJP003, an inducible ccdB gene was introduced into the E. coli genome using established lambda Red recombineering [26]. The ccdB gene under pBAD with the lacY (A177C) gene was amplified by PCR from the p11-LacY-wtx1 (Addgene #69056) [27]. This fragment was subcloned into the pTSC29 vector backbone, a gift from Joseph E. Peters, proximal to the spectinomycin resistance (SmR) gene via Gibson assembly. The entire gene fragment, including SmR, was amplified with a homology arm to the downstream region of the glmS, the location known as a “safe haven” for Tn7 transposons [28]. This linear DNA fragment was electroporated into the Lac+ derivative of BW27783 strain (PO619) [29] that harbors pKD46 plasmid, which enables site-specific integration of the linear DNA fragment. After the transformation and the integration of a linear DNA fragment containing ccdB and SmR genes, the engineered cells were selected on an agar plate with 25 μg/mL spectinomycin. Correct integration was confirmed by sequencing the PCR amplicons of the junctions between the insert and target genomic sites. The selected colony was re-streaked on the spectinomycin plates and incubated at 42°C overnight to remove the heat-sensitive pKD46 plasmids. Strain PO603, another gift from Joseph E. Peters (unpublished), is derived from strain BW20767 and contains a donor plasmid, which carries a kanamycin resistance gene, R6K origin of replication, and an origin of transfer. PO603 was used as the donor strain for mate-in transposition assay to individually test each ShCAST variant.
Generation of ShCAST variant library
To generate the gRNA library, the spacer site of gRNA expressing cassette in pHelper_ShCAST was replaced with SapI flanking sites. Oligo pools containing spacer variants were purchased from TWIST Bioscience. The gRNA library was then assembled via Golden Gate assembly using SapI enzyme (NEB, R0569L).
For TnsB, TnsC, and TniQ screens, the spacer site of pHelper_ShCAST was replaced with the ccdB-01 spacer, generating pHelper_ShCAST_ccdB01. To generate the TnsB library, a TnsB entry vector was constructed by replacing the TnsB from pHelper_ShCAST_ccdB01 with a BbsI flanking site. Another BbsI flanking site was introduced into a non-coding region between T7 terminator and the promoter of the ampicillin resistance gene (AmpR) as a barcode entry site. TnsB single mutant fragments with BbsI sites at both ends were purchased from TWIST Bioscience. Barcode fragments were generated by PCR using primers designed to amplify a non-coding sequence, with the forward primer containing N10 degenerate nucleotides (N = A, T, G, or C). The TnsB entry vector was then mixed with the TnsB single-mutant fragments and barcode fragments, followed by digestion with BbsI-HF (NEB, R3539L) and assembly using T4 DNA ligase (NEB, M0202L) in a one-step Golden Gate assembly. The barcoded TnsB library was digested with AflIII (NEB, R0541L) and sent to the Hartwell Center in St. Jude Children’s Research Hospital for Revio PacBio sequencing to assign variant-barcode pairs. Each TnsB variant and its corresponding barcode were aligned from reads with a quality score of ≥Q49. Variants with fewer than 10 read counts or redundant barcodes were excluded, resulting in 99.8% (1897 out of 1901) of variants being retained for screening.
A TnsC entry vector was also constructed to generate the TnsC library. The TnsC site of pHelper_ShCAST_ccdB01 was replaced with an SapI flanking site. Additionally, a PaqCI flaking site was introduced in a non-coding region between T7 terminator and the promoter of AmpR for barcoding. TnsC single mutant fragments with SapI sites at both ends were purchased from TWIST Bioscience. Barcode fragments that contain customized 29 bp N3WS barcodes [30] were generated by PCR using primers with degenerate nucleotides (W = A or T, S = G or C). A two-step Golden Gate assembly was performed: First, the entry vector was assembled with the TnsC fragment using SapI and T4 DNA ligase. Next, barcode fragments were assembled using PaqCI (NEB, R0745L) and T4 DNA ligase. The barcoded TnsC library was digested with PacI (NEB, R0547L) and sent to the Hartwell Center in St. Jude Children’s Research Hospital for Revio PacBio sequencing. Each TnsC variant and its corresponding barcode were aligned from reads with a quality score of ≥Q49. Variants with fewer than 10 read counts or redundant barcodes were excluded, covering 95.3% (5502 out of 5796) of all possible TnsC single mutants.
The TniQ library was constructed using codon-tiling NNK degenerate primers (K = T or G), designed using the established pipeline [31, 32]. First, a TniQ entry vector was constructed by replacing the TniQ site of pHelper_ShCAST_ccdB01 with a SapI flanking site. Next, the full-length DNA fragment of wild-type (WT) TniQ was prepared as a template for PCR. Forward or reverse NNK degenerate primers, containing an NNK sequence at each codon, were then paired with a universal reverse or forward primer to amplify mutated amplicons. These amplicons were subsequently joined through joining PCR to generate full-length TniQ variant fragments. The final products were assembled into the TniQ entry vector using Gibson assembly, generating the TniQ library with two mutations per gene on average, which follows a Poisson distribution. The TniQ library was not barcoded since the entire TniQ fragment was sequenced using next-generation sequencing (NGS).
All assembled libraries were electroporated into 10-beta electrocompetent cells (NEB, C3020K) and spread on agar plates with 100 μg/mL carbenicillin. Selected colonies were scraped from the plates with ∼100-fold coverage per variant and then midi-prepped using ZymoPURE™ II Plasmid Midiprep Kit (Zymo Research, D4201) to extract plasmids of the library.
The sequences of entry vectors, gRNAs used in the gRNA screen, and NNK degenerate primers used to build the TniQ library are listed in Supplementary Tables S7–S9, respectively.
High-throughput screening of ShCAST variants
The plasmid concentration of each library was measured using the Qubit™ 1X dsDNA HS Assay Kit (Thermo Fisher, Q33231). Each library was electroporated into cJP003 at a 0.5:1 plasmid-to-cell ratio to minimize transforming multiple plasmids per cell. Colonies were scraped from the carbenicillin plates and then stored as 25% glycerol stocks (OD600 = 2.0) at −80°C. The stock was thawed and diluted 50-fold in LB medium supplemented with 100 µg/mL carbenicillin and 0.1 mM isopropylthio-β-galactoside (IPTG), and grown until the OD600 reached ∼0.6. Cells were then harvested, washed, and resuspended with 10% ice-cold glycerol three times to make them electrocompetent. pDonor-Kan was electroporated into the resuspended cells at a 0.5:1 plasmid-to-cell ratio. Remaining electrocompetent cells were midi-prepped to generate input libraries before the activity or specificity screen. The electroporated cells were then recovered for 1 h with vigorous shaking at 37°C and incubated for another 2 h without shaking before the screen. For activity screen, cells were spread on agar plates with 100 μg/mL carbenicillin, 25 μg/mL spectinomycin, and 50 μg/mL kanamycin. For specificity screen, cells were spread on agar plates with 100 μg/mL carbenicillin, 25 μg/mL spectinomycin, 50 μg/mL kanamycin, and 0.1% w/v L-arabinose. The resulting colonies from each screen were collected separately with ∼100-fold coverage per variant and then midi-prepped to extract screened pHelper plasmids.
For the gRNA screen, fragments of the gRNA-coding region from extracted pHelper plasmids were amplified by amplicon PCR, followed by index PCR using Nextera XT Index Kit v2 Set A (Illumina, FC-131-2001). The amplicons were then sequenced on MiSeq using MiSeq Reagent Kit v2 (150-cycle) (Illumina, MS-102-3001).
For TnsB and TnsC screens, barcode amplicons were amplified from extracted plasmids by amplicon PCR, followed by index PCR using Nextera XT Index Kit v2 Set A. The amplicons were then sequenced on NovaSeq X using NovaSeq™ X Series 1.5B Reagent Kit (200-cycle) (Illumina, 20104704) at the Hartwell Center in St. Jude Children’s Research Hospital. For TniQ screen, TniQ fragments from extracted pHelper plasmids were amplified by amplicon PCR, followed by index PCR using Nextera XT Index Kit v2 Set A. The amplicons were then sequenced on MiSeq using MiSeq Reagent Kit v3 (600-cycle) (Illumina, MS-102-3003). Primers used for amplicon PCRs are listed in Supplementary Table S10.
Mate-in transposition assay
Each pHelper variant was cloned and transformed into cJP003 individually. A glycerol stock of donor cell, PO603, was freshly streaked on agar plates with 50 μg/mL kanamycin and 8 μg/mL tetracycline. Donor cells can replicate pDonor-Kan with R6K origin of replication because they have PIR gene, whereas cJP003 cannot. cJP003 and donor cells were cultured in 2×YT medium supplemented with corresponding antibiotics and 0.1 mM IPTG until the OD600 reached ∼0.7–0.8. Cells were then washed by pelleting down the cells and resuspending the cells with fresh 2×YT medium (+ 0.1 mM IPTG). Resuspended cJP003 and donor cells were mixed at a 1000:200 OD600 * volume (μL) ratio, and LB medium (+ 0.1 mM IPTG) was added to make a final volume of 150 μL. Cells were vigorously vortexed for 10 s, centrifuged for 1 min at 15 000 × g, and incubated for 2 h at 37°C. Cells were then spread on agar plates with 100 μg/mL carbenicillin, 25 μg/mL spectinomycin, and 50 μg/mL kanamycin, with or without 0.1% w/v L-arabinose. Selected colonies from each variant group (n = 3) were counted for analysis.
Unbiased profiling of integration sites using NGS
Transposition target sites were analyzed using tagmentation-based transposon insertion sequencing (TagTn-seq) [17]. For non-replicable pDonor condition, we followed the protocol for the high-throughput transposition assay. To evaluate integration specificity of replicable pDonor condition, cJP003 was transformed with both pHelper and replicable pDonor-Kan and plated on agar plates with 100 μg/mL carbenicillin, 30 μg/mL chloramphenicol, and 50 μg/mL kanamycin. In both cases, >500 colonies were collected, and their genomic DNA was purified using Wizard® Genomic DNA Purification Kit (Promega, A1125). The extracted genomic DNA was tagmented using Nextera XT DNA Library Prep Kit (Illumina, FC-131-1024). The tagmented fragments were then amplified by PCR using ShCAST LE-specific primers and i7 primers (Supplementary Table S10), followed by index PCR using Nextera XT Index Kit v2 Set A. The resultant amplicons were sequenced by MiSeq system using MiSeq reagent kit v2 (Illumina) and then analyzed. Sequences from intact pDonor-Kan or replicable pDonor-Kan were excluded, and the sequences adjacent to LE were mapped to the genome of cJP003 to identify integration sites.
Droplet digital PCR
cJP003 cells carrying pHelper with ccdB-01 or ccdB-04 gRNA were electroporated with pDonor-Kan and then spread on agar plates containing 100 μg/mL carbenicillin, 25 μg/mL spectinomycin, and 50 μg/mL kanamycin. Genomic DNA of E. coli from the resulting colonies was extracted using Wizard Genomic DNA Purification kit (Promega, A1120). Genomic DNA samples were diluted to 5–20 μg/μL to set up a 20 μL droplet digital PCR (ddPCR) reaction in triplicate. The reaction mixture contained 1× ddPCR Supermix for probes (no dUTP) (Bio-Rad, 1863024), 900 nM ccdB- or insertion-specific probes, and 250 nM forward and reverse primers. Droplets generated using droplet-generating oil for probes (Bio-Rad, 1863005) were cycled through the following PCR program in a thermocycler: 1 cycle at 95°C for 10 min; 40 cycles of 94°C for 30 s and 60°C for 1 min (ramp rate: 2°C/s); 1 cycle at 98°C for 10 min; followed by 4°C hold. Droplets were analyzed using Qx 200 Droplet Reader (Bio-Rad), and the absolute quantification of the insertion events was determined using Qx Manager software 2.1 Standard edition. The percentage of on-target insertions was calculated using the formula: insertion event / (insertion event + target count). Sequence information about the primers and probes used for ddPCR can be found in Supplementary Table S10.
Analysis of relative integration activity and specificity from high-throughput screen
To compute the enrichment scores for each variant, we followed Enrich2 from Rubin et al. [33]. First, the enrichment score for each variant is computed as:
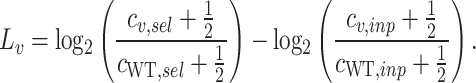 |
Here,  and
and 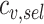 represent the number of counts of the variant before and after screening, respectively. The score is independent of the total number of variants, as it is normalized by the WT counts (
represent the number of counts of the variant before and after screening, respectively. The score is independent of the total number of variants, as it is normalized by the WT counts (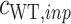 and
and 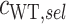 ). To account for variants with zero counts after screening, we add a factor of
). To account for variants with zero counts after screening, we add a factor of 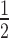 to each count. We compute the standard error (
to each count. We compute the standard error ( ) for enrichment scores of each variant under the assumption that the number of counts follows a Poisson process.
) for enrichment scores of each variant under the assumption that the number of counts follows a Poisson process.
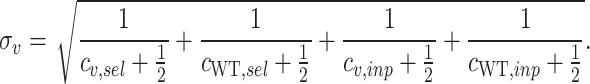 |
To combine the scores obtained from synonymous barcodes (TnsB and TnsC screen) or synonymous codons (TniQ screen) of each biological replicate, we also followed the analysis from Enrich2 [33], where the scores and errors are fitted using Fisher scoring iterations of restricted maximum likelihood estimates. For a set of replicate scores (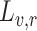 ) and estimated errors (
) and estimated errors (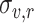 ) where
) where  , the combined score is given by:
, the combined score is given by:
 |
The combined error ( ) is computed by following the fixed-point solution given by:
) is computed by following the fixed-point solution given by:
 |
To initialize the fitting of the error, the starting value is set to:
 Similar to Enrich2, we performed 50 Fisher scoring iterations, which were sufficient to converge the scores and errors of all variants. The same calculation was performed to combine the scores obtained from different biological replicates.
Similar to Enrich2, we performed 50 Fisher scoring iterations, which were sufficient to converge the scores and errors of all variants. The same calculation was performed to combine the scores obtained from different biological replicates.
Multiple sequence alignment
Multiple sequence alignments of TnsC and TniQ were performed using MAFFT [34] using the homolog sequences from a prior study [35]. Conservation scores were calculated based on residue weights generated by WebLogo [36], where a weight of 0.8 at a position indicates that 80% of the sequences have the same amino acid at that position.
Energetic frustration profile
For each variant we first threaded the sequence onto the WT structure (PDB:8RDU, chain R for TnsB, chain K for TnsC, and chain C for TniQ) and computed the energy of the system using the AWSEM Hamiltonian [37]. Next, we calculated the frustration index for the i-th residue as the Z-score of the variant's energy, relative to the energy distribution of energy of a set of decoys [38, 39]. For mutational frustration at the i-th residue, the decoys consist of the variant sequence with the i-th residue mutated to every possible amino acid. The frustration index for the i-th residue is then given by:
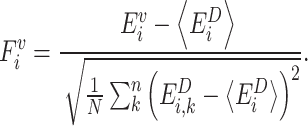 |
Where 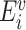 represents the energy of the variant sequence,
represents the energy of the variant sequence, 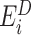 denotes the set of decoy energies.
denotes the set of decoy energies. 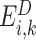 is the energy of the k-th decoy and N is the number of decoys. Given that the AWSME Hamiltonian computes energy based on the neighboring particles, the energy of the system is an addition of local behavior. Consequently, the frustration profile highlights energy changes in the local environment of the system. Based on this definition, a higher frustration index indicates that i-th residue of the variant is among the amino acids that result in the lowest energy (highest local stability). We refer to the frustration profile of a variant with L amino acids as the frustration index of all the residues:
is the energy of the k-th decoy and N is the number of decoys. Given that the AWSME Hamiltonian computes energy based on the neighboring particles, the energy of the system is an addition of local behavior. Consequently, the frustration profile highlights energy changes in the local environment of the system. Based on this definition, a higher frustration index indicates that i-th residue of the variant is among the amino acids that result in the lowest energy (highest local stability). We refer to the frustration profile of a variant with L amino acids as the frustration index of all the residues:
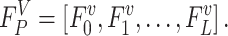 |
To quantify the change in local stability of the variant sequence relative to the WT sequence, we compute the difference between frustration profile of the variant and the WT sequence (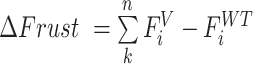 ).
).
Results
High-throughput screen quantifies relative integration activity and specificity
We developed a dual screen in E. coli that can characterize integration activity and specificity of a large pool of transposon variants (Fig. 1B). The dual screen can measure all genomic integration events (on- and off-target) or on-target integration events, using positive selection under different conditions. Briefly, we generated a recipient E. coli strain (cJP003) containing a toxin gene (ccdB) under the control of an arabinose-inducible promoter (pBAD) [27]. Transposon variants are expressed from a plasmid (pHelper), encoding Cas12k, TnsB, TnsC, and TniQ, along with a gRNA targeting the ccdB gene. A non-replicating donor plasmid (pDonor) contains transposon ends and a kanamycin resistance gene (kanR) as cargo (Fig. 1B). Bacteria are transformed with both pHelper and pDonor and plated on kanamycin (Kan) or kanamycin and arabinose (Kan/Ara) plates. To minimize the probability of multiple integration events per cell, we used a low multiplicity of transformation for pDonor (see the “Materials and methods” section for details). Cell growth on Kan reports on all genomic integration events (activity screen), while growth on Kan/Ara requires on-target integration events where the ccdB gene is disrupted (specificity screen).
To quantify activity and specificity of individual variants in a pool, we defined activity and specificity scores using the read counts from NGS. Generally, these scores are defined as the log2 fold change in the fraction (f) of each variant (v) after the screen (fv,after) compared to before (fv,before), normalized with respect to a chosen baseline. This baseline serves to define a reference point for comparison, which we specify in the relevant sections.
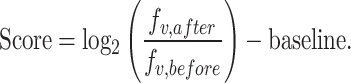 |
To assess the overall integration activity (both on- and off-target integration), we defined the activity score as shown below. fv,kan represents the frequency of the variant from the kanamycin screen (Kan), whereas fv,input represents the frequency of the variant from the input library prior to screening.
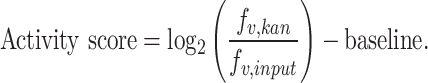 |
To address confounding effects of integration activity and specificity, we defined specificity scores as log2-enrichment of the variant's frequency from Kan/Ara screen (fv,kan+ara) over the frequency from Kan (fv,kan), as shown below. This normalizes the impact of changes in integration activity, allowing a direct evaluation of the integration specificity of each variant.
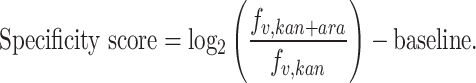 |
Analyzing variants in this two-dimensional space of activity and specificity allows us to identify variants with different functional properties. For example, a promiscuous variant with increased off-target activity can be readily identified by its high activity score, but negative specificity score. On the other hand, a desirable variant with improved on-target activity will have high scores for both activity and specificity.
As a proof of principle, we tested whether our dual screen can reliably quantify the relative activity and specificity of a small pooled library consisting of WT V-K CAST proteins and 108 different gRNAs: 99 targeting lacZ, 8 targeting ccdB, and 1 non-targeting (NT) as a negative control. Here, baseline corresponds to the average activity of the gRNA pool. Both activity and specificity scores were highly reproducible between the two biological replicates (Pearson correlation r = 0.99 and 0.90, Supplementary Fig. S1A and B), supporting the robustness of our dual screen. We found that activity scores have a wide dynamic range (from −5 to +3) (Fig. 1C). We also measured integration activity for a subset of the gRNAs using a well-established mate-in assay [29] (see the “Materials and methods” section). In this assay, a non-replicable pDonor carrying the KanR gene is delivered via bacterial conjugation, and the resulting number of colonies serves as a direct measure of integration activity. Here, we found a strong correlation between colony counts and activity scores (r = 0.82) (Fig. 1D), validating our pooled screen. Some gRNAs have little to no activity, because measured enrichment scores are close to that of the non-targeting gRNA (activity scores <−4 Fig. 1C), which represents the basal activity of the off-target pathway. Although activity variation across gRNA spacers has been observed previously [8], the mechanistic basis of their activity difference remains elusive. The results obtained here indicate that our dual screen correlates with measurements obtained from standard tests (i.e. mate-in transposition assays); however, the limited size of the gRNA library and corresponding target sites is not comprehensive enough to identify a clear sequence pattern that explains the activity differences observed here.
As expected, activity scores from Kan/Ara selection (ccdB-activity score) were negative across all lacZ‐targeting gRNAs, while most of the ccdB-targeting gRNAs were enriched (6 out of 8, Supplementary Fig. S1C). Specificity scores for ccdB target (ccdB-specificity score) were negative across all lacZ-targeting gRNAs and positive in all the ccdB-targeting gRNAs (Supplementary Fig. S1D). Notably, two ccdB-targeting gRNAs with lowest activity scores (ccdB-02 and ccdB-08) showed positive specificity scores, confirming that our assay measures on-target integration preference even for variants with low overall activity. To quantitatively characterize targeting behavior, we conducted unbiased profiling of integration sites under kanamycin conditions for four selected ccdB-targeting gRNAs, sampling a wide range of activity scores from −4.2 to 1.8. This profiling uses Tn5-based tagmentation and NGS to directly measure the proportion of ccdB-targeted integrations relative to all observed integrations [17]. Three of the four tested gRNAs showed remarkably high specificity, with ≥88% on-target integration (Fig. 1E), which is in contrast with the widely reported promiscuity of the same V-K system [8, 14, 17].
We reasoned that the discrepancy between our observations and previous targeting specificity measurements could be due to differences in donor DNA availability. Our screen used the non-replicable pDonor to minimize multiple transposition events per cell, whereas prior studies often used replicating donor plasmids. A replicating pDonor can persist in cells, conferring kanamycin resistance without requiring a genomic integration event. Consistent with this, when we subcloned the donor DNA into a replicable vector, on-target integration percentage substantially decreased (8%–49%, Fig. 1E), confirming that the preference for the RNA-guided target site strongly depends on the availability of the pDonor.
Integration frequency assessed using ddPCR was overall consistent. As expected, in the non-replicable pDonor condition, 93%–100% of the RNA-guided target sites showed an integration event (Supplementary Fig. S2). In contrast, the replicable pDonor condition resulted in integration at only 26% of target sites (Supplementary Fig. S2). This suggests that even with readily available pDonor, most RNA-guided target sites were not used for integration, while pervasive off-site integrations were detected. This complex behavior likely stems from a combination of factors, including target site immunity [40] and cell-state-dependent activity changes in the replicable pDonor condition. Although the detailed kinetics and the mechanism of these target site selection behaviors remain unclear, we were interested to find that using a non-replicable pDonor clearly yields promising integration accuracy. These results also suggest that the previously reported promiscuity of V-K CASTs may have been a consequence of experimental conditions and that they can have higher targeting specificity under limited donor DNA availability.
Transposase mutations can substantially improve integration activity without impacting specificity
With our screen validated, we investigated how mutations in core transposition components (TnsB, TnsC, and TniQ) would alter activity and specificity using a gRNA (ccdB-01) that exhibits close to average activity levels (asterisk, Fig. 1C). We began with a small-scale screen with variants of the transposase TnsB (Supplementary Fig. S3A), as we hypothesized that stabilizing STC interfaces could enhance catalytic activity without affecting integration specificity. We defined 7 TnsB segments that are involved in protein–protein interactions and DNA binding, based on existing structures (Fig. 2A and Supplementary Fig. S3B) and spanning 95 residues in total (Supplementary Table S1). We built a site-saturation variant library (see the “Materials and methods” section) that included all possible single mutations for those residues and stop codons, and subjected it to our screen. We observed a very strong correlation between activity scores obtained from two independent biological replicates (r = 0.98, Fig. 2B), indicating robust reproducibility. We assessed activity and specificity scores for each variant with respect to WT values; thus, WT score = 0. We found that activity of the TnsB variants ranged from strongly depleted (−6) to strongly enriched (+2) and overall clustered into two general regions: neutral effect on integration activity (between −1 and +1) or strongly deleterious (<−4) (Fig. 2C). Specificity scores for a minor subset (27.3% of variants) could not be obtained due to their decreased activity and therefore low NGS read counts after Kan selection. Nevertheless, consistent with our initial hypothesis, single mutations on TnsB did not have a significant impact on integration specificity—most specificity scores were between −1 and +1 with poor correlation between replicates (r= 0.18, Fig. 2D). Thus, consistent with our model of V-K CAST transposition [10], we do not observe a correlation between the integration activity and specificity in the TnsB single mutation library (Fig. 2E).
Figure 2.

Transposase mutations improve integration activity with little impact on specificity. (A) The oligomeric assembly of TnsB STC (left) and its atomic model (PDB: 8RDU) (right) are shown in surface representation, with nucleic acids depicted in cartoon representation. Each TnsB subunit is numbered and shown in a different color; target DNA is in blue, donor DNA in green, and the strand transfer site is in red. The screened loop regions are highlighted with boxes and labeled with the panels (F–J) where they are shown in detail. (B) Activity scores from two biological replicates are shown as scatter plots. The red dotted line represents the fit obtained from linear regression. n = number of observed variants; r = Pearson coefficient. (C) The frequency (y-axis) is plotted as a function of the activity scores (x-axis). (D) Scatter plot of specificity scores from two biological replicates; numbers in inset shown are the same as in panel (B). (E) Scatter plot of activity (x-axis) versus specificity scores (y-axis). (F–J) Heatmaps of activity scores for the screened TnsB loop regions are shown on the left, while the structural context of each region is depicted on the right. Each mutation (y-axis) at each residue position (x-axis) for each heatmap is represented by a color scale based on enrichment scores. * indicates stop codon. The color scale common to all heatmaps is shown on panel (F), on the left. Blue indicates negative scores, red indicates positive scores, and white indicates scores near 0. WT amino acid is indicated with a dot, and gray boxes indicate data were excluded due to insufficient read counts or absence from the input library. The maximum activity score at each screened residue is mapped onto the structure using the same color scale. The position and WT amino acid labels residues that are mentioned in the main text. Interactive versions of the heatmaps, scatter plots, and raw data are provided in supplementary materials.
We analyzed the mutations based on their location and function. As expected, DNA binding residues were critical for transposase activity. For example, W178 (Fig. 2F) and R380 (Supplementary Fig. S3D), previously identified as critical for transposition [19], did not tolerate any other substitution (Supplementary Table S2). In the DNA binding loop spanning residues 96–105, R99 and R106 directly interact with DNA bases (Fig. 2G) [19, 41] and were strictly preferred. D101 was also strictly preferred; while it does not contact DNA directly, D101 forms stabilizing hydrogen bonding interactions with R106 (Fig. 2G), suggesting an indirect role in stabilizing TnsB–DNA interactions. On the other hand, we did not find amino acid preferences at TnsB-TnsB interfaces in the tested regions. For example, substitutions in residues at TnsB1–TnsB2 interface (Fig. 2H) showed minimal impact on activity, which is consistent with that interface being reliant on backbone interactions. Residues in other protein–protein interface regions exhibited similar trends, with the exception of DNA-binding residues such as R223 and R380 (Supplementary Fig. S3C and D).
We identified three TnsB mutants with substantially improved activity scores (between 1.8 and 2.3) and no impact on specificity scores (Supplementary Fig. S3E): P131K (activity score: 1.9 ± 0.6, specificity score: 0.0 ± 0.5, Fig. 2I and Supplementary Fig. S3E), L183M (1.8 ± 0.5, 0.0 ± 0.5, Fig. 2F and Supplementary Fig. S3E), and R355C (2.3 ± 0.6, 0.0 ± 0.6, Fig. 2J and Supplementary Fig. S3E). Each of these substitutions would have a stabilizing effect on the protein structure. L183M fills a hydrophobic pocket (Fig. 2F), while P131 is located at the beginning of an α-helix and proximal to donor DNA (Fig. 2I), so substitution with a lysine would simultaneously relieve backbone strain and enhance TnsB–DNA interactions. For R355, we observed enhanced activity for several variants (Fig. 2J), suggesting that the WT residue is non-optimal; the substitutions with highest activity scores are small hydrophobic residues (R355C: 2.3 ± 0.6, R355I: 1.3 ± 0.8, and R355V: 1.7 ± 0.5, Fig. 2J and Supplementary Fig. S3E) that may stabilize TnsB through improved hydrophobic packing. Taken together, our dual screen successfully identified promising variants from the limited sampling of 95 out of the 584 positions (16%) in TnsB.
TnsC sequence is constrained by its multiple functional roles during transposition
The 276-residue AAA+ protein TnsC forms a filament on target DNA in an ATP-dependent manner, which is required for V-K CAST transposition and target-site selection [40, 42] (Fig. 1A). In addition, its interactions with target DNA and interfaces with both TnsB and TniQ are also critical for RNA-guided DNA integration [10, 40]. Given TnsC’s multiple interactions and functional requirements, we constructed a deep mutational scanning library spanning the full length of the protein using the same approach used for TnsB. Activity and specificity scores from two independent biological replicates are well correlated (r= 0.97 and 0.92, respectively) (Fig. 3A and B). Both scores ranged between −6 and +1 for both screens (Fig. 3C and D), indicating that no single point mutation considerably enhanced activity or specificity.
Figure 3.

Deep mutational scanning of TnsC identifies distinct regions important for integration activity and specificity. (A) Scatter plots of activity and (B) specificity scores from two biological replicates (x-axis and y-axis). n = number of observed variants; r = Pearson coefficient. The red dotted line represents line obtained from linear regression. (C) Frequency (y-axis) is plotted as a function of activity scores (x-axis) and (D) specificity scores (x-axis). (E) Heatmaps of activity and (F) specificity scores. Each mutation, including stop codon (*) and deletion (Δ), is depicted on the y-axis. Residue position is indicated on the x-axis. WT amino acid is indicated as a dot, and gray boxes indicate data that were excluded due to insufficient read counts or absence from the library. Right, color scale indicates enrichment score. Positive values are red, neutral values are white, and negative values are blue. Conservation scores at each position are shown above each heatmap as a line plot. Colored bars under the line plot show positions categorized according to function, as depicted in panel (G). Black arrows under line plot in panel (E) mark residues in the N-terminus of TnsC mentioned in the main text. (G) TnsC hexamer (PDB 8RDU) is shown in surface representation from the TniQ-interacting face, colored white. A single TnsC protomer (shown for reference) is colored gray. Target DNA is colored white and shown in cartoon representation. ATP is shown in spacefill and colored red. Categories are colored on the surface and defined as follows: TnsC–TnsC interface (green), TnsB–TnsC interface (purple), TniQ–TnsC interface (orange), target DNA binding (blue), or ATP binding (red). (H) Frequency of activity scores for the interface residues of TnsC. Color scheme is same as panel (G). (I) Scatter plot shows correlation between activity and specificity scores for TnsC single-mutation variants. Groups mentioned in the main text are highlighted: group 1 (blue), group 2 (purple), and group 3 (orange). (J) Frequency of specificity scores for the categorized interface residues of TnsC. Each line on the density plot represents a different category, with the color scheme defined in panel (G). Interactive versions of the heatmaps, scatter plots, and raw data are provided in supplementary materials.
Systematic mutational analysis of TnsC identified key regions where substitutions substantially impact integration activity and/or specificity (Fig. 3E and F). For integration activity, we observed strong depletion across all substitutions for select residues (A4, I7, A8, L11, G12, and D17) in the N-terminus of TnsC (black arrows, Fig. 3E). These residues are predicted to form an α-helix yet are not visualized in prior structures (Supplementary Fig. S4A) [10, 40, 42]. Even though these residues are not ordered in the structure, our results point to an important functional role for this segment. Other strongly depleted mutations were found in the residues buried within the hydrophobic core of TnsC, which is expected for positions involved in the protein fold maintenance (Supplementary Fig. S4B).
We systematically examined the mutations based on their location and function (Fig. 3G and Supplementary Table S3): TnsC–TnsC interface, ATP binding, target DNA binding, TniQ interacting, and TnsB interacting. For residues in the target DNA binding (blue), TnsB interacting (purple), and TniQ interacting (orange) categories, the distribution of activity scores resembles that of all TnsC variants (compare Fig. 3C–H), suggesting that these positions do not experience stronger selection pressure to retain the WT residue compared to the rest of the protein. In contrast, for positions involved in ATP binding (red) and the TnsC–TnsC interface (green), the distribution of activity scores skews toward strongly depleting values (Fig. 3C and H), indicating the mutational intolerance of these positions.
Our results are overall consistent with previously reported TnsC mutants, with two exceptions, G84A (activity score: −0.1 ± 0.7) and R85A (−0.7 ± 0.7), which were previously reported to improve integration activity but showed neutral impact in our screen (Supplementary Table S4) [11]. Mutations in TnsC could also substantially change specificity scores, consistent with the critical role of this protein in target-site selection [40]. We observed a particularly strong depletion pattern of specificity scores for positions 103–104 and 120–124 (Fig. 3F), which are located in two target DNA-interacting loops. These results are in agreement with our previous observation that TnsC double mutant K103A + T121A had significantly decreased specificity [40].
The absence of mutants with substantially higher activity and/or specificity suggests that the WT TnsC is constrained in sequence, possibly due to its many functional requirements. Plotting activity scores versus specificity scores for all mutations revealed a complex relationship between activity and specificity in TnsC (Fig. 3I). Mutations can be categorized into three distinct groups based on their effects: (i) strongly depleted activity and near-WT specificity (blue highlight); (ii) near-WT activity and specificity (purple highlight); and (iii) near-WT activity and strongly depleted specificity (orange highlight). Mutations in groups 1 and 2 can be explained by their locations and functional roles, as described earlier. In contrast, mutations in group 3 appear to be mostly associated with target DNA binding (Fig. 3J and Supplementary Fig. S5) or, to a lesser extent, with ATP binding or the TnsC–TnsC interface (Supplementary Fig. S5).
Thus, our screen and analyses were able to identify separation-of-function mutants in TnsC and reveal the contributions of its different functional regions to integration activity and specificity. Domains associated with ATP binding and TnsC oligomerization are required for both integration activity and specificity. Domains associated with target DNA binding appear devoted to targeting specificity, indicating that those residues may serve as on-target sensors. Finally, these results suggest that screens focused solely on optimizing overall activity may have unintended consequences on integration specificity, potentially resulting in active but promiscuous transposon variants.
TniQ mutagenesis uncovers variants with improved activity or specificity
Given TniQ’s small size (167 residues), we used a PCR-based mutagenesis method [31] to generate higher mutational complexity and a richer mutational landscape (see the “Materials and methods” section). The resulting library contained ∼10 000 protein variants, covering 3281 out of the 3320 possible single mutations, 1701 double mutations, and 97 triple mutations. Activity and specificity scores were highly correlated across biological replicates (r = 0.86 and 0.92, respectively, Fig. 4A and B), with a larger dynamic range compared to the previous saturation mutagenesis libraries: −8 to +4 (activity) and −10 to +2 (specificity). Furthermore, individual tests of select variants—W10A (–6.7 ± 0.4), G89E (3.7 ± 1.5), C103G (–0.2 ± 2.1), and W120A (–3.6 ± 1.1)—confirmed the robustness of the scores obtained from our screen (Supplementary Fig. S6A and B).
Figure 4.

TniQ mutagenesis reveals a rich landscape and opportunities to improve integration activity with multiple mutations. (A) Activity and (B) specificity scores from two biological replicates are shown as scatter plots. Each dot on the scatter plot represents an individual TniQ variant, with different colors indicating single (black), double (blue), or triple mutants (red). n = number of observed variants; r = Pearson coefficient. Red dotted line indicates linear regression fit. (C) TniQ interactions with Zn (cyan), TnsC (green), and nucleic acids (yellow) are functionally categorized and highlighted on the structure (PDB: 8RDU). TnsC promoters (shades of green) are numbered starting from the one closest to TniQ (orange). tsDNA indicates target-strand DNA (blue), and sgRNA is colored gray. Regions for the close-up view are indicated with numbered boxes. Insets depicting structural details are shown to the right; dotted lines indicate strong non-covalent interactions. Zinc ion is shown as a pink sphere, and positions shown in sticks are colored according to their functional groups: carbon (orange), oxygen (red), nitrogen (blue), and sulfur (yellow). Heatmaps of (D) activity and (E) specificity scores of TniQ single mutants. Each mutation (y-axis) at each residue (x-axis) is represented by a color scale based on enrichment scores. Stop codon is labeled with an asterisk (*). Red colors indicate positive scores, white indicates near 0, and blue indicates negative scores. WT amino acid is indicated with a dot, and gray boxes indicate data that were excluded due to insufficient read counts or absence from the library. Conservation scores at each position are also shown at the top, with the color scheme defined in panel (C). In panel (D), W10 and N33 are indicated by black arrows. (F) Scatter plot shows a correlation between activity and specificity scores of TniQ mutants. Variants with both activity and specificity score values are plotted. Interactive versions of the heatmaps, scatter plots, and raw data are provided in supplementary materials.
In general, the landscape of TniQ activity scores conforms to our expectations. We observed strict preference for the eight cysteine residues in zinc finger domain (ZnF) that coordinate two zinc ions in the C-terminal portion of TniQ (Fig. 4C and D and Supplementary Table S5), consistent with their structural role. There was also strict preference for residues in the TniQ–TnsC interface (Fig. 4C), such as W10 and N33 (black arrows in Fig. 4D). The result was also consistent with previously reported TniQ mutants, with some exceptions, such as W120A and P60A, which were previously reported to have no effect on integration activity but were depleted in our activity screen (Supplementary Table S6) [11, 18].
We used two-dimensional plot of activity and specificity score to identify enhanced TniQ variants (Fig. 4F). While we did not identify mutations that substantially improved both activity and specificity, there were several outliers that improved either activity or specificity (score >1), with no clear pattern for their locations on the protein. Some mutations improved one aspect at the expense of the other. For example, I129M substantially boosts activity score (2.0 ± 0.3) but compromises specificity score (−4.2 ± 0.9), whereas M92K increases specificity (1.6 ± 0.6) while strongly decreasing activity (−3.0 ± 0.7) (Fig. 4D and E). Other mutations improved in one aspect without compromising the other: A102D and Q115K improve specificity (1.8 ± 0.2 and 2.0 ± 0.6, respectively) (Fig. 4E) with no appreciable effect on activity (−0.7 ± 0.2 and −0.9 ± 0.1, respectively). G89E and A88D substantially increase activity scores (3.7 ± 1.5 and 1.2 ± 0.2, respectively) without changing specificity (−0.1 ± 1.3 and −0.2 ± 0.9, respectively). The vastly different effects of the point mutations highlighted here illustrate the importance of considering each mutation on a case-by-case basis.
Plotting activity and specificity scores for single, double, and triple mutations in TniQ (Fig. 4F) highlighted the potential to improve activity via multiple mutations. Most single mutants that substantially increase activity without compromising specificity were found at G89, a linker residue that connects the N- and C-terminal domains of TniQ [40]. Double mutants were far more likely to substantially increase activity compared to single mutants, but increases in activity were often accompanied by decreases in specificity. Indeed, the variants with the highest activity scores (>2) typically exhibited poor specificity scores (<−2). Most double mutants with enhanced activity (and reduced specificity) included substitutions in positions 90 and 96 (see supplementary materials). This enrichment is likely due to non-uniform sampling of the double-mutant set. Unfortunately, we did not obtain a sufficient subset of triple mutants (n= 3) to observe their effects on both TniQ activity and specificity (Fig. 4F). Nevertheless, these results suggest that synergistic effects of higher-order mutants will compound and are worth exploring more comprehensively in future work.
Mutations resulting in higher local stability are associated with more active variants
A consistent thread throughout our observations here is that mutations that increase integration activity may do so by stabilizing the integration complex. To test this hypothesis, we used a statistical mechanics-based method called frustration analysis [38, 39], which measures the local stability around each amino acid based on its interactions with its neighbors. We computed the frustration profile (frustration indexes for all positions) across all mutants from TnsB, TnsC, and TniQ screens by threading their sequences onto the WT structure (PDB: 8RDU) using the Frustratometer (see the “Materials and methods” section) [39]. As an example, we calculated the frustration profile of TniQ W10K, one of the most strongly depleted variants in the activity screen (activity scores = −7.9 ± 0.1). The W10K variant has a lower frustration index for residues around position 10 (blue line, Fig. 5A) than WT (black line), a signature of lower local stability. These observations are consistent with the idea that the reduced local stability negatively impacts integration activity.
Figure 5.

Local stability estimates are correlated with activity scores. (A) Representative frustration profile of the TniQ WT sequence (black) and TniQ W10K variant (blue), shown for a subset of residues from positions 3 through 30 (x-axis). A lower frustration index indicates reduced local stability. Aggregated changes in frustration index (ΔFrust) of beneficial (red) and deleterious (blue) mutants relative to WT in (B) TnsB, (C) TnsC, and (D) TniQ are shown as violin plots. A positive ΔFrust indicates an increase in local stability. n is determined by the number of variants with activity scores >0.5. The top and bottom n variants were then selected and analyzed. P-values between two groups are indicated by asterisks. ** P < .01; ***** P < .00001; n.s. = not significant. Statistical analysis was performed using a two-tailed Student’s t-test.
We next computed the difference in frustration (ΔFrust) between mutant and WT sequences by summing the differences in frustration profiles. ΔFrust is positive if the mutant is more stable than WT or negative if the mutant is less stable. We classified variants from each screen with activity scores >0.5 as beneficial mutants and compared the average ΔFrust to the same number of variants with the worst activity scores (classified as deleterious mutants). For TnsB (Fig. 5B) and TniQ (Fig. 5D), the most deleterious variants showed, on average, decreased stability relative to WT; conversely, the most beneficial variants tended to exhibit increased or neutral stability. In contrast, the frustration profiles for TnsC mutants revealed no significant trends associated with activity scores (Fig. 5C). This might be because of the many functional constraints of TnsC and its interaction with DNA and ATP, which are not taken into account in the frustration analysis.
These observations also support our hypothesis that further stabilizing the integration complex would increase the integration activity. This trend was most prominent in TniQ (P-value < 1e−6) (Fig. 5D), potentially because the TniQ library was more comprehensive than those for TnsB or TnsC. The inclusion of synonymous codon substitutions in the TniQ library likely contributed to more robust activity score estimates. However, our findings could also be indicative of an intrinsic feature of TniQ: considering its low conservation and its dynamic mutational landscape [35], it is also plausible that TniQ is highly malleable in the face of evolutionary pressure.
Combining mutations identified through screening results in significantly improved CAST variants
We investigated whether mutations identified in our screen could be stacked to achieve synergistic effects. We selected and tested the top-performing TnsB and TniQ variants that enhanced integration activity without compromising specificity (Supplementary Fig. S6C). In the mate-in assay, identified TnsB variants (P131K, L183M, and R355C) had slightly higher integration activity than WT. However, combining these mutations generally resulted in significantly improved activity over both single mutants and WT. The best combination, P131K + R355C ( +
+ ), showed a four-fold improvement in integration activity (Fig. 6A), and the best TniQ variant, G89E, also showed approximately two-fold increase in activity compared to WT (Fig. 6B).
), showed a four-fold improvement in integration activity (Fig. 6A), and the best TniQ variant, G89E, also showed approximately two-fold increase in activity compared to WT (Fig. 6B).
Figure 6.

Mutant combinations improve overall activity without compromising specificity. Mate-in transposition assays were performed to individually measure the integration activity of (A) TnsB variants (purple) and (B) TniQ variants (orange), identified to enhance the activity in the screen. Each mutant is numbered and referred to by its number in subsequent panels. Bars with black diamonds indicate colony counts from Kan selection, while red diamonds indicate colony counts from Kan/Ara selection. P-values for the best TnsB and TniQ variants were calculated by comparing them to WT under Kan (black bracket) or Kan/Ara selection (red bracket) and are indicated by asterisks. (C) Bar plots of on-target integration ratio, as determined by unbiased NGS profiling. The combination of TnsB and TniQ variants is in burgundy. On-target ratio values are shown above each bar. (D) The best TnsB mutant pair (P131K + R355C, +) and the TniQ mutant (G89E,  ) were combined and tested for integration activity. The statistical analysis indicates that the difference in activity between the TnsB pair alone and the combined TnsB + TniQ variant is not significant (n.s.), suggesting no additive effect. All data shown represent the mean ± standard error; n= 3 for each bar. WT, wild-type pHelper; NT, WT pHelper with a non-targeting gRNA; ** P < .01; *** P < .001. Statistical analysis was performed using a two-tailed Student’s t-test.
) were combined and tested for integration activity. The statistical analysis indicates that the difference in activity between the TnsB pair alone and the combined TnsB + TniQ variant is not significant (n.s.), suggesting no additive effect. All data shown represent the mean ± standard error; n= 3 for each bar. WT, wild-type pHelper; NT, WT pHelper with a non-targeting gRNA; ** P < .01; *** P < .001. Statistical analysis was performed using a two-tailed Student’s t-test.
To assess the integration specificity of these enhanced variants, we next profiled their integration target sites using the non-replicable pDonor (Fig. 6C). We found that the selected TnsB and TniQ variants did not compromise integration specificity but instead substantially increased it compared to the WT. TnsB variants P131K and R355C individually increased on-target percentage to 96.9% and 96.4%. Remarkably, combining these two mutations further enhanced specificity, achieving near-perfect on-target integration of 99.7%. The TniQ G89E variant also showed enhanced on-target specificity of 90.7%. Notably, this trend held true under a more permissive condition with a replicable pDonor (Supplementary Fig. S6D), where all the enhanced variants maintained or improved their on-target accuracy. These results highlight the effectiveness of our pipeline that considers both activity and specificity scores when selecting mutations to combine.
Next, we tested whether the top-performing variants in different transposon components could be combined to further enhance activity. The combination of the TniQ G89E and the best TnsB mutation pair (P131K + R355C) still improved activity compared to WT, without compromising specificity (Fig. 6D and Supplementary Fig. S6D). However, this stacked variant did not outperform the TnsB pair alone. Several factors may explain this finding. First, the screen measures relative changes within a variant pool, which can be influenced by both the mutational landscape of the target and the library size. Therefore, enrichment scores obtained from different screens may not be directly comparable on the same absolute scale (i.e. an enrichment score of +2 from the TniQ screen may not be equivalent to the score of +2 from the TnsB screen). Furthermore, it is often inaccurate to assume that single-point mutations would be purely additive, as epistatic interactions can lead to unpredictable functional outcomes [43]. However, it is curious that mutations in components that are physically separated would not improve overall activity when combined. Clearly, more work here is needed to investigate the consequences of sampling combinations of mutations across multi-component systems. Overall, we were encouraged to find that the screen identifies mutations that, when combined, improve the activity of V-K CAST without compromising targeting specificity. This also indicates that a more systematic profiling of mutant combinations is likely to yield significantly more active variants that would be useful for many downstream engineering applications.
Discussion
CASTs are known for their general programmability and coordinated, one-step integration mechanism [7]. These desirable attributes are offset by complications associated with delivering large, multi-gene systems to the appropriate genomic site for donor DNA integration. Despite extensive bioinformatics mining of genomic and metagenomic data, CAST diversity is limited, with only a handful of subtypes and varying levels of integration activity and targeting specificity [35, 44]. This stands in stark contrast to the extensive diversity of CRISPR nucleases [45], which provides numerous candidates for developing novel genome-editing tools.
The limited efficiency of CASTs in heterologous systems such as human cells has motivated several engineering attempts [15, 16, 46]. Typical workflows seek to optimize integration efficiency and do not explicitly take into account targeting specificity. To explore the implications of this strategy and to investigate how activity and specificity are related, we developed a dual genetic screen and conducted the first (to our knowledge) comprehensive and systematic profiling of the core transposition components conserved across all CAST systems: transposase TnsB, AAA+ protein TnsC, and target-site-associated protein TniQ. We find no general relationship between activity and specificity that applies across the entire system, since each transposon component had a distinct mutational and functional landscape, consistent with their roles during transposition. As the catalytic component, TnsB mutagenesis primarily affects activity, with no significant impact on specificity. Given the limited sampling in the TnsB screen, more comprehensive screens of TnsB may offer deeper and more promising insights. In contrast, TnsC is known to control target site selection, and indeed its mutagenesis identifies regions that are associated with either activity or specificity. In particular, mutations in TnsC residues involved in target DNA binding have a strong anti-correlation between activity and specificity, with several variants significantly enhancing activity at the expense of targeting specificity. These findings demarcate clear functional roles associated with integration activity and specificity in TnsC, consistent with our prior work [40]. Finally, we reveal a rich activity-specificity landscape for TniQ, which we believe is partially due to larger library complexity, allowing us to probe the effect of multiple mutations. At the same time, analysis of single mutations identified TniQ variants with increased activity without compromising specificity. Computational estimates of local stability (based on structural information) suggest that integration complex stability correlates with enrichment/depletion trends.
The sparseness of mutations with increased activity and specificity suggests that the underlying functional landscapes for both properties are rugged, with sharp local maxima, consistent with existing theories of protein sequence-function landscapes [47, 48]. Nevertheless, we were able to identify single mutations in TnsB and TniQ that increase the integration activity without compromising targeting specificity, and stacking beneficial TnsB mutations increased the integration activity by up to four-fold compared to WT TnsB. These findings suggest that exploring larger mutational steps in sequence space could result in substantial activity gains with no impact on targeting. The potential of this strategy is strongly supported by a recent directed evolution study on type I-F TnsB in E. coli, which substantially increased activity in human cells and reduced dependency on a bacterial host factor [49]. This highlights that findings from bacterial screens, like the one described here, can be effectively translated into future therapeutic applications.
While promising, systematically sampling even double mutant variants across the entire CAST system remains infeasible for any existing screen because of the combinatorial explosion of sequence space from the four transposon-encoded proteins. To circumvent this limitation, activity and specificity scores obtained from our dual screen could be used as input for training recently developed ML-based protein engineering pipelines. These approaches have been shown to be more efficient in sampling sequences than purely experimental directed evolution approaches [50]. In addition to the directed evolution of CASTs, our dual genetic screen presented here is broadly applicable to rational design approaches on CASTs, such as protein fusions or de novo design. Furthermore, this screen is generalizable beyond CAST engineering and could be extended to any site-specific integrase. For example, the screen could be used to modify and assess targeting behavior of retroelements [51] or large serine recombinases [52], systems that integrate into defined genomic locations with high specificity. Therefore, we anticipate that our screen will be generally useful, both for understanding complex integration behavior and for engineering novel site-specific transposons.
Supplementary Material
Acknowledgements
The authors would like to thank Ines Chen for providing valuable feedback on the manuscript, Adam Smiley for assisting with analysis, the staff at the Hartwell Center of St. Jude Children’s Research Hospital for conducting NGS, and the members of the Kellogg lab for their insightful discussions. We also appreciate Joseph E. Peters and Shan-Chi Hsieh for sharing reagents and discussions, and the members of David Savage’s lab for their advice on the library barcoding strategy.
Author contributions: Seong Guk Park (Data curation [lead], Formal analysis [lead], Investigation [lead], Methodology [lead], Validation [lead], Visualization [lead], Writing—original draft [lead], Writing—review & editing [lead]), Jung-Un Park (Conceptualization [lead], Data curation [lead], Formal analysis [lead], Investigation [lead], Methodology [lead], Validation [lead], Visualization [lead], Writing—original draft [lead], Writing—review & editing [lead]), Esteban Dodero-Rojas (Data curation [supporting], Formal analysis [supporting], Investigation [supporting], Methodology [supporting], Software [supporting], Writing—review & editing [supporting]), John A. Bryant Jr (Investigation [supporting], Writing—review & editing [supporting]), Geetha Sankaranarayanan (Investigation [supporting], Writing—review & editing [supporting]), and Elizabeth H. Kellogg (Conceptualization [lead], Data curation [lead], Formal analysis [lead], Funding acquisition [lead], Project administration [lead], Resources [lead], Supervision [lead], Validation [lead], Writing—original draft [lead], Writing—review & editing [lead]).
Contributor Information
Seong Guk Park, Department of Structural Biology, St. Jude Children’s Research Hospital, Memphis, TN 38105, United States.
Jung-Un Park, Department of Structural Biology, St. Jude Children’s Research Hospital, Memphis, TN 38105, United States; Innovative Genomics Institute, University of California, Berkeley, CA 94720-5230, United States; Howard Hughes Medical Institute, University of California, Berkely, CA 94720-5230, United States.
Esteban Dodero-Rojas, Department of Structural Biology, St. Jude Children’s Research Hospital, Memphis, TN 38105, United States.
John A Bryant, Jr, Department of Structural Biology, St. Jude Children’s Research Hospital, Memphis, TN 38105, United States.
Geetha Sankaranarayanan, Department of Structural Biology, St. Jude Children’s Research Hospital, Memphis, TN 38105, United States.
Elizabeth H Kellogg, Department of Structural Biology, St. Jude Children’s Research Hospital, Memphis, TN 38105, United States.
Supplementary data
Supplementary data is available at NAR online.
Conflict of interest
None declared.
Funding
This research is supported by the Pew Charitable Trusts, NIH NIGMS 5R01GM144566-02, and the Cystic Fibrosis Foundation (E.H.K.). J.P. is an HHMI Fellow of the Jane Coffin Childs Memorial Fund. S.G.P. was supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI19C1095). This research also included experiments conducted by the Hartwell Center for Bioinformatics & Biotechnology, which is supported in part by ALSAC and the National Cancer Institute (grant P30 CA021765).
Data availability
NGS reads files are accessible on NCBI SRA (accession PRJNA1238918). The script is available at Zenodo (DOI: 10.5281/zenodo.16915585). Sequence information for plasmids and NGS primers is provided in the Supplementary Tables. Reagents and cell strains are also available from the corresponding author upon request.
References
- 1. Anzalone AV, Koblan LW, Liu DR Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020; 38:824–44. 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 2. Scully R, Panday A, Elango R et al. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019; 20:698–714. 10.1038/s41580-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anzalone AV, Gao XD, Podracky CJ et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat Biotechnol. 2022; 40:731–40. 10.1038/s41587-021-01133-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yarnall MT, Ioannidi EI, Schmitt-Ulms C et al. Drag-and-drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases. Nat Biotechnol. 2023; 41:500–12. 10.1038/s41587-022-01527-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pandey S, Gao XD, Krasnow NA et al. Efficient site-specific integration of large genes in mammalian cells via continuously evolved recombinases and prime editing. Nat Biomed Eng. 2025; 9:22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peters JE, Makarova KS, Shmakov S et al. Recruitment of CRISPR–Cas systems by Tn7-like transposons. Proc Natl Acad Sci USA. 2017; 114:E7358–66. 10.1073/pnas.1709035114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hsieh S-C, Peters JE Natural and engineered guide RNA–directed transposition with CRISPR-associated Tn7-like transposons. Annu Rev Biochem. 2024; 93:139–61. 10.1146/annurev-biochem-030122-041908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Strecker J, Ladha A, Gardner Z et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science. 2019; 365:48–53. 10.1126/science.aax9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klompe SE, Vo PL, Halpin-Healy TS et al. Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature. 2019; 571:219–25. 10.1038/s41586-019-1323-z. [DOI] [PubMed] [Google Scholar]
- 10. Park J-U, Tsai AW-L, Rizo AN et al. Structures of the holo CRISPR RNA-guided transposon integration complex. Nature. 2023; 613:775–82. 10.1038/s41586-022-05573-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tenjo-Castaño F, Sofos N, Stutzke LS et al. Conformational landscape of the type VK CRISPR-associated transposon integration assembly. Mol Cell. 2024; 84:2353–67. 10.1016/j.molcel.2024.05.005. [DOI] [PubMed] [Google Scholar]
- 12. Rubin BE, Diamond S, Cress BF et al. Species-and site-specific genome editing in complex bacterial communities. Nat Microbiol. 2022; 7:34–47. 10.1038/s41564-021-01014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gelsinger DR, Vo PLH, Klompe SE et al. Bacterial genome engineering using CRISPR RNA-guided transposases. Nat Protoc. 2024; 19:752–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vo PLH, Acree C, Smith ML et al. Unbiased profiling of CRISPR RNA-guided transposition products by long-read sequencing. Mobile DNA. 2021; 12:13. 10.1186/s13100-021-00242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lampe GD, King RT, Halpin-Healy TS et al. Targeted DNA integration in human cells without double-strand breaks using CRISPR-associated transposases. Nat Biotechnol. 2024; 42:87–98. 10.1038/s41587-023-01748-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tou CJ, Orr B, Kleinstiver BP Precise cut-and-paste DNA insertion using engineered type VK CRISPR-associated transposases. Nat Biotechnol. 2023; 41:968–79. 10.1038/s41587-022-01574-x. [DOI] [PubMed] [Google Scholar]
- 17. George JT, Acree C, Park J-U et al. Mechanism of target site selection by type VK CRISPR-associated transposases. Science. 2023; 382:eadj8543. 10.1126/science.adj8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schmitz M, Querques I, Oberli S et al. Structural basis for the assembly of the type V CRISPR-associated transposon complex. Cell. 2022; 185:4999–5010. 10.1016/j.cell.2022.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park J-U, Tsai AW-L, Chen TH et al. Mechanistic details of CRISPR-associated transposon recruitment and integration revealed by cryo-EM. Proc Natl Acad Sci USA. 2022; 119:e2202590119. 10.1073/pnas.2202590119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hew BE, Gupta S, Sato R et al. Directed evolution of hyperactive integrases for site specific insertion of transgenes. Nucleic Acids Res. 2024; 52:e64. 10.1093/nar/gkae534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sclimenti CR, Thyagarajan B, Calos MP Directed evolution of a recombinase for improved genomic integration at a native human sequence. Nucleic Acids Res. 2001; 29:5044–51. 10.1093/nar/29.24.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kovalev MA, Davletshin AI, Karpov DS Engineering Cas9: next generation of genomic editors. Appl Microbiol Biotechnol. 2024; 108:209. 10.1007/s00253-024-13056-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fanton A, Bartie LJ, Martins JQ et al. Site-specific DNA insertion into the human genome with engineered recombinases. bioRxiv3 November 2024, preprint: not peer reviewed 10.1101/2024.11.01.621560. [DOI]
- 24. Gaj T, Sirk SJ, Barbas CFIII Expanding the scope of site-specific recombinases for genetic and metabolic engineering. Biotechnol Bioeng. 2014; 111:1–15. 10.1002/bit.25096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saito M, Ladha A, Strecker J et al. Dual modes of CRISPR-associated transposon homing. Cell. 2021; 184:2441–53. 10.1016/j.cell.2021.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Datsenko KA, Wanner BL One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000; 97:6640–5. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Z, Zhao H A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Res. 2005; 33:e154. 10.1093/nar/gni148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peters JE, Craig NL Tn7: smarter than we thought. Nat Rev Mol Cell Biol. 2001; 2:806–14. 10.1038/35099006. [DOI] [PubMed] [Google Scholar]
- 29. Hsieh S-C, Peters JE Discovery and characterization of novel type ID CRISPR-guided transposons identified among diverse Tn7-like elements in cyanobacteria. Nucleic Acids Res. 2023; 51:765–82. 10.1093/nar/gkac1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johnson MS, Venkataram S, Kryazhimskiy S Best practices in designing, sequencing, and identifying random DNA barcodes. J Mol Evol. 2023; 91:263–80. 10.1007/s00239-022-10083-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bloom JD An experimentally determined evolutionary model dramatically improves phylogenetic fit. Mol Biol Evol. 2014; 31:1956–78. 10.1093/molbev/msu173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dingens AS, Haddox HK, Overbaugh J et al. Comprehensive mapping of HIV-1 escape from a broadly neutralizing antibody. Cell Host Microbe. 2017; 21:777–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rubin AF, Gelman H, Lucas N et al. A statistical framework for analyzing deep mutational scanning data. Genome Biol. 2017; 18:150. 10.1186/s13059-017-1272-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Katoh K, Misawa K, Kuma Ki et al. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002; 30:3059–66. 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Faure G, Saito M, Benler S et al. Modularity and diversity of target selectors in Tn7 transposons. Mol Cell. 2023; 83:2122–36. 10.1016/j.molcel.2023.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Crooks GE, Hon G, Chandonia J-M et al. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–90. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davtyan A, Schafer NP, Zheng W et al. AWSEM-MD: protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J Phys Chem B. 2012; 116:8494–503. 10.1021/jp212541y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bryngelson JD, Wolynes PG Spin glasses and the statistical mechanics of protein folding. Proc Natl Acad Sci USA. 1987; 84:7524–8. 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ferreiro DU, Komives EA, Wolynes PG Frustration in biomolecules. Quart Rev Biophys. 2014; 47:285–363. 10.1017/S0033583514000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park J-U, Tsai AW-L, Mehrotra E et al. Structural basis for target site selection in RNA-guided DNA transposition systems. Science. 2021; 373:768–74. 10.1126/science.abi8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tenjo-Castaño F, Sofos N, López-Méndez B et al. Structure of the TnsB transposase-DNA complex of type VK CRISPR-associated transposon. Nat Commun. 2022; 13:5792. 10.1038/s41467-022-33504-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Querques I, Schmitz M, Oberli S et al. Target site selection and remodelling by type V CRISPR-transposon systems. Nature. 2021; 599:497–502. 10.1038/s41586-021-04030-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miton CM, Tokuriki N How mutational epistasis impairs predictability in protein evolution and design. Protein Sci. 2016; 25:1260–72. 10.1002/pro.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rybarski JR, Hu K, Hill AM et al. Metagenomic discovery of CRISPR-associated transposons. Proc Natl Acad Sci USA. 2021; 118:e2112279118. 10.1073/pnas.2112279118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Makarova KS, Wolf YI, Iranzo J et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020; 18:67–83. 10.1038/s41579-019-0299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lampe GD, Liang AR, Zhang DJ et al. Structure-guided engineering of type IF CASTs for targeted gene insertion in human cells. bioRxiv19 September 2024, preprint: not peer reviewed 10.1101/2024.09.19.613948. [DOI] [PMC free article] [PubMed]
- 47. Onuchic JN, Luthey-Schulten Z, Wolynes PG Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997; 48:545–600. 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 48. Ferreiro DU, Komives EA, Wolynes PG Frustration, function and folding. Curr Opin Struct Biol. 2018; 48:68–73. 10.1016/j.sbi.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Witte IP, Lampe GD, Eitzinger S et al. Programmable gene insertion in human cells with a laboratory-evolved CRISPR-associated transposase. Science. 2025; 388:eadt5199. 10.1126/science.adt5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wittmann BJ, Johnston KE, Wu Z et al. Advances in machine learning for directed evolution. Curr Opin Struct Biol. 2021; 69:11–8. 10.1016/j.sbi.2021.01.008. [DOI] [PubMed] [Google Scholar]
- 51. Zhang X, Van Treeck B, Horton CA et al. Harnessing eukaryotic retroelement proteins for transgene insertion into human safe-harbor loci. Nat Biotechnol. 2025; 43:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smith MC Phage-encoded serine integrases and other large serine recombinases. Microbiol Spectr. 2015; 3: 10.1128/microbiolspec.mdna3-0059-2014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NGS reads files are accessible on NCBI SRA (accession PRJNA1238918). The script is available at Zenodo (DOI: 10.5281/zenodo.16915585). Sequence information for plasmids and NGS primers is provided in the Supplementary Tables. Reagents and cell strains are also available from the corresponding author upon request.